
Abstract
Secretory intestinal IgA can protect from re-infection with rotavirus (RV), but very little is known about the mechanisms that induce IgA production during intestinal virus infections. Classical dendritic cells (cDCs) in the intestine can facilitate both T cell-dependent and -independent secretory IgA. Here, we show that BATF3-dependent cDC1, but not cDC2, are critical for the optimal induction of RV-specific IgA responses in the mesenteric lymph nodes. This depends on the selective expression of the TGFβ-activating integrin αvβ8 by cDC1. In contrast, αvβ8 on cDC1 is dispensible for steady state immune homeostasis. Given that cDC2 are crucial in driving IgA during steady state but are dispensable for RV-specific IgA responses, we propose that the capacity of DC subsets to induce intestinal IgA responses reflects the context, as opposed to an intrinsic property of individual DC subsets.
Similar content being viewed by others


Introduction
Rotavirus (RV) is a double-stranded RNA virus that replicates in mature enterocytes at the tips of the small intestinal villi1,2 and was first identified by electron microscopy in intestinal biopsies of children diagnosed with acute gastroenteritis.3 It continues to be the leading cause of life-threatening diarrheal diseases among young children in the developing world.4 Intestinal RV-specific IgA is the major correlate of protection against natural infection,5,6 but the mechanisms inducing protective RV-specific IgA are not well understood. RV-specific secretory IgA arises as early as three days after primary infection, directly preceding RV-clearance.6 This rapid response suggests at least some T-cell independent B cell activation, an idea supported by the fact that α/β TCR deficient or nude mice lacking all T cells shed much less RV than RAG-deficient mice lacking both T and B cells.7 Nevertheless, most of the anti-RV response in intact animals requires T cell help.8
Classical dendritic cells (cDCs) are the major antigen-presenting cells in the body and integral to the differentiation of helper T cells. Several populations of cDCs are located throughout the intestinal lamina propria (LP), as well as within gut-associated lymphoid tissues including Peyer’s Patches and isolated lymphoid follicles. Most cDCs in the LP express the integrin αE(CD103)β7 and can be divided into two distinct populations by the expression of CD11b: Interferon regulator factor 8 (IRF8)- and Basic leucine zipper transcription factor ATF-like 3 (BATF3)-dependent CD103+CD11b− DCs (cDC1) and IRF4-dependent CD103+CD11b+ DCs (cDC2).9,10,11,12 Although both subsets of intestinal DCs are implicated in intestinal immune homeostasis via the induction and maintenance of regulatory T cells (Tregs),13 they engulf different types of antigens and differ in their ability to induce specific types of T cell responses. Thus, cDC2 are implied in supporting Th2 and Th17 responses against parasites and extracellular bacteria,14,15,16 while cDC1 excel at priming CD8+ T cell responses and Th1 responses11,17 to viral and intracellular bacteria. Importantly, cDC1 play the dominant role in cross-presenting intestinal epithelium-derived cell-associated antigen to CD8+ T cells both in steady state18 and during rotavirus infection.19
cDCs also participate in the initiation of B cell responses and several groups have documented a role for DCs in facilitating IgA production in the intestine (reviewed in20,21). However, much less is known on the division of labour between the different cDC subsets for the induction of B cell responses. IgA class-switch recombination in mice can occur via both T cell-dependent and T cell-independent pathways, with T cell-dependent IgA class-switching usually occurring within germinal centers of the mLNs and Peyer’s Patches.22,23 In contrast, T cell-independent IgA class switching depends on myeloid cells and plasmacytoid DCs (pDC) expressing BAFF and APRIL,24 potentially located in the LP itself.25,26 In any case, TGFβ is critical for both TI and TD IgA responses in vivo, as mice with defective TGFβ signalling in B cells show complete IgA deficiency.27 In addition, TGFβ and retinoic acid provided by mucosal cDCs synergize in the induction of IgA.28
TGFβ is produced in a latent form that requires conversion into an active form before it can bind to its receptor. By binding to an Arg-Gly-Asp (RGD) tripeptide in latent TGFβ, αvβ8 integrin is an important pathway for TGFβ activation in vivo, with mutation of the αv integrin-binding site in the RGD sequence of latent TGFβ recapitulating many of the phenotypes of TGFβ knockout mice.29 αvβ8 integrin on CD11c+ mononuclear phagocytes (MNPs), including DCs, is important for intestinal immune homeostasis, as CD11c-targeted αvβ8 deficiency leads to spontaneous colitis in mice.30 Previously, we and others have shown that expression of αvβ8 on CD11c+ MNPs activates TGFβ, facilitating the generation of intestinal regulatory T cells (Treg)31,32 and Th17 cells.33,34 A recent study suggests that αvβ8 on cDC2 in Peyer’s Patches is important for driving IgA responses to commensals in steady state mice,35 but it is not known whether αvβ8-mediated TGFβ activation by cDCs is required for the induction of IgA responses during infections such as RV.
In this context, protective IgA responses in the lung induced by polyinosinic:polycytidylic acid (poly(I:C)) adjuvanted vaccination against influenza virus require cDC1 in the nasal tissues.36 In contrast, cDC2 induce IgA responses to commensal bacteria in the steady state35 or after immunization with flagellin derived from Salmonella typhimurium.37 Here, we show that BATF3-dependent cDC1, but not cDC2, are required for optimal RV-specific IgA responses in the mLN of adult mice infected orally with RV. This requires TGFβ-activating αvβ8 integrin on cDC1 even though αvβ8 on cDC1 is dispensable for immune homeostasis in the absence of infection.
Results
BATF3-dependent cDC1 are required for the optimal induction of anti-RV-specific IgA responses in mLN
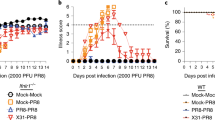
Because antibodies play a major role in protection of infants from RV-reinfection,38 we sought to explore the cellular requirements for B cell activation during primary RV infection. cDC1 efficiently sense and coordinate the immune response towards viral infections.17,18,39 The transcription factor BATF3 is essential for the development of cDC1, and BATF3-deficient mice lack both CD8α+ and CD103+ cDC1 in mLN12 and small intestinal lamina propria cDC1 in steady-state and upon RV infection (Supplementary Fig. S1A). In order to assess the role of cDC1 in inducing humoral anti-RV responses in adult mice, we infected Batf3Het and Batf3KO littermates orally with RV. As expected from a previous study,19 and in line with the prominent role of cDC1 in driving immunity towards viruses, Batf3KO mice cleared the infection with a delay of one day (Fig. 1a). This correlated with fewer total and RV-specific CD8+ T cells in the mLNs 7 days post infection (Fig. 1b) together with significantly delayed secretion of fecal RV-specific IgA (Fig. 1c). Fecal RV-specific IgA in BATF3KO mice reached wild-type levels around day 17 after infection and plateaued for at least 8 weeks (Fig. 1d).

a–c, f Batf3WT/Het and Batf3KO mice were orally infected with RV. a Levels of RV antigen shedding in feces was measured by ELISA. b The total number of total CD8+ T cells (left) and RV-specific CD8+ T cells (right) was assessed at 7 days post infection. c The levels of anti-RV IgA secreted into the feces after primary infection at indicated time-points was measured by ELISA. AUC = area under the curve, p value determined using an unpaired T test. d The levels of anti-RV IgA secreted into the feces before re-challenge at 8 weeks after primary infection (d0) and 4 days after rechallenge were measured by ELISA. e, f show kinetics for the total number of different B cells subsets accumulating in the mLNs after infection in C57Bl/6 mice (total B cells: live CD19+B220+, antigen-experienced: live CD19+B220+IgD−, plasmablasts: live CD19+IgD−CD138+) in BATF3 mice at 7 days post infection. Data pooled from a, b, d–f two (n = 4), or g six (n = 3–5) independent experiments, and c one representative experiment out of three (n = 4 mice each). a, c: unpaired T test, b, d–g: two-way ANOVA, Tukey post hoc. *p < 0.05, **p < 0.01, ***p < 0.001. White symbols: HET; black symbols: KO; shaded bars: RV-infected. AUC Area under the curve.
To explore the basis of the defective IgA levels in the absence of cDC1 early after infection, we first examined the kinetics of B cell proliferation and differentiation in wild type C57Bl/6 mice. As reported previously,40 RV infection triggered a massive temporary increase in total B cell numbers in the mLNs (Fig. 1e), with the number of naïve IgD+ B cells peaking at day 5 post infection, followed by a peak in Ag-experienced (IgD−) B cells on days 6–7. This was accompanied by significantly increased numbers of total and IgA+ CD138+ B cells at these times (Fig. 1f). Further characterization of CD138+ B cells showed that they expressed the proliferation marker Ki67 and CXCR4, indicating that they were plasmablasts (Supplementary Fig. S1B). All subsets of B cells in the mLNs returned to baseline levels by day 9 after infection. cDC1-deficient Batf3KO mice showed the same increases in the numbers of total B cells and a minor decrease in Ag-experienced (IgD−) B cells 7 days post infection (Fig. 1g), showing that cDC1 might influence B cell activation, but are dispensible for the general B cell expansion occurring in the context of RV infection.
In order to better understand the reduced levels of RV-specific IgA in the intestine of Batf3KO mice, we identified specific B cells using RV-like particles (VLPs) constructed from the viral proteins 2 and 6 bound to GFP.41 Rotavirus triggered a mild increase in the generally low abundance of IgA+ B cells in the mLNs. Among those, we detected significantly fewer RV-specific VLP+IgA+ B cells in Batf3KO mice when compared with their Batf3Het littermates (Fig. 2a and Supplementary Fig. S1C), despite not significantly decreased total IgA+ B cells. There was also a non-significant trend towards fewer VLP+IgM+ B cells (Fig. 2a), which also showed significance when assessing cumulative percentage (Supplementary Fig. S1C). In parallel, Batf3KO mice had reduced numbers of CD138+ VLP+IgA+ and to a minor extent VLP+IgM+ plasmablasts in mLN, while total IgA+ and IgM+ plasmablast were also reduced (Fig. 2b). Lower numbers of RV-specific IgA producing B cells was specific to the mLNs, as there were no differences in any B cell subsets in the Peyer’s Patches (Supplementary Fig. S1D–F). We were unable to enumerate RV-specific B cells in the small intestinal lamina propria, due to loss of staining caused by the enzymatic digestion process involved in isolating these cells. All B cell subsets in Batf3Het and Batf3KO mice expressed identical levels of the gut homing receptors CCR9 and α4β7 integrin, suggesting that differential homing to the small intestine is unlikely to explain lower RV-specific IgA secretion in the intestinal secretion of cDC1-deficient mice (Fig. 2c).

a–c Batf3Het and Batf3KO mice were orally infected with RV. a Representative flow cytometry plots for RV-VLP staining in the B220+CD138− B cells (pre-gated on CD19+IgD−) and the total number of IgA+ and IgM+ and RV-specific B cells in the mLNs. b Representative flow cytometry plots for RV-VLP staining in plasmablasts (B220−CD138+; pre-gated on CD19+IgD−) and the total number of IgA+ and IgM+ and RV-specific plasmablasts at 7 days post infection. c Representative flow cytometry plots for CCR9 and α4β7 expression on the different IgA+ plasmablasts (concatenated data from three mice, representative of two independent experiments). d huCD207-DTA and wildtype mice were orally infected with RV. The total number of IgA+ and RV-specific B cells (top) or plasmablasts at 7 days post infection. Cells were pre-gated as mentioned in a, b above. Dots represent data from individual mice. Data were collected from a, b six independent experiments, or d four independent experiments with 3–6 infected mice per experiment. Two-way ANOVA with Tukey post hoc test was performed for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001. White symbols: HET; black symbols: KO; shaded bars: RV-infected.
Anti-RV-specific IgA responses in the mLN do not require cDC2
cDC2 coordinate IgA class switching in the Peyer’s Patches at steady state35 and are required for IgA induction in the mLNs in response to flagellin.37 We therefore tested whether cDC2 were also required for RV-specific IgA responses. huCD207.DTA mice specifically lack small intestinal CD103+ cDC2 and Langerhans cells42 and when infected orally with RV, these mice generated total and VLP+IgA+ B cell responses in mLNs that were comparable to controls (Fig. 2d). huCD207.DTA mice also, as previously published,19 did not differ from wild type mice in shedding of RV or in the levels of fecal RV-specific IgA (data not shown). Together, these results indicate that Batf3-dependent cDC1 are required for the optimal induction of anti-RV-specific IgA+ plasmablasts in the mLNs, while cDC2 are dispensable.
Optimal IgA+ B cell generation in the mLN requires CD4+ T Cells, but not CD8+ T Cells
To examine whether the cDC1-dependent, RV-specific B cell responses in the mLNs were T cell dependent, we first depleted CD4+ T cells in infected Batf3Het and Batf3KO mice. Administration of anti-CD4 antibody depleted >90% of CD4+ mLN T cells (Supplementary Fig. S2A) and ablated the expansion of total IgA+ and VLP+IgA+ B cells and plasmablasts that normally occurred during RV infection (Fig. 3a). In contrast, administration of anti-CD8 antibody to Batf3Het and Batf3KO mice did not reduce the numbers of VLP+ or total IgA+ B cells in the mLN of infected mice (Fig. 3b), despite causing >90% reduction in conventional CD8αβ+ T cells within the intraepithelial lymphocyte compartment, mLN or PPs (Supplementary Fig. S2B). Indeed, there was a consistent trend towards more IgA+ B cells in CD8+ T cell-depleted Batf3Het mice compared with control Batf3Het mice, probably reflecting the higher virus burden in CD8+ T cell-deficient mice.

a Batf3Het and Batf3KO mice treated with either anti-mouse CD4 (GK 1.5) Mab or Rat IgG2b,ĸ isotype control and oral RV infection. b Batf3Het and Batf3KO mice treated with either anti-mouse CD8β Mab or IgG1,ĸ isotype control and oral RV infection. The graphs depict the total number of B cells (live CD19+B220+) and antigen-experienced (live CD19+B220+IgD−) cells (left panels), the total number of IgA+ and RV-specific B cells (live CD19+B220+IgD−IgA+VLP+/−) (middle panels) and the total number of IgA+ and RV-specific plasmablasts (live CD19+IgD−CD138+IgA+VLP+/−) (right panels) at 7 days post infection. Dots represent data from individual mice. Data was collected from two independent experiments. Two-way ANOVA was performed for statistical analysis with Tukey post hoc testing. Statistics are depicted for within genotype comparisons only. *p < 0.05, **p < 0.01, ***p < 0.001.
Thus, CD4+ T cells are required to generate RV-specific IgA responses, whereas CD8+ T cells do not contribute to this process. Furthermore, our results argue that the differences in IgA induction observed in cDC1-deficient mice do not reflect a secondary effect of altered CD8 T cell induction in these mice.
Type 1 IFN receptor signalling on cDC1 is dispensible for the induction of antibody responses during Rotavirus infection
Type I interferons (IFNs) are produced rapidly during viral infections43,44 and type I IFN signalling in DCs has been implicated in their ability to drive the generation of TFH cell in response to systemic vaccination in a model using OVA and LPS.45 Furthermore, we have shown previously that pDCs and type I IFN signalling play an important role in early B cell activation, RV-specific IgA secretion and RV clearance.46 Therefore, to examine whether type I IFN sensing in cDC1 was needed for their role in RV-specific IgA responses, we generated a novel mouse model expressing cre recombinase under the control of the cDC1-specific XCR1 promoter crossed to ifnarflox/flox mice.47 The resulting XCR1-IFNARKO mice that lack IFNAR specifically on cDC1 generated comparable numbers of VLP+ and total IgA+ B cells and plasmablasts in mLN to those seen in ifnarflox/lfox mice used as controls (Fig. 4a). Similar results were obtained in CD11c-IFNARKO mice that lack the IFNAR on all DC subsets, with unaltered VLP+ and total IgA+ B cell and plasmablast induction in the mLNs in response to RV infection, together with normal viral clearance (Fig. 4b and data not shown). Together, these experiments show that IFNAR signalling on DCs is dispensable for the induction of IgA antibody responses during RV infection.

a XCR1-IFNARKO and b CD11c-IFNARKO mice were orally challenged with RV. The total number of total or RV-specific IgA+ B220+CD138− B cells (top; pre-gated on CD19+IgD−) and total or RV-specific IgA+ plasmablasts (bottom; B220−CD138+; pre-gated on CD19+IgD−) at 7 days post infection. Dots represent data from individual mice. Data were collected from a two and b four independent experiments with three mice per group. Two-way ANOVA was performed for statistical analysis with Tukey post hoc testing. *p < 0.05, **p < 0.01, ***p < 0.001. White symbols: HET; black symbols: KO; shaded bars: RV-infected.
αvβ8 integrin expression by cDC1 is not required for steady-state mucosal immune responses
As discussed above, TGFβ is one of the key factors for the induction of IgA responses in vivo27 and the TGFβ-activating integrin αvβ8 expressed on CD11b+ PP DCs has been implicated in the generation of IgA responses towards commensal bacteria.35 However, αvβ8 is expressed preferentially by migratory cDC1 in steady state mLN, where it is required for their ability to induce Tregs via TGFβ activation.48 Therefore, we examined whether αvβ8-mediated TGFβ activation by cDC1 could also account for their ability to drive IgA class-switching during viral infection.
We generated a novel mouse model in which a tdTomato fluorescent reporter is expressed under the control of the Itgb8 promoter (Itgb8-tdTomato mice) (Fig. 5a, b). Analysis of DC subsets in these mice confirmed that Itgb8-tdTomato expression was confined mostly to migratory cDC1 in the mLN (Fig. 5c–e), with 65–70% of XCR1+ migratory cDC1 expressing β8 integrin (Fig. 5c,d). Thus, while migratory cDC1 represent 23.7 % of total mLN DCs, they represent 80% of the Itgb8-tdTomato+ mLN DCs (Fig. 5e). In contrast, <10% of XCR1+SIRPα− cDC1 in the PPs were tdTomato+ and expression of the transgene was absent from other myeloid populations in PPs (Fig. 5c, d), which recapitulates Immgen-derived RNA-seq data on Itgb8 expression at the cell population level (Fig. 5f). Thus, αvβ8 is expressed preferentially by migratory cDC1 in the mLNs in steady state and not in the PPs.

a, b Genetic construction (a) and model validation (b) of the new Itgb8-tdTomato reporter mouse for Itgb8 expression. a Black rectangles represent Itgb8 coding sequences, grey rectangle indicates non-coding exon portions, solid line represents introns. Start (ATG) and Stop codons are indicated, and yellow and red rectangles represent the IRES and the reporter (tdTomato) sequences, respectively. b Tomato+ and Tomato− CD3+ T cells were sorted from the spleen of naive Itgb8-tdTomato mice. β8 integrin gene (Itgb8) and protein expression was assessed by qRT-PCR and Western Blot. The bar graph shows Itgb8 expression expressed relative to Gapdh. c Mononuclear phagocyte subsets from mLN or PP of Itgb8-tdTomato mice were analyzed for Itgb8 expression as indicated. Cells are pre-gated on live CD45+CD19−CD3−CD11c+ (mLN) or live CD45+CD19−CD3− (PP). d Percentage Itgb8-tdTomato+ cells for each subset. Note that the CD11b+ cDC2 gate contains both PP and DAV (Dome-associated villus) cDC2. (data pooled from three experiments with at least two mice each). e Percentage of each subset among total mLN DCs (left) or among Itgb8-tdTomato+ DCs (right). f Itgb8 expression value normalized by DESeq2 from ImmGen Open Source Mononuclear Phagocytes RNA-seq data. g, h Itgb8-tdTomato mice were infected orally with RV. Itgb8 expression in mLN and PP was monitored by flow cytometry 3 days post infection (data pooled from 2 experiments with 2 mice each). g Percentages of parent (DC subset, left) and number (right) of Itgb8-tdTomato+ DCs were plotted for the different mLN DC subsets. h Histograms of Itgb8-tdTomato expression in mLN and PP DC subsets. i Histogram shows the analysis of XCR1 expression in mLN DCs subsets from C57Bl/6 mice gated as in c. Two-way ANOVA was performed for statistical analysis with a Tukey post hoc test. **p < 0.01.
We have previously shown that αvβ8 expression by DCs is tightly regulated and depends on a combination of lineage and tissue-microenvironment factors, as well as activation status.48 Interestingly, upon in vitro TLR3 engagement by the synthetic analog of dsRNA, polyinosinic-polycytidylic acid (poly(I:C)), splenic cDC1 not expressing β8 in the steady state48 get activated, up-regulate CD86 expression and induce expression of β8 integrin (Supplementary Fig. S3B,C). This is not the case for cDC2. In order to determine whether β8 expression levels change during infection, Itgb8-tdTomato mice were orally infected with RV. Importantly, three days post infection, the percentage but not the number of β8+ migratory cDC1 was slightly decreased in the mLN (Fig. 5g), while total percentage and number of DCs remained stable (Supplementary Fig. 3D). We observed no induction of β8 in other mLN or any PP DC subsets (Fig. 5h). Therefore, the β8 expression pattern is conserved during RV infection with the maintenance of preferential expression in migratory cDC1 mLN DCs.
In order to study the role of αvβ8 on cDC1 in regulating intestinal IgA responses, we generated mice lacking αvβ8 integrin expression specifically on cDC1 (Xcr1Cre.Itgb8flox/−, called XCR1-β8KO thereafter). As expected, only cDC1 (migratory CD103+CD11b− and resident CD8α+ mLN DCs expressing XCR1 (Fig. 5i)), but not cDC2, deleted the floxed Itgb8 allele in these mice (Fig. 6a). XCR1-β8KO mice developed normally, did not develop wasting disease or colitis (Fig. 6b, c) and had normal cellularity of total lymphocytes (Fig. 6d) and populations of Tregs and TH17 cells in the colonic lamina propria (Fig. 6e). Numbers of total IgA producing B cells and plasma cells in spleens, PPs, mLNs and colonic lamina propria were also unaltered (Fig. 6f), translating to normal total IgA levels in the serum and intestinal washes from XCR1-β8KO mice (Fig. 6g) and in line with normal TFH and TFR fractions in the mLNs and PPs (Fig. 6h). This was in sharp contrast to mice in which αvβ8 is deleted in all DCs (CD11c-β8KO), which developed spontaneous colitis and dysregulated intestinal B and T cell responses (Fig. 6b–e and Supplementary Fig. S4). Thus, expression of αvβ8 integrin by cDC1 is dispensable for immune homeostasis in the intestine, including the generation of steady state IgA responses.

XCR1-β8KO mice were generated to specifically delete αvβ8 in cDC1. a Genomic PCR for the Itgb8 locus in sorted mLN DC subpopulation as gated in Fig. 5a. b, c XCR1-β8KO mice do not develop colitis. Weight (at least seven individual mice per timepoint and per genotype) (b) and representative histology of the colon (c) of XCR1-β8KO, CD11c-β8KO and littermate controls. Statistics in b consistently relate to every time point from 12 weeks onward and were assessed using a two-way ANOVA between genotypes followed by Sidak correction for multiple comparison test for each individual timepoint. Scale bar in c: 500 µm. d, e Number of total CD45+ cells (d) and fraction of Treg and TH17 (e) were quantified in the Colon Lamina Propria (CLP) of XCR1-β8KO and CD11c-β8KO mice by flow cytometry. f Percentage of IgA+ B cells (CD45+CD3−B220+CD19+CD138−IgA+) and plasma cells (CD45+CD3−B220−CD138+IgA+) from indicated organs from XCR1-β8KO mice measured by flow cytometry. g Serum and intestinal wash IgA levels in XCR1-β8KO versus control littermates measured by ELISA. h TFH and TFR percentages of total CD45+ live cells from PPs and mLNs analyzed by flow cytometry. Cell percentages are plotted as a percent of CD45+ cells. Data were collected from 2-4 independent experiments (total n = 7–15). Statistical analysis for d–h: two-way ANOVA was performed for statistical analysis with a Tukey post hoc test, ***p < 0.001.
The TGFβ-activating αvβ8 integrin on cDC1 is essential for optimal RV-specific IgA responses
As XCR1-β8KO mice did not develop colitis and had normal total IgA production, we were able to use them to assess the impact of cDC1-restricted deletion of αvβ8 deletion on RV-specific IgA responses without the confounding effects of inflammation. As we had found in Batf3KO mice, mice lacking αvβ8 specifically on cDC1 had normal numbers of total and IgA+ B cell numbers 7 days after infection with RV (Fig. 7a), but had decreased numbers of IgA+ (both total and VLP+) plasmablasts in mLN compared with XCR1-β8het littermate controls (Fig. 7a). This did not affect the number of IgM+ B cells in mLN (Supplementary Fig S5A). Thus, the TGFβ-activating αvβ8 integrin on cDC1 is required for optimal induction of RV-specific IgA in the mLN at 7 days post infection.

a XCR1-β8KO and littermate control (XCR1-β8het) mice were orally challenged with RV. The total number of B cells, antigen-experienced IgD− cells (left), total or RV-specific IgA+B220+CD138− B cells (pre-gated on CD19+IgD−) (middle) and total or RV-specific IgA+ plasmablasts (B220−CD138+; pre-gated on CD19+IgD−) at 7 days post infection is plotted. Dots represent data from individual mice. Data were collected from three independent experiments with 4–5 mice per group each. b Splenic QM B cells were isolated by magnetic purification and cultured alone or with total CD11c+MHC+ DCs (+mLN DCs) isolated from mLN of C57Bl/6 mice in serum-free medium and in the presence of NP-Ficoll, anti-CD40 Ab and RA. Active TGFβ (act), latent TGFβ (lat) and/or a synthetic RGD (or RAD)-containing peptide were added as indicated. After 5 days cells were harvested and analysed for IgA expression by flow cytometry. Additional statistics: a1-a2, b1-b2, c1-c2, d1-d2 and e1-e2: *** (total n = 4-17) (c, d) CD103+ CD11b− migratory cDC1 (+mig cDC1, (c, d)) or CD103+ CD11b+ migratory cDC2 (+mig cDC2, (c)) were sorted from the mLN of XCR1-β8WT (C,D) or XCR1-β8KO (d) mice as previously described and cultured with FACS-sorted QM B cells (gated as CD19+ B220+). Data is normalized to control cDC1 cultured with latent TGFβ + RAD for each replicate (total n = 4–7). Additional statistics: a1-a2, b1-b2, b1-b3:***, a1-a3:** Dots represent data from independent biological replicates. Data were collected from four (b) or three (c, d) independent experiments. Two-way ANOVA with Tukey post hoc was performed for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001.
αvβ8-mediated TGFβ activation is required for cDC1 to induce optimal IgA class-switching in vitro
To confirm directly that αvβ8 on cDC1 plays a role in IgA class-switching via TGFβ activation, we switched to an in vitro model. Splenic NP-specific B cells from QM mice were cultured in vitro in serum-free medium to minimize TGFβ contamination and stimulated with anti-CD40 Ab and NP-Ficoll. Total mLN DCs from WT mice stimulated IgA class-switch in vitro and this was significantly enhanced by addition of either active or latent TGFβ (Fig. 7b). Binding and activation of latent TGFβ by αvβ8 occurs via an Arg-Gly-Asp (RGD) tripeptide in the latency-associated peptide and a cyclic form of the RGD peptide inhibits the binding and activation of latent TGFβ by αvβ8 integrins in the context of T cell-dependent responses.33,48 This RGD mimetic blocked the IgA class switching enhancing effect of added latent TGFβ in DC-B cell co-cultures in these cultures as well as in cultures containing keyhole limpet hemocyanin-NP instead of NP-Ficoll (Fig. 7b and Suppl. Fig. 5B,C). Both cDC1 and cDC2 subsets were able to induce IgA class switching in this in vitro model efficiently but blocking αvβ8 with RGD only affected the IgA induction efficiency by cDC1s, while cDC2s were unaltered (Fig. 7c). Likewise, only migratory mLN cDC1 but not cDC2 sorted from XCR1-β8KO mice were significantly impaired in their capacity of inducing IgA class switching (Fig. 7d and Suppl. Fig. S5C). Thus, mLN cDC1s appear to activate latent TGFβ to promote IgA class switching via αvβ8, a process that could be replicated in vitro using FACS-sorted migratory CD103+CD11b− mLN cDC1. Taken together, these in vitro experiments support the idea that intestine-derived cDC1 can induce IgA switching in B cells via αvβ8 integrin-mediated activation of latent TGFβ (for a graphical summary see Fig. 8).

While subepithelial dome cDC2 are required in the PP for induction of IgA responses to extracellular bacteria, we show that mLN cDC1 promote both cellular and humoral immune responses to RV infection. cDC1 in the LP take up viral antigen in apoptotic bodies, activate and migrate to the mLN, where they induce RV-specific cytotoxic T cells. We here show that they also contribute to the induction of RV-cDC1-driven T cell-dependent IgA responses in the mLN. αvβ8 expression, uniquely prominent on migratory cDC1 in the mLN is required to instruct the class switch towards IgA through provision of bioactive TGFβ. CD8+ T cells and plasma cells eventually migrate to the LP to kill infected enterocytes and produce secretory IgA, respectively.
Discussion
Specific IgA production is the major correlate of protection from reinfection with rotavirus,5,6 but very little is known about the mechanisms that induce IgA responses against intestinal viruses. Here we show that the cDC1, but not cDC2 subset of DCs, is critical for generating optimal T-cell dependent RV-specific IgA responses in the mLNs early during primary infection. Type I IFN signalling in DCs is dispensible for their role in B cell priming upon infection. Instead, induction of optimal IgA responses depends, at least in part, on αvβ8 expression by cDC1, driving activation of TGFβ.
As we confirm here, intestinal cDC1 are crucial for the optimal immune response towards RV, with RV-specific CD8+ T cells depending on this subset for their priming.19 This finding is consistent with the ability of RV to infect the intestinal epithelium and cause epithelial cell death, as well as the fact that cDC1 are the primary DC subset that presents epithelial-derived antigens to CD8+ T cells in the mLN.18 Importantly, we and others19 found that some RV-specific CTLs remained present in the absence of cDC1, suggesting that other cells can also prime CD8+ T cell responses to RV. It is not known whether RV can infect DCs directly in vivo, but if this occurs, the residual CTL response to RV may also reflect direct presentation by infected non-cDC1 DCs. Alternatively, the few remaining cDC1 that are known to be present in BATF3-deficient mice on the C57Bl/6 genetic background49 could suffice to prime RV-specific CTLs. This is thought to reflect the ability of other BATF transcription factors to take over the role of BATF3 in an IL-12 dependent manner in the presence of inflammation,49 although it is notable that we did not find remaining cDC1 in Batf3KO mice during RV infection (Supplementary Fig. S1).
Given these findings, we hypothesized that cDC1 might also influence B cell priming and IgA class-switching in response to RV. Indeed, we found delayed secretion of fecal RV-specific IgA in infected Batf3KO mice, together with a marked defect in the expansion of antigen-specific IgA+ plasmablasts in the mLNs. The numbers of total IgA+ and IgM+ plasmablasts, but not those of total IgA+ or IgM+ non-plasmablasts were also slightly affected by the absence of cDC1, indicating that the initial massive increase in B cells that occurs in response to RV is independent of cDC1 and that the defect is biased towards RV-specific IgA. Strikingly, the absence of intestinal cDC2 had no effect on RV-specific IgA+ B cells numbers in the mLN, contrasting with the known role of this DC subset in intestinal IgA production in the steady-state and in response to extracellular bacteria.35,37 cDC1s excel at transporting apoptotic intestinal epithelial cells to the draining mLN,18,50 and RV infection induces ample cell death.51,52 Together, this suggests that the induction of IgA class-switching is not restricted to intestinal cDC2, but rather depends on the context and nature of the antigen.
The defective generation of RV-specific IgA B cell responses in Batf3KO mice also appears to be an anatomically restricted phenomenon, as we found no effect in the PPs. This might be due to a redundancy in DC subsets at sites such as PPs that are likely heavily loaded with antigen or could reflect the relative absence of αvβ8 integrin expression by cDC1 in PPs (see below). It might also reflect the specific ability of cDC1 to deliver epithelial-derived antigen to the mLN.18
To our knowledge this is the first demonstration that cDC1 play a role in the generation of antigen-specific IgA responses in the intestine. However, previous studies have shown a similar requirement for cDC1 in IgA responses in the lungs after administration of an influenza hemagglutinin subunit with poly(I:C), a synthetic mimic of double-stranded RNA.36 This effect depended on a functional TLR3-TRIF pathway and correlated with the presence of T follicular helper cells. As we found in the intestines, there were no differences in total B cell numbers in the lung in the absence of cDC1, suggesting a specific defect in IgA class switching.
Having established a role for cDC1 in the induction of RV-specific IgA in the mLN, we then explored the mechanisms involved. cDC1 have been reported to drive germinal center-derived IgA responses in the respiratory tract36 and consistent with previous observations,8 we found that the RV-specific B cell recruitment and IgA responses in the mLN were dependent on CD4+ α/β T cells. As published earlier by Sun et al., IFNγ production by virus-specific CD4+ T cells was, however, not altered (ref. 19 and own observations). While total numbers of mLN TFH cells were also not altered in the absence of cDC1 (data not shown), the requirement for germinal center reactions for early IgA plasmablast induction upon RV infection requires further investigation. As Batf3KO mice lacking cDC1 have markedly reduced CD8+ T cell responses during RV infection (our data and ref. 19) and IFNγ produced by CD8+ T cells can induce class switching in B cells,53 we assessed whether CD8+ T cells contributed to RV-specific IgA production and whether that could explain the decrease in RV-specific IgA in BATF3-deficient mice. However, depletion of CD8+ T cells did not reduce IgA+ B cell responses in RV-infected mice, indicating that CD8+ T cells do not contribute to IgA class switching in this model.
Rotavirus elicits type I IFN responses54 and stimulation of DCs with type I IFN enhances humoral immunity, promotes isotype switching55 and increases the development of lymph node-resident TFH cells.45 Using a complete knock-out for the type I IFN receptor, we have previously reported a role for type I IFN in the early induction of bulk B cell responses during RV infection.46 Given the similar viral load in type I IFN deficient and wild type mice infected with homologous RV strains,56 this is however unlikely to contribute to viral clearance. Here, we show that deletion of the type I IFN receptor specifically on DCs or cDC1 did not alter RV-specific IgA responses. This negative finding is consistent with other work examining cDC1-dependent IgA production in the lung in response to poly(I:C)36; adaptive immunity to norovirus infection also does not require type I IFN receptor signalling on DCs.57 Thus, the requirement for type I IFN signalling in bulk B cell activation early after infection does not appear to involve the DC compartment and is more likely to reflect a direct role for the type I IFN receptor on B cells.46
The initiation of class switch recombination towards IgA requires activation of the cytokine TGFβ,58,59 with binding of its latent form to cell surface αvβ8 integrin being an important mechanism of TGFβ activation.29,30 In the gut, αvβ8 integrin on mononuclear phagocytes (MNPs) is critical to maintain mucosal immune homeostasis, as mice selectively lacking β8 in all CD11c-expressing cells develop severe spontaneous colitis,30 associated with loss of intestinal Treg and Th17 cells. Subsequently, we and others have shown that induction of intestinal TGFβ-dependent T cell responses is under the strict control of DC αvβ8, which licenses TGFβ signaling to T cells via local activation of TGFβ.31,32,33,34 As we confirm here, αvβ8 is expressed preferentially by migratory cDC1 in the normal mLN,13,48 but in contrast to its deletion in all CD11c+ cells, selective β8 deletion in cDC1 under the control of the XCR1 promoter does not disrupt mucosal immune homeostasis. The reasons for the discrepancy between these models are unclear, but may suggest that the low abundance of β8+ mLN cDC2 (~12%) and PP DCs (1–4%) can compensate for αvβ8-deficiency on cDC1 to maintain steady state conditions. Along those lines, we have previously shown that stimulation in vitro can reveal αvβ8 expression on cells other than cDC1.48 Importantly, together with targeted deletion of β8 integrin in specific myeloid cell populations, our Itgb8-tdTomato reporter mice will serve as a valuable tool to dissect the contribution of different cell subsets to αvβ8-dependent immune homeostasis in the future.
Here, we show that β8 expression in any other PP or mLN MNPs is minor compared to β8 expression in migratory cDC1 in the mLN, both by FACS and by qRT-PCR or RNA-seq (Fig. 5 and data not shown). A recent study reported that both CD11b+ and CD8α−CD11b− DCs in PP express αvβ8.35 In their study, Reboldi et al.35 examined β8 expression at the population level only in the PPs, while our analysis both at the single-cell level using Itgb8-specific reporter mice and in sorted cell populations (ImmGen data, Fig. 5f) revealed much higher proportions of β8+ cells and higher levels of β8 transcripts on DCs in mLNs compared with PPs. This did not change in the context of RV infection. As discussed above, the low percentage of β8+ cells in DC subsets other than mLN cDC1 does not preclude them from playing a critical role in mucosal immune homeostasis, which will need to be explored in the future. In addition, as we have shown previously that β8 expression by mLN DC can be influenced by several factors in the intestinal microenvironment such as dietary metabolites, the microbiota and the presence of inflammation,48,60 we cannot exclude that the apparent discrepancies in β8 expression profiles may reflect differences in these factors. Furthermore, it remains possible that other triggers such as bacterial or fungal infections might induce β8 expression by cDC2 in mLN and/or PP, allowing them to participate in TGFβ activation for the induction of IgA responses. For example, ATP promotes αvβ8 expression in colonic LP CD103−CX3CR1+CD11b+ MNP.61 An alternative possibility in that cDC2 may be able to activate TGFβ via other, αvβ8-independent, mechanisms such as proteases, ROS or thrombospondin-1.62,63 Finally, several studies have suggested that DC may promote IgA class switching in vivo by non-TGFβ-dependent mechanisms, such as retinoic acid, TNF/iNOS or APRIL.25,64,65 Therefore, the molecular mechanisms driving IgA responses may be complex and context dependent.
A novel finding of the current study was the significant reduction in IgA+ plasmablasts in the mLN of mice lacking αvβ8 specifically in cDC1, providing a mechanism for the dependency of RV-specific IgA on the presence of this subset of DC consistent with the ability of αvβ8 to activate this cytokine.66 Thus, αvβ8 expressing cDC1 may play a critical and specific role in the generation of IgA responses to viruses and other antigens that require cross-presentation in the intestine. It has to be noted that the defect in IgA plasmablasts in XCR1-β8KO mice was not as dramatic as was seen in cDC1-deficient mice. Intestinal cDC1 excel in carrying epithelium-derived antigens to the mLN,18 possibly explaining the pronounced phenotype in mice lacking cDC1. The difference in phenotypes could also reflect a role for other molecules capable of converting TGFβ into its bioactive form, such as other integrins or matrix metalloproteinases (MMPs)67 and this requires further study. In addition, as noted above, TGFβ is not the only mediator that can facilitate class-switching towards IgA at mucosal sites. These include retinoic acid, which can act synergistically with TGFβ in this role.65,68 Migrating intestinal DCs including cDC1 possess all the enzymes necessary to convert vitamin A into RA, indicating that lack of RA might help explain the larger defect in IgA production in Batf3KO mice. However, the induction of the gut homing molecules CCR9 and α4β7 on B cells was normal in Batf3KO mice, suggesting that overall levels of RA may be relatively unaffected even in the absence of cDC1. In addition, this result is also likely to exclude defective B cells migration from the mLN to the intestinal mucosa as a mechanism of the lower levels of secreted IgA we observed in these animals.
In summary, we show here that BATF3-dependent cDC1, but not cDC2, are required for optimal T-cell dependent RV-specific IgA responses in the mLNs during primary RV infection in adult mice. This strongly suggests that both major DC subsets in the intestines can induce IgA production but vary in their contribution according to the nature of the trigger. While type I IFN receptor signalling in DCs is dispensable for their role in IgA induction, their expression of the TGFβ activating αvβ8 integrin is in part accountable for their capacity of inducing IgA during RV infection. Given the prominent role of RV-specific IgA in protection against re-infection, we hypothesize that strong RV-specific IgA induction in the mLNs may account for functionally relevant immune memory. Of note, BATF3-deficient mice are fully protected from re-infection eight weeks after primary infection (data not shown). This is in line with our data showing that RV-specific IgA titers in the feces eventually reach wildtype levels in the absence of cDC1 (Fig. 1c, d), which are maintained for at least eight weeks and probably accounting for the lack of recall following this timeline (data not shown). Further analysis of the RV-specific affinity and longevity of the B cell memory pool remains to be performed in order to define whether targeted engagement of cDC1 is a superior strategy for future vaccine design against rotavirus and other mucosal viral pathogens.
Methods
Mice
All animals were housed under specific pathogen-free conditions at Lund/Malmo (Sweden) or Lyon (France). All mice were on the C57Bl/6 background. Littermate or age-matched mice were used for all experiments. Male and female mice were used between 8 and 16 weeks of age. Xcr1-IRES-iCre-2A-mTFP1 gene-targeted mice (also known B6-Xcr1tm2Ciphe mice and called XCR1.cre mice here) mice permit to specifically delete floxed gene in cDC1.69 Batf3KO and Het (B6.129S(C)-Batf3tm1Kmm/J), CD11c-IFNARKO ((B6.Cg-Tg(Itgax-cre)1-1Reiz/J70 crossed to IFNAR floxed mice (obtained from U. Kalinke47)), XCR1-IFNARKO (XCR1.cre crossed to IFNAR floxed mice) and huCD207-DTA 42 mice were bred and maintained in the Clinical Research Center at Lund University. β8 integrin-reporter mice were generated on a C57Bl/6 background by expression of an internal ribosomal entry site-fluorescent tandem dimer (td)-Tomato cassette (IRES-Td-Tomato) immediately following the stop codon in exon 14 of Itgb8 (Fig. 5a) (GenOway, Lyon, France). Itgb8-TdTomato mice had no gross abnormalities and were born at the expected female:male ratios. Itgb8-TdTomato, Quasi-monoclonal (QM) mice71 (kindly provided by Dr. M. Cascalho, University of Michigan, Medical School, Ann Arbor, MI, USA) and XCR1-β8KO mice (XCR1.cre crossed to Itgb8flox/− 30) were bred at Plateau de Biologie Expérimentale de la Souris (PBES, ENS Lyon, France). Animal experiments were performed under appropriate licenses within local and national guidelines for animal care.
Rotavirus infections
The virulent wildtype ECw strain of RV was obtained from cleared intestinal homogenates of suckling mice orally gavaged with RV at 3 days of age and sacrificed 2 days after infection. For infection of adult mice, RV was inoculated orally at a dose of 3 × 103 DD50, with Peyer’s patches and mLNs being collected and lymphocytes isolated at intervals as described below.
Cell Isolation and flow cytometry
Single-cell suspensions from spleen, mLNs and Peyer’s Patches were generated by mechanical disruption through 70 µm cell strainer in FACS buffer (5% FBS, 500 ml PBS (Gibco)). Non-specific binding was blocked with normal rat serum (Sigma) and rat anti-mouse CD16/CD32 Fc block (2.4G2, BD Biosciences) for 20 min at 4 °C. To stain for RV-specific B cells, cells were incubated with rotavirus-like particles (VLP2/6) containing RV VP2 and VP6 bound to GFP (obtained from Didier Poncet, I2BC CNRS Gif sur Yvette, France)41 for 45 min at 4 °C. The following antibodies were used: anti-CD19 (1D3), anti-CD138 (281-2), anti-B220 (RA3-6B2), anti-IgM (R6-60.2) (BD Horizon), anti-GL-7 (GL7), anti-IgD (11-26c.2a), anti-CD45.2 (104) (BioLegend), anti-IgA (mA-6E1) (eBioscience), and anti-CD95 (Jo2) (BD Biosciences). The Live/DeadTM Fixable Near-IR Dead cell stain kit (L10119, Life Technologies) was used to assess cell viability, while VP7VGPVFPPGM tetramers coupled to BV421 obtained from the NIH Tetramer core facility were used at a concentration of 12 µg/ml to stain for RV-specific CD8+ T cells. B cells were sorted from splenocytes of QM mice71 either by magnetic separation (R&D) or by flow cytometry as indicated using the following Ab: anti-CD19 (1D3), anti-B220 (RA3-6B2), anti-CD11c (Hl3), anti-CD3 (145-2C11) (all from BD Biosciences, Pont de Claix, France). DCs subsets from spleen, mLN and PP were isolated as described previously.48 Briefly, after tissue digestion, cell suspensions were enriched for DCs using an OptiPrep gradient. Total spleen DCs were then isolated by positive magnetic separation using anti-CD11c beads (Miltenyi). DC subsets from mLN and spleen were FACS-sorted using a BD FACSAriaII cytometer and the following antibodies: anti-CD11c (HL3), anti-I-A/I-E (2G9), anti-CD8α (53-6.7), anti-CD45RB/B220 (RA3-6B2), anti-CD103 (M290) (all from BD Biosciences, Pont de Claix, France), anti-CD11b (M1/70; eBioscience, Paris, France) and anti-XCR1 (ZET; Biolegend). Data acquisition was performed using LSRII or LSR-Fortessa cytometers and DIVA software (BD Bioscience). The analysis was performed using FlowJo software 10 (Treestar Inc).
Measurement of rotavirus antigen shedding and IgA in feces
Fecal samples from infected mice were weighed and soaked in PBS, 1% BSA, 1 mM EDTA, soybean trypsin inhibitor (0.05 mg/ml), 2 mM PMSF (phenylmethylsulfonyl fluoride) (Sigma) and 0.025% azide for 2 h at 100 mg/ml, homogenized and centrifuged at 13000 rpm for 10 min. The supernatant was analysed for the presence of RV antigen and RV-specific IgA as previously described in ref. 38 using guinea pig anti-RV hyperimmune serum, rabbit anti-RV hyperimmune serum (both from the Greenberg laboratory), goat anti-rabbit IgG-HRP (Millipore) and TMB Substrate Reagent Set (RUO, 555214, BD Biosciences).
Depletion of CD8+ T cells and CD4+ T cells in vivo
Mice were depleted of CD8+ T cells by administration of 300 µg InVivoMAb anti-mouse CD8β (53-5.8, Nordic BioSite) intraperitoneally on days −2, −1 before RV infection, on the day of infection and days 3 and 5 post RV infection. Non-depleted mice received purified mouse IgG1ĸ isotype as a control (MOPC-21, Nordic BioSite). For depletion of CD4+ T cells, mice received 50 µg InVivoMAb anti-mouse CD4 (GK 1.5, Nordic Biosite) on day -1 before RV infection and day 3 post RV infection. Non-depleted mice received purified Rat IgG2b, ĸ isotype as a control.
β8 integrin expression analysis
Itgb8 gene expression analysis by quantitative RT-PCR was performed as previously described.48 β8 integrin protein expression was measured by Western blot using an anti-β8 antiserum generated in rabbit against the C-terminal tail of the human β8 cytoplasmic region (provided by Joseph McCarty72) as previously described.48
IgA class switch by B cells in vitro
B cells were isolated from QM mice71 either by magnetic isolation (MagCellect Mouse B Cell Isolation Kit, R&D) or sorted by FACS as indicated in Figure legends. B cells (2 × 104) were co-cultured with cDCs (2 × 104) with 4-Hydroxy-3-nitrophenylacetic (NP) conjugated to Ficoll (NP-Ficoll, Biosearch Technologies, 15 ng/mL), anti-CD40 (Enzo Life Sciences, 2 µg/mL) and RA (Sigma, 2 µM) in the presence or absence of latent TGFβ (R&D, 4 ng/mL), active TGFβ (R&D, 1 ng/mL), cRGD (Enzo Life Sciences, 20 µg/mL), cRAD (Enzo Life Sciences, 20 µg/mL) and anti-TGFβ Ab (BD, 40 µg/mL) for 5 days in X-vivo 15 medium (Lonza) supplemented with sodium pyruvate (1 mM), MEM NEAA, HEPES buffer (10 µM), 2-mercaptoethanol (50 µM) and L-glutamine (2 mM). IgA+ B cells were identified by flow cytometry using the following antibodies: anti-IgA-FITC (11-44-2; SouthernBiotech), anti-CD19-PECF594 (1D3), anti-B220-BV786 (RA3-6B2) (BD Biosciences). Viability was assessed using Thermoscience Fixable Viability Dye FVD-ef780.
DC stimulation in vitro
DCs isolated from the spleen of Itgb8-tdTomato were stimulated in vitro as previously described.48 Briefly, spleen DCs were cultured at 37 °C in complete RPMI medium in the presence of 1 µg/ml high molecular weight poly(I:C) (InvivoGen) for 8 h. Itgb8-tdTomato expression in DC subsets was then assessed by flow cytometry using the following antibodies: anti-CD11c (HL3), anti-I-A/I-E (2G9), anti-CD45RB/B220 (RA3-6B2), anti-CD19 (1D3), anti-CD3 (145-2C11) (all from BD Biosciences, Pont de Claix, France), anti-CD11b (M1/70; eBioscience, Paris, France), and anti-XCR1 (ZET) and anti-CD86 (GL-1; both from Biolegend).
Statistical analysis
Statistics were performed using two-way ANOVA (post hoc: Tukey) considering treatment and experiment as factors for the analysis. Kruskal–Wallis test was applied to compare more than two groups (e.g.: different genotypes (n = 3) within the same treatment) (host hoc: Dunn73) when the experiment was performed only once. Statistical significance was estimated by using R Studio.
R Script:
-
Two-way ANOVA: aov(value ~ genotype + day, data = Data)
-
○
Post-Hoc test Tukey: TukeyHSD(Data_anova2, which = “genotype”)
-
○
-
Kruskal–Wallis test: kruskal.test(value ~ treatment, data = Data)
-
○
Post-Hoc test Dunn: dunnTest(value ~ treatment, data = Data, method = “bh”)
-
○
Where genotype accounts for analysis of different genotypes within the same treatment. Using treatment instead of genotype allows for analysis of different treatments within a genotype. Data refers to the data to be analysed.
Area under the curve calculation (unpaired T test as statistical method) and two-way ANOVA with Sidak correction were performed using Graphpad Prism Software.
References
Lundgren, O. & Svensson, L. Pathogenesis of rotavirus diarrhea. Microbes Infect. 3, 1145–1156 (2001).
Ramig, R. F. Pathogenesis of intestinal and systemic rotavirus infection MINIREVIEW pathogenesis of intestinal and systemic rotavirus infection. J. Virol. 78, 10213–10220 (2004).
Bishop, R. F., Davidson, G. P., Holmes, I. H. & Ruck, B. J. Virus particles in epithelial cells of duodenal mucosa from children with acute non-bacterial gastroenteritis. Lancet 302, 1281–1283 (1973).
Troeger, C. et al. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 17, 909–948 (2017).
Angel, J., Franco, M. A. & Greenberg, H. B. Rotavirus immune responses and correlates of protection. Curr. Opin. Virol. 2, 419–425 (2012).
Blutt, S. E., Miller, A. D., Salmon, S. L., Metzger, D. W. & Conner, M. E. IgA is important for clearance and critical for protection from rotavirus infection. Mucosal Immunol. 5, 712–719 (2012).
VanCott, J. L. et al. Role of T cell-independent B cell activity in the resolution of primary rotavirus infection in mice. Eur. J. Immunol. 31, 3380–3387 (2001).
Franco, M. A. & Greenberg, H. B. Immunity to rotavirus in T cell deficient mice. Virology 238, 169–179 (1997).
Bogunovic, M. et al. Origin of the lamina propria dendritic cell network. Immunity 31, 513–525 (2009).
Persson, E. K. et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38, 958–969 (2013).
Luda, K. M. et al. IRF8 transcription-factor-dependent classical dendritic cells are essential for intestinal T cell homeostasis. Immunity 44, 860–874 (2016).
Edelson, B. T. et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J. Exp. Med 207, 823–836 (2010).
Esterházy, D. et al. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral Treg cells and tolerance. Nat. Immunol. 17, 545–555 (2016).
Persson, E. et al. IRF4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 38, 958–969 (2013).
Schlitzer, A. et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38, 970–983 (2013).
Mayer, J. U. et al. Different populations of CD11b + dendritic cells drive Th2 responses in the small intestine and colon. Nat. Commun. 8, 15820 (2017).
Hildner, K. et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008).
Cerovic, V. et al. Lymph-borne CD8α+ dendritic cells are uniquely able to cross-prime CD8+ T cells with antigen acquired from intestinal epithelial cells. Mucosal Immunol. 8, 38–48 (2015).
Sun, T. et al. Intestinal Batf3 -dependent dendritic cells are required for optimal antiviral T-cell responses in adult and neonatal mice. Mucosal Immunol. 10, 1–14 (2016).
Gutzeit, C., Magri, G. & Cerutti, A. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev. 260, 76–85 (2014).
Schulz, O. & Pabst, O. Antigen sampling in the small intestine. Trends Immunol. 34, 155–161 (2013).
Cerutti, A. The regulation of IgA class switching. Nat. Rev. Immunol. 8, 421–434 (2008).
Bemark, M., Boysen, P. & Lycke, N. Y. Induction of gut IgA production through T cell-dependent and T cell-independent pathways. Ann. N. Y. Acad. Sci. 1247, 97–116 (2012).
Litinskiy, M. B. et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 3, 822–829 (2002).
Tezuka, H. et al. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 448, 929–933 (2007).
Tezuka, H. et al. Prominent role for plasmacytoid dendritic cells in mucosal T cell-independent IgA induction. Immunity 34, 247–257 (2011).
Cazac, B. B. & Roes, J. TGF-β receptor controls B cell responsiveness and induction of IgA in vivo. Immunity 13, 443–451 (2000).
Ruane, D. et al. Microbiota regulate the ability of lung dendritic cells to induce IgA class-switch recombination and generate protective gastrointestinal immune responses. J. Exp. Med. 213, 53–73 (2015).
Yang, Z. et al. Absence of integrin-mediated TGFβ1 activation in vivo recapitulates the phenotype of TGFβ1-null mice. J. Cell Biol. 176, 787–793 (2007).
Travis, M. A. et al. Loss of integrin αvβ8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449, 361–365 (2007).
Païdassi, H. et al. Preferential expression of integrin αvβ8 promotes generation of regulatory T cells by mouse CD103+ dendritic cells. Gastroenterology 141, 1813–1820 (2011).
Worthington, J. J., Czajkowska, B. I., Melton, A. C. & Travis, M. A. Intestinal dendritic cells specialize to activate transforming growth factor-β and induce Foxp3+ regulatory T cells via integrin αvβ8. Gastroenterology 141, 1802–1812 (2011).
Acharya, M. et al. αv Integrin expression by DCs is required for Th17 cell differentiation and development of experimental autoimmune encephalomyelitis in mice. J. Clin. Invest. 120, 4445–4452 (2010).
Melton, A. C. et al. Expression of αvβ8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J. Clin. Invest. 120, 4436–4444 (2010).
Reboldi, A. et al. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 352, aaf4822 (2016).
Takaki, H. et al. Toll-like receptor 3 in nasal CD103 + dendritic cells is involved in immunoglobulin A production. Mucosal Immunol. 11, 82–96 (2018).
Flores-Langarica, A. et al. CD103+CD11b+ mucosal classical dendritic cells initiate long-term switched antibody responses to flagellin. Mucosal Immunol. 11, 1–12 (2017).
Franco, M. A. & Greenberg, H. B. Role of B cells and cytotoxic T lymphocytes in clearance of and immunity to rotavirus infection in mice. J. Virol. 69, 7800–7806 (1995).
Fujimoto, K. et al. A new subset of CD103+CD8alpha+ dendritic cells in the small intestine expresses TLR3, TLR7, and TLR9 and induces Th1 response and CTL activity. J. Immunol. 186, 6287–6295 (2011).
Blutt, S. E., Warfield, K. L., Lewis, D. E. & Conner, M. E. Early response to rotavirus infection involves massive B cell activation. J. Immunol. 168, 5716–5721 (2002).
Charpilienne, A. et al. Individual rotavirus-like particles containing 120 molecules of fluorescent protein are visible in living cells. J. Biol. Chem. 276, 29361–29367 (2001).
Welty, N. E. et al. Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. J. Exp. Med. 210, 2011–2024 (2013).
Levy, D. E. & García-Sastre, A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 12, 143–156 (2001).
Katze, M. G., He, Y. & Gale, M. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2, 675–687 (2002).
Cucak, H., Yrlid, U., Reizis, B., Kalinke, U. & Johansson-Lindbom, B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity 31, 491–501 (2009).
Deal, E. M. et al. Plasmacytoid dendritic cells promote rotavirus-induced human and murine B cell responses. J. Clin. Invest. 123, 2464–2474 (2013).
Kamphuis, E., Junt, T., Waibler, Z., Forster, R. & Kalinke, U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood 108, 3253–3261 (2006).
Boucard-Jourdin, M. et al. β8 integrin expression and activation of TGF-β by intestinal dendritic cells are determined by both tissue microenvironment and cell lineage. J. Immunol. 197, 1968–1978 (2016).
Tussiwand, R. et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490, 502–507 (2012).
Cummings, R. J. et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 539, 565–569 (2016).
Zhu, S. et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670 (2017).
Dou, Y., Yim, H. C., Kirkwood, C. D., Williams, B. R. & Sadler, A. J. The innate immune receptor MDA5 limits rotavirus infection but promotes cell death and pancreatic inflammation. EMBO J. 36, 2742–2757 (2017).
Mohr, E. et al. IFN- produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine. Proc. Natl Acad. Sci. 107, 17292–17297 (2010).
Sen, A. et al. Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proc. Natl Acad. Sci. USA 109, 20667–20672 (2012).
Bon, A. Le et al. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14, 461–470 (2001).
Lin, J. Da et al. Distinct roles of type I and type III interferons in intestinal immunity to homologous and heterologous rotavirus infections. PLoS Pathog. 12, 1–29 (2016).
Nice, T. J. et al. Type I interferon receptor deficiency in dendritic cells facilitates systemic murine norovirus persistence despite enhanced adaptive immunity. PLoS Pathog. 12, 1–19 (2016).
Islam, K. B., Nilsson, L., Sideras, P., Hammarström, L. & Smith, C. I. E. TGF-β1 induces germ-line transcripts of both IgA subclasses in human B lymphocytes. Int. Immunol. 3, 1099–1106 (1991).
Stavnezer, J. & Kang, J. The surprising discovery that TGF specifically induces the IgA class switch. J. Immunol. 182, 5–7 (2009).
Chanab, D., Boucard-Jourdin, M. & Paidassi, H. Gut Inflammation in Mice Leads to Reduction in αvβ8 Integrin Expression on CD103+CD11b- Dendritic Cells. J. Crohns Colitis 11, 258–259 (2017).
Atarashi, K. et al. ATP drives lamina propria TH17 cell differentiation. Nature 455, 808–812 (2008).
Annes, J. P., Munger, J. S. & Rifkin, D. B. Making sense of latent TGFβ activation. J. Cell Sci. 116, 217–224 (2003).
Ahamed, J. et al. In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor- 21. Blood 112, 3650–3660 (2008).
Castigli, E. et al. Impaired IgA class switching inn APRIL-deficient mice. Proc. Natl Acad. Sci. USA. 101, 3903–3908 (2004).
Mora, J. R. et al. Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314, 1157–1160 (2006).
Worthington, J. J., Klementowicz, J. E. & Travis, M. A. TGFβ: a sleeping giant awoken by integrins. Trends Biochem. Sci. 36, 47–54 (2011).
Jenkins, G. The role of proteases in transforming growth factor-β activation. Int. J. Biochem. Cell Biol. 40, 1068–1078 (2008).
Seo, G.-Y. et al. Retinoic acid, acting as a highly specific IgA isotype switch factor, cooperates with TGF- 1 to enhance the overall IgA response. J. Leukoc. Biol. 94, 325–335 (2013).
Wohn, C. et al. Absence of MHC class II on cDC1 dendritic cells triggers fatal autoimmunity to a cross-presented self-antigen. Sci. Immunol. 5, eaba1896 (2020).
Caton, M. L., Smith-Raska, M. R. & Reizis, B. Notch–RBP-J signaling controls the homeostasis of CD8—dendritic cells in the spleen. J. Exp. Med 204, 1653–1664 (2007).
Cascalho, M., Ma, A., Lee, S., Masat, L. & Wabl, M. A quasi-monoclonal mouse. Science 272, 1649–1652 (1996).
McCarty, J. H. Selective ablation of v integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132, 165–176 (2004).
Ogle D. H., Wheeler P., D. A. FSA: Fisheries Stock Analysis. R package version 0.8.26, https://github.com/droglenc/FSA. (2019).
Acknowledgements
We thank Prof. Dan Kaplan for sharing huCD207-DTA mice and Prof. Ulrich Kalinke for sharing the IFNARflox mice. We thank Thibault Andrieu and Sébastien Dussurgey for technical assistance for flow cytometry and cell sorting (AniRA – flow cytometry platform, SFR BioSciences, UMS34444/US8) and Céline Anglereaux for help at the animal facility (PBES, SFR BioSciences). We thank Ningguo Feng for continuing technical guidance. The following reagent was obtained through the NIH Tetramer Core Facility: H-2K(b) VGPVFPPGM BV421 (anti-RV VP7 tetramer). This work benefited from data assembled by the ImmGen consortium. We thank the Lahl, Paidassi, Defrance and Agace laboratories for many fruitful discussions and Prof. Allan Mowat for critical reading of the manuscript. Servier Medical Art illustrations (under Creative Commons Attribution 3.0 Unported License) were used and modified to realize the graphical abstract (https://smart.servier.com/). K.L. was supported by the Crohn’s and Colitis Foundation of America, a Vetenskaprådet Young Investigator Award (2014-3595), the Ragnar Söderberg Foundation Fellowship in Medicine, a Lundbeck Foundation Research Fellowship, the Åke Wiberg Foundation, the Carl Trygger Foundation and the Crafoord Foundation. HP was supported by the Agence Nationale de la Recherche (ANR-13-PDOC-0019) and the Marie Sklodowska-Curie Actions for People Program of the European Union (PIIF-GA-2012-330432). S.T. was supported by a PhD fellowship from the French Ministry of Higher Education and by a technique sharing travel grant from the Society for Mucosal Immunology. J.H. was supported by a MARIE CURIE Fellowship as part of the EU-funded project HC Ørsted Postdoc.
Author information
Authors and Affiliations
Contributions
J.N., S.T., J.H., H.P., and K.L. conceived and designed the project; J.N., S.T., J.H., I.U., A.G.L., H.P., and K.L. designed experiments; J.N., S.T., J.H., M.B-J., V.B., K.G.M., L.J.G., K.F.T., H.P., I.U., A.G.L., and K.L. performed experiments and analysed the data; D.P., B.M., and H.G. provided essential tools; O.T. and T.D. provided advice on conceptualization and experimental design, and participated in editing the paper; J.N., S.T., H.P., and K.L. wrote the paper; H.P. and K.L. supervised the study and acquired funding; all authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Nakawesi, J., This, S., Hütter, J. et al. αvβ8 integrin-expression by BATF3-dependent dendritic cells facilitates early IgA responses to Rotavirus. Mucosal Immunol 14, 53–67 (2021). https://doi.org/10.1038/s41385-020-0276-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-0276-8
This article is cited by
-
Concise review: The heterogenous roles of BATF3 in cancer oncogenesis and dendritic cells and T cells differentiation and function considering the importance of BATF3-dependent dendritic cells
Immunogenetics (2024)
-
Establishment of isotype-switched, antigen-specific B cells in multiple mucosal tissues using non-mucosal immunization
npj Vaccines (2023)
-
APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFβ-mediated checkpoints
Nature Immunology (2023)
-
Dendritic cell functions in the inductive and effector sites of intestinal immunity
Mucosal Immunology (2022)
-
Novel antigen-presenting cell imparts Treg-dependent tolerance to gut microbiota
Nature (2022)
