
Abstract
Thymic stromal lymphopoietin (TSLP), an epithelial cell-derived cytokine, exhibits both pro-inflammatory and pro-homeostatic properties depending on the context and tissues in which it is expressed. It remains unknown whether TSLP has a similar dual role in the airways, where TSLP is known to promote allergic inflammation. Here we show that TSLP receptor (TSLPR)-deficient mice (Tslpr−/−) and mice treated with anti-TSLP antibodies exhibited increased airway inflammation and morbidity rates after bleomycin-induced tissue damage. We found that signaling through TSLPR on non-hematopoietic cells was sufficient for TSLP’s protective function. Consistent with this finding, we showed that TSLP reduces caspase-1 and caspase-3 activity levels in primary human bronchial epithelial cells treated with bleomycin via Bcl-xL up-regulation. These observations were recapitulated in vivo by observing that Tslpr−/− mice showed reduced Bcl-xL expression that paralleled increased lung caspase-1 and caspase-3 activity levels and IL-1β concentrations in the bronchial-alveolar lavage fluid. Our studies reveal a novel contribution for TSLP in preventing damage-induced airway inflammation.
Similar content being viewed by others
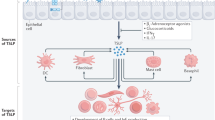


Introduction
Thymic stromal lymphopoietin (TSLP) is a member of the IL-2 cytokine family and is a distant paralog of IL-7. TSLP signals via a receptor which includes the IL-7 receptor α-chain (IL-7Rα CD127) and the unique TSLPR chain. TSLPR-deficient mice1 have been extensively used to determine the contribution of TSLP-TSLPR signaling to homeostasis and disease. By using this genetic approach, it has been shown that TSLP can play a dual role depending on the context and tissues in which it is expressed: it can be pro-inflammatory in the context of lung and skin allergic disorders2 and pro-homeostatic in others, such as colitis3. We recently found that patients with sepsis-induced acute respiratory distress syndrome (ARDS) exhibited increased plasma TSLP levels4. ARDS is associated with lung epithelial cell loss as well as increased neutrophil recruitment, survival, and mediator release, which are all thought to contribute to poor ARDS outcomes. Despite our knowledge about ARDS pathophysiology, the immunologic processes that can cause and/or prevent these changes are incompletely understood. Based on the evidence that highlights TSLP’s pivotal role in the regulation of inflammation, we were prompted to investigate TSLP’s contribution to the airway inflammatory process associated with tissue damage, a potential mechanism of injury amplification in ARDS5.
Using mice with a deficiency in TSLPR1 and mice treated with anti-TSLP neutralizing antibodies in a model of bleomycin-induced acute tissue damage and airway inflammation, we discovered that TSLP can reduce inflammation and morbidity rates after bleomycin administration. We further used in vitro and in vivo approaches to show that TSLP’s protective function is mediated by its ability to inhibit apoptosis and caspase-1 activity and, consequently, the production of the caspase-1 substrate and pro-inflammatory cytokine, IL-1β. Collectively, these data are the first to demonstrate that TSLP can play a protective role in airway inflammation by regulating the epithelium’s response to damage.
Results
TSLP protects mice from bleomycin-induced airway inflammation
It has been shown that injury is one of the main triggers for TSLP release from damaged cells6. Therefore, we decided to use a model of bleomycin-induced lung injury, which causes acute tissue damage and airway inflammation7,8 to assess the contribution of TSLP to the outcomes of this airway insult. First, we assessed TSLP expression levels in mice at 7 days after oropharyngeal (o.p.) bleomycin administration. A significant increase in TSLP mRNA expression and protein levels in lung tissue and bronchioalveolar fluid (BALF), respectively, was observed in bleomycin-treated mice when compared with saline-treated mice (Fig. 1a, b).

a–n Mice were administered either sterile saline (pyrogen-free 0.9% NaCl) or bleomycin (100ug) (BLM) on days 1, 3, and 5, and euthanized at day 7. a, b Tslp mRNA expression levels in the lung (a) and protein amounts in the BALF (b) of C57BL/6 mice. Data were pooled from three independent experiments (n = 10–13 mice). Tslp mRNA was detectable in 3 out of 10 mice treated with saline. Data are shown as mean + SEM with squares representing values from individual mice. c–g Total cell numbers in the BALF (c), representative BALF cell cytocentrifuge preparations stained with diff-quik (neutrophils are indicated by yellow arrows) (d), BALF neutrophil numbers (e), representative flow cytometry profile of neutrophils (Gr-1+ CD11b+) in dissociated lung tissues (f) and numbers of neutrophils (Gr-1+ CD11b+) in the lungs (g) of Tslpr+/+ mice (n = 10–18) and Tslpr−/− mice (n = 19-20). h–m Hematoxylin and eosin (HE) histopathology showing that a Tslpr+/+ mouse treated with saline (h, k) had no changes in its bronchioles or bronchiolar lumens (Br), vessels (V), or alveolar spaces (a). Tslpr+/+ mice treated with bleomycin (i, l) had multifocal mild to moderate perivascular and peribronchiolar inflammation and lower numbers of inflammatory cells within the alveoli. Tslpr−/− mice treated with bleomycin (j, m) had moderate to regionally severe multifocal inflammation with more severe alveolar neutrophilic inflammation. h–j bar = 200 µm; Figures k–m, bar = 50 µm. n Weight loss in Tslpr+/+ (n = 16) and Tslpr−/− mice (n = 16) are plotted as a percentage of starting weight. Data in h–m are representative of similar results that were obtained in three independent experiments. Data in c, e, g and n were pooled from the five experiments and are shown as mean + SEM with squares representing values from individual mice. P value was calculated by Mann–Whitney test. In a, b, *P < 0.05 and **P < 0.01 versus corresponding values for saline-treated mice. In c, e, g and n, *P < 0.05, ***P < 0.005, and ****P < 0.001 versus corresponding values for Tslpr+/+ mice (controls).
We then assessed the role of TSLP in bleomycin-induced airway inflammation using a genetic approach. For this purpose, we used Tslpr−/− mice. In comparison with wild type (Tslpr+/+) mice, which had a BALF cellularity of 5.3 × 105 ± 6.8 × 104 7-days post-bleomycin instillation, Tslpr−/− mice exhibited a marked increase in BALF cellularity (7.2 × 105 ± 3.7 × 104, n = 16–20 mice/group, P < 0.05) (Fig. 1c). Examination and differential quantification of cyto-centrifuged BALF preparations stained with a modified Wright Giemsa stain revealed increased neutrophilia in Tslpr−/− mice when compared with Tslpr+/+ mice (Fig. 1d, yellow arrows; and Fig. 1e). Moreover, the frequency and total number of neutrophils (Gr-1+ CD11b+) observed by flow cytometric analysis of dissociated lung tissue was significantly greater in Tslpr−/− mice (Fig. 1f, g). Pathology lung injury scores in Tslpr+/+ mice were significantly lower (range = 8–12; median 9.5, n = 6) than in Tslpr−/− mice (range = 11–17; median 13.5, n = 6) after bleomycin administration. Tslpr−/− mice exhibited more widespread and severe inflammation (Fig. 1j, m) than Tslpr+/+ mice (Fig. 1i, l), with a more pronounced neutrophilic component to the inflammatory infiltrate and greater disruption of the underlying architecture, including expansion of the interstitium by inflammatory cells in the more severely affected mice. In contrast, saline-treated Tslpr+/+ and Tslpr+/+ mice showed only rare perivascular inflammatory cells (Fig. 1h, k). In concordance with the increased neutrophil numbers in the airways, Tslpr−/− mice exhibited significantly higher BALF and lung expression of the neutrophil chemokine KC when compared with Tslpr+/+ mice at 7 day after bleomycin administration (fig. S1). Body weight loss, which is associated with increased inflammation and is a good indicator of disease severity, was greater in Tslpr−/− mice than in Tslpr+/+ mice at 7 day after bleomycin administration (Fig. 1n).
Consistent with the phenotype observed in Tslpr−/− mice, bleomycin-treated C57BL/6 mice that were treated with anti-TSLP neutralizing antibodies exhibited an increase in airway neutrophilia (Fig. 2a, b) and significantly increased body weight loss (Fig. 2c) when compared with bleomycin exposed C57BL/6 mice treated with isotype control.

Mice were administered bleomycin (100 μg) (BLM) on days 1, 3, and 5, and euthanized at day 7. Mice were administered once i.p. with 200 μg anti-TSLP antibody or isotype 24 h prior to bleomycin administration. a–c BALF neutrophil numbers (a), numbers of neutrophils (Gr-1+ CD11b+) in the lungs (b), and weight loss plotted as a percentage of starting weight (c) in C57BL/6 mice treated with either isotype (n = 10) or anti-TSLP (n = 6–8). Data, shown as means + SEM with triangles representing values from individual mice, were pooled from the three independent experiments, each of which gave similar results. P value was calculated by Mann–Whitney test. *P < 0.05 versus corresponding values for isotype-treated mice.
Numerous studies have shown that the fibrotic phase in the bleomycin-induced acute lung injury model can be observed at 14 day after bleomycin last administration9. Accordingly, we scored lung fibrosis on Masson’s trichrome stained lung tissues from mice harvested 14 day following bleomycin administration (Fig. S2A–D). Overall, a trend toward lower fibrosis scores was observed in Tslpr−/− mice (range = 3–6; mean = 4.5, n = 6) when compared with Tslpr+/+ mice (range = 4–6; mean = 5.2, n = 5) (Fig. S2E), although this difference was not statistically significant. This observation suggests that TSLP–TSLPR interactions may have a limited impact on the fibrosis phase of the bleomycin-induced acute lung injury model.
To determine whether TSLP–TSLPR interactions can influence acute inflammatory responses in other models, we examined the effects of intranasal (i.n.) LPS administration in Tslpr−/− mice. In contrast to the bleomycin model, Tslp+/+ and Tslp−/− mice did not exhibit an increase in TSLP mRNA expression in lung tissues at 6 hours after LPS i.n. administration (Fig. S3A). Moreover, LPS-induced airway inflammation was not influenced by TSLP-TSLPR interactions (Fig. S3B–D). We think that these data are in concordance with previous studies that have shown that primary human airway epithelial cells express low levels of TLR4 and therefore do not release TSLP in response to LPS10. In agreement with these studies, we observed that human bronchial epithelial cells (HBECs) obtained from three different donors up-regulate TSLP expression in response to bleomycin or TLR2 and TLR3 ligands used as positive controls, but not LPS (Fig. S4). Based on this evidence, we decided to evaluate the effects of oropharyngeal polyinosine-polycytidylic acid (Poly I:C) administration, a TLR3 agonist, in Tslpr−/− mice. In comparison with Tslpr+/+ mice, Tslpr−/− mice exhibited a marked increase in BALF neutrophil numbers and the number of neutrophils in the lungs (Fig. S5A, B). These observations suggest that the down-regulatory effects of TSLP–TSLPR signaling on airway inflammation is restricted to models in which TSLP production is induced, such as the bleomycin and Poly I:C models.
Collectively, these findings indicate that TSLP reduces airway neutrophilic inflammation induced by bleomycin and Poly I:C and protects from bleomycin-induced body weight loss.
TSLP–TSLPR signaling induces an up-regulation in Bcl-xL expression but does not influence the expression of Th1 and Th2 cytokines in bleomycin-treated mice
Serum albumin concentrations in BALF from Tslpr−/− mice increased at 7 day after bleomycin oropharyngeal administration but were similar to those found in Tslpr+/+ mice (fig. S6). Therefore, the increased airway inflammatory response in Tslpr−/− mice cannot be explained by an increase in vascular leak.
It has been shown that the TSLP-mediated skewing of the T cell response toward a Th2 phenotype and/or suppression of exacerbated Th1 responses can be protective in certain parasitic infections and colitis models3,11. Importantly, there is evidence that Th2 cytokines, such as IL-13 and IL-4, whose production can be triggered by TSLP12,13, can contribute to reduced bleomycin-induced airway inflammation14,15. Based on this evidence, we decided to investigate whether TSLP-TSLPR interactions induce an increase in the Th2 response in bleomycin-treated mice. Our data showed no significant differences in IL-13 and IL-4 mRNA (Fig. 3a, b) and protein concentrations in the BALF (Fig. 3d, e) of Tslpr+/+ and Tslpr−/− mice treated with bleomycin. Moreover, IFNγ mRNA (Fig. 3c) and protein concentrations in the BALF and lung (Fig. 3f, g) were also similar in Tslpr+/+ and Tslpr−/− mice. These data suggest that TSLP-TSLPR interactions do not protect from bleomycin-induced inflammation by altering the Th1/Th2 response to injury. Based on these results, we decided to investigate alternative pathways by which TSLP may protect from bleomycin-induced inflammation. Mouse studies showed that TSLP–TSLPR interactions can contribute to CD8 T cell homeostasis and natural helper cell resistance to corticosteroids16,17. However, we did not observe a significant reduction in lymphocyte numbers in the BALF from Tslpr−/− mice when compared with Tslpr+/+ mice (Fig. S7) suggesting that the effects of TSLP on the lymphoid compartment do not contribute to protection from bleomycin-induced airway inflammation.

a–f Il13, Il4, and Ifng mRNA expression levels in the lung (a–c) and protein concentrations in the BALF (d–f); IFNγ protein concentrations in lung (g); (H-I) Bcl2l1 (Bcl-xL) (g) and Bcl2 (h) mRNA expression levels in the lung in Tslpr+/+ (n = 9–15) and Tslpr−/− mice (n = 9–14) 7-days following bleomycin administration (100 μg) (BLM), which was given on days 1, 3 and 5. Lung mRNA expression data in a–c and h–i for Tslpr+/+ and Tslpr−/− bleomycin-treated mice are expressed in relation to their respective saline-treated controls. Data shown as means + SEM with squares representing values from individual mice, were pooled from the three independent experiments, each of which gave similar results. P value was calculated by Mann–Whitney test. *P < 0.02 versus corresponding values for Tslpr+/+ mice (controls).
There is extensive literature supporting a protective role for Bcl-2 and Bcl-xL in models of acute lung injury, including bleomycin-induced damage and inflammation18,19,20,21,22. In concordance with this evidence, Tslpr−/− mice exhibited significantly lower mRNA expression levels for Bcl-xL (Fig. 3h) while a slight, but not significant, increase in Bcl-2 mRNA expression levels (Fig. 3i) was observed when compared with Tslpr+/+ mice treated with bleomycin. These observations indicate that TSLP–TSLPR signaling may mediate its protective effects during bleomycin-induced airway inflammation via Bcl-xL up-regulation.
TSLP reduces caspase-1 and caspase-3 activity in bleomycin-treated human primary bronchial epithelial cells via Bcl-xL
Accordingly, we adopted a reductionist approach to dissect mechanisms by which the TSLP-Bcl-xL axis may protect from bleomycin-induced inflammation. For this purpose, we set out to identify potential TSLP target cells during bleomycin-induced inflammation. We have shown that TSLP acts on myeloid cells to down-regulate the inflammatory response in the cecal ligation and puncture (CLP) model of sepsis4. Therefore, we used mice in which TSLP–TLSPR signaling is impaired in myeloid cells (Lys-Cre+; Tslprfl/fl mice) to examine whether TSLP reduces inflammation by directly influencing myeloid cell function. As shown in Fig. S8A–C, Lys-Cre+; Tslprfl/fl mice did not show increased bleomycin-induced inflammation and morbidity. Next, we performed studies with mixed bone marrow chimeric mice that included lethally irradiated Tslpr+/+ and Tslpr−/− mice that were adoptively transferred with bone marrow cells obtained from either Tslpr+/+ or Tslpr−/− mice. Our analysis of peripheral blood chimerism indicates robust hematopoietic reconstitution of Tslpr+/+ and Tslpr−/− mice with Tslpr+/+ bone marrow cells (Fig. 4a, b). Moreover, Tslpr+/+ recipient mice could be successfully reconstituted with Tslpr−/− bone marrow cells (Fig. 4c).

a–c Peripheral blood analyzed from (a) Tslpr+/+ (CD45.1) and (b) Tslpr−/− (CD45.2) recipient mice shows blood chimerism (red box) arising from Tslpr+/+ (CD45.2 or CD45.1) transplanted bone marrow cells. c Blood chimerism (red box) in Tslpr+/+ (CD45.1) recipient mice arising from Tslpr−/− (CD45.2) transplanted bone marrow cells. Data in a–c are representative of similar results obtained in two independent experiments with 3–5 mice per group. d Mice were administered bleomycin (100 μg) (BLM) on days 1, 3, and 5, and euthanized at day 7. Numbers of neutrophils (Gr-1+ CD11b+ ) in the lungs in Tslpr+/+ (n = 4–10) and Tslpr−/− (n = 4–5) mice that received total body irradiation with two exposures of 550 cGy separated by 3–4 h prior to receiving 5 × 106 bone marrow cells from either Tslpr+/+ or Tslpr−/− mice by i.v. within 4 h of secondary irradiation. Data in d, shown as means + SEM with squares representing values from individual mice, were pooled from the two independent experiments, each of which gave similar results. P value was calculated by Mann–Whitney test. *P < 0.05 and **P < 0.005 vs. Tslpr+/+ + BM cells from Tslpr+/+; #P < 0.05 vs. Tslpr+/+ + BM cells from Tslpr−/−.
The robust hematopoietic reconstitution in Tslpr+/+ and Tslpr−/− mice prompted us to evaluate disease severity after bleomycin administration. As shown in Fig. 4d, Tslpr−/− mice engrafted with either Tslpr+/+ or Tslpr−/− bone marrow cells exhibited increased neutrophil recruitment in the lung tissue when compared with Tslpr+/+ mice engrafted with either Tslpr+/+ or Tslpr−/− bone marrow cells suggesting that TSLP mainly targets radio-resistant resident lung cells during bleomycin-induced airway inflammation. It has been shown that human lung epithelial cells constitutively express TSLPR, and stimulation with TSLP can promote epithelial cell growth and wound repair12.
These observations led us to hypothesize that TSLP may influence airway epithelial cell function to promote homeostasis and thereby protect against bleomycin-induced inflammation. To test this hypothesis in vitro, we decided to use primary human bronchial epithelial cells (HBECs). HBECs can respond to TSLP stimulus by up-regulating Bcl-xL protein expression levels (Fig. S9) indicating that HBECs are a relevant in vitro system to examine how TSLP-induced Bcl-xL contributes to reduce the effects of bleomycin-induced stress on airway epithelium.
An association between increased Bcl-xL expression and decreased caspase-3 has been observed in an acute lung injury model that measured lung epithelial cell apoptosis20. Importantly, we observed that the addition of TSLP inhibited caspase-3 activity in bleomycin-treated HBECs (Fig. 5a). As shown in Fig. 5b, the inhibitory effect of TSLP on caspase-3 activity levels was partially reversed with the addition of WEHI-539, a potent and selective Bcl-xL inhibitor23. It has been shown that Bcl-xL can bind and inhibit the NACHT, LRR, and PYD domains-containing protein (NLRP)1, a key component of the inflammasome complex24,25. Notably, NLPR1 promotes caspase-1-mediated IL-1β and IL-18 maturation, which are key pro-inflammatory cytokines in bleomycin-induced lung injury in humans and mice26. Based on this evidence we tested whether the protective effects of TSLP in bleomycin-induced inflammation are linked to its ability to inhibit caspase-1 activity via Bcl-xL. Our data indicate that pre-incubation with TSLP for 48 h can reduce caspase-1 activity levels in HBECs treated with bleomycin for 24 h (Fig. 5c), an effect that was significantly inhibited by WEHI-539 addition (Fig. 5d).

Caspase-3 (a, b) and caspase-1 (c, d) activity levels, and extracellular ATP (eATP) amounts (e, f) in HBECs treated with or without recombinant human TSLP (100 ng/ml) for 48 h and bleomycin (250 μg/ml) for 24 h (a, c, e); and HBECs treated under the same conditions in the presence of a selective Bcl-xL inhibitor (100 nM) (WEHI-539) or vehicle (Veh) (b, d, f). Data are shown as means + SEM from five independent experiments with squares showing values for individual cell treatments. P values were calculated by one-way ANOVA followed by Tukey’s multiple comparisons test.
A dual role for the NLRP1 and NLRP3 inflammasomes in bleomycin-induced airway inflammation has been reported27. The contribution of the NLRP3 inflammasome to caspase-1 activation in bleomycin-induced inflammation is triggered by the release of danger signals, such as ATP, from apoptotic airway epithelial cells28. As shown in Fig. 5e, TSLP treatment reduced the extracellular levels of ATP (eATP) generated by HBECs when treated with bleomycin, but this protective effect was significantly attenuated with WEHI-539 addition (Fig. 5f). Overall, these data suggest that TSLP-induced Bcl-xL can inhibit cell apoptosis and prevent caspase-1 activation by reducing the release of danger signals that trigger the activation of the NLRP3 inflammasome.
The TSLP–TSLPR pathway reduces caspase-1 and caspase-3 activity levels and airway inflammation in bleomycin-treated mice
Our in vitro data seem to indicate that TSLP’s protective effects in bleomycin-induced airway inflammation may be explained by its ability to up-regulate Bcl-xL that in turn can prevent cell apoptosis and limit caspase-1 activity and the consequential generation of pro-inflammatory cytokines26. To test this hypothesis, we then proceeded to examine whether the suppressive effects of TSLP–TSLPR have an impact on the outcome of bleomycin-induced inflammation. First, we observed that TSLPR-deficient mice were impaired in their ability to up-regulate Bcl-xL protein expression levels in the lung at 1 day after bleomycin administration (Fig. 6a, b). Immunohistochemistry studies for the cleaved or active form of caspase-3 (CC-3) showed a significantly increased ratio of positive CC-3 to total tissue area in lung tissues from Tslpr−/− mice treated with bleomycin (n = 6) compared with Tslpr+/+ mice (n = 6) (Fig. 6c–g). Moreover, caspase 3 activity levels in the lung tissue of Tslpr−/− mice were significantly increased when compared with Tslpr+/+ mice treated with bleomycin (Fig. 6h).

a, b Western blot analysis (a) and densitometry values for Bcl-xL expression (b) in lungs from Tslpr+/+ (n = 7) and Tslpr−/− mice (n = 6) at 1 day after either sterile saline or bleomycin (100 μg) (BLM) administration Data in a are representative of similar results that were obtained in three independent experiments. Data in b, shown as means + SEM with squares representing values from individual mice, were pooled from the three independent experiments, each of which gave similar results. c–g Cleaved caspase 3 (CC-3) immunohistochemistry (c–f) and ratio of positive CC-3 expression relative to total tissue area in saline-treated Tslpr+/+ (c) and Tslpr−/− mice (d) and at 7 day following bleomycin administration in Tslpr+/+ (e) and Tslpr−/− mice (f). Positive anti-cleaved caspase 3 staining is brown (indicated by arrows), with hematoxylin counterstain. Minimal to mild background staining of the bronchiolar epithelium (indicated by “b”) is present. c–f bar = 50 µm. h Caspase-3 activity levels in lungs from Tslpr+/+ (n = 8) and Tslpr−/− mice (n = 10) at 7 day following bleomycin administration. i, j Caspase-1 activity levels in lungs (i), and IL-1β amounts in the BALF (j) of Tslpr+/+ (n = 6–8) and Tslpr−/− mice (n = 6–7) at 1 day after bleomycin administration. Data in g–i were pooled from the three independent experiments and are shown as means + SEM with squares representing values from individual mice. P value was calculated by Mann–Whitney test. In b, g, h, i and j, *P < 0.05, **P < 0.01, and ***P < 0.005 versus corresponding values for Tslpr+/+ mice (controls).
Reduced Bcl-xL expression levels were also associated with increased caspase-1 activity levels in the lungs of Tslpr−/− mice when compared with Tslpr+/+ mice at 1 day after bleomycin administration (Fig. 6i). In bleomycin-challenged mice, it has been shown that increased caspase-1 activity results in increased mature IL-1β levels26. Importantly, IL-1β is required for the increased neutrophil recruitment into the bronchoalveolar space of bleomycin-challenged mice29. In agreement with these reports, we found that IL-1β levels were increased in Tslpr−/− mice that also exhibited increased caspase-1 activity levels at 1d after bleomycin administration (Fig. 6j).
Together, these data suggest that TSLP-TSLPR signaling induces Bcl-xL up-regulation, which reduces cell apoptosis and bleomycin-induced airway inflammation caused by increased caspase-1 and caspase-3 activity in this setting.
Discussion
TSLP has been studied extensively in the context of lung and skin allergic disorders where it promotes inflammatory Th2 responses30; however, it is becoming clear that TSLP also influences other disorders and multiple organ systems, including accelerated wound healing in inflammatory bowel disease (IBD), promotion of cancer, and exacerbation of autoimmunity30. Importantly, mouse models of these disorders such as IBD, have shown that TSLP can play a beneficial role by limiting inflammation in response to damage and/or by promoting wound healing3,11. ARDS is characterized by potent inflammatory responses within the lungs that result in widespread damage to the alveolar-capillary barrier, flooding of the airspaces with protein-rich edema fluid, and severe gas-exchange abnormalities31. Recently, we observed increased plasma TSLP levels in sepsis-induced ARDS patients4. Based on our own research that TSLP can reduce inflammation and morbidity in the CLP model of sepsis4, we hypothesized that TSLP may also play a protective role in a mouse model of damage-induced inflammation that resembles the early inflammatory stages of ARDS. For this purpose, we treated mice with a deficiency in the TSLP-specific TSLPR subunit required for TSLP signaling1 with bleomycin, a medication that can induce lung tissue damage and inflammation. We found that TSLPR-deficient mice exhibited increased lung injury, inflammation in the airways and morbidity after bleomycin administration (Fig. 1). A similar phenotype was observed with the administration of anti-TSLP antibodies to C57BL/6 mice (Fig. 2) confirming that TSLP-TSLPR ligation confers protection in a model of damage-induced airway inflammation.
As a Th2 cytokine, TSLP might well be expected to alter matrix deposition and fibrosis. However, we did not observe evidence for a significant TSLP-mediated pro-fibrotic effect at 14 day after bleomycin administration (Fig. S2). This observation suggests that TSLP-TSLPR interactions can reduce inflammation with a reduced impact on the fibrosis phase of the bleomycin-induced acute lung injury model. A similar phenotype has been recently reported for mice deficient in the inducible T cell costimulatory molecule (ICOS) involved in type 2 inflammatory responses32. Like TSLP, ICOS protects against bleomycin-induced acute lung injury without having a pro-fibrotic effect32. Based on this evidence, we think that Th2-associated mediators/molecules can down-regulate inflammation without promoting lung fibrosis after bleomycin administration.
In contrast to our observations, Herro et al. reported a significant reduction in the fibrosis phase of bleomycin-induced acute lung injury in Tslpr−/− mice when compared with Tslpr+/+ mice33. We can only speculate at this point that well-known factors that can influence bleomycin potency such as the route of administration (i.t. vs. o.p.)34 contributed to the development of acute lung injury models of different severities and TSLP concentrations in the airways that may or may not be enough to induce fibrosis in tissues.
The next question in our study was to understand the mechanisms by which TSLP exerts its protective role in the airways upon bleomycin-induced damage. It is well-known that TSLP can trigger the production of Th2 cytokines, such as IL-13 and IL-4. Despite the critical role of these cytokines in the promotion of allergic inflammation, IL-13 and IL-4 can reduce bleomycin-induced airway inflammation14,15. However, we did not observe that TSLP–TSLPR signaling influences IL-4 and IL-13 expression in the airways (Fig. 3a, b), instead, we found that TSLP–TSLPR signaling induces an up-regulation in the expression of the anti-apoptotic molecule Bcl-xL (Fig. 3d). By using a genetic approach, we showed that Bcl-xL up-regulation is associated with reduced apoptosis and caspase-1 activity levels. By reducing caspase-1 activity and the consequential generation of pro-inflammatory cytokines such as IL-1β (Fig. 6), TSLP–TSLPR signaling can limit the magnitude airway inflammatory response triggered by bleomycin. This finding is consistent with Bcl-xL’s known ability to bind and inhibit NLRP1, a component of the inflammasome that leads to pro-IL-1β cleavage into mature IL-1β via caspase-1 activation. Interestingly, TSLP production can be induced by IL-1β35,36, suggesting the existence of a TSLP-mediated negative feedback loop that can limit the magnitude of the inflammatory response to injury.
TSLP’s ability to protect against bleomycin-induced inflammation by limiting the amounts of mature IL-1β generated by caspase-1 is particularly relevant to our understanding of ARDS pathogenesis considering the extensive amount of literature supporting a detrimental role for this pro-inflammatory cytokine in this disorder37,38,39,40,41. It has been shown in vitro that IL-4 can suppress NLRP3-dependent caspase-1 activation and the subsequent IL-1β secretion by macrophages in a transcription-independent manner42. However, there is no evidence that IL-4 or other Th2-associated mediators use a similar mechanism of protection to what we report here for TSLP in in vivo models of damage-induced inflammation. In fact, there is very limited information on the cellular and molecular mechanisms by which Th2-associated mediators protect in these models. For example, it has been shown that IL-4 and IL-5 are anti-inflammatory in models of bleomycin-induced airway inflammation by regulating T cell and myeloid cell expansion, respectively15,32 but the molecular mechanisms are unknown.
By using mixed bone marrow chimeric mice, we found that neutrophil recruitment in the lung tissue in response to bleomycin largely depends on TSLP–TSLPR signaling in radio-resistant resident lung cells (Fig. 4e). It is important to point out, though, that we did observe a significant increase in lung neutrophilia in Tslpr+/+ mice that received Tslpr−/− bone marrow cells when compared with Tslpr+/+ mice that received Tslpr+/+ bone marrow cells. This suggests a partial contribution of hematopoietic cells expressing TSLPR to protection from bleomycin-induced airway inflammation. Studies are under way to attempt to identify these hematopoietic cells targeted by TSLP in our model.
The fact that human lung epithelial cells constitutively express TSLPR and stimulation with TSLP promotes epithelial cell growth and wound repair12 led us to hypothesize that airway epithelial cells may be one of the main targets for TSLP upon bleomycin-induced damage. We provided evidence that supports our hypothesis by showing that the TSLP-Bcl-xL axis reduced caspase-1 activity in HBECs upon bleomycin treatment (Fig. 5c). Interestingly, our in vitro studies also showed that TSLP-induced Bcl-xL can reduce the release of danger signals from apoptotic cells that can mediate the activation of the NLRP3 inflammasome and the consequent increase in caspase-1 activity levels (Fig. 5c–f). However, our in vivo data showed that TSLP–TSLPR signaling does not significantly influence the BALF levels of danger signals, such as eATP and uric acid (Fig. S10). These data suggest that the in vivo environment may provide additional pro-homeostatic signals that may reduce airway epithelial cell susceptibility to bleomycin-induced damage and apoptosis, and hence excessive caspase-1 activation via the NLRP3 inflammasome. For example, endothelial cells have been shown to significantly contribute to homeostasis in acute lung injury by secreting mediators with known anti-apoptotic effects on the endothelium itself and epithelium, such as vascular endothelial growth factor (VEGF)43. Despite this evidence, we cannot rule out a more significant contribution of TSLP to the inhibition of caspase-1 activation via the NLRP3 inflammasome in other models of airway inflammation where the NLRP3 may play a more dominant role, such as with rhinovirus44 and Pseudomonas aeruginosa45 infection, as well as exposure to cigarette and biomass fuel smoke46.
Overall, this study provides additional novel evidence to the growing body of work reporting that mediators and cells typically associated with the development of airway inflammation in the context of chronic Th2 disorders, such as allergy and asthma, can be protective in sepsis and damage-induced airway inflammation4,32,47,48,49. Like other Th2-associated mediators with protective effects in bleomycin-induced inflammation, it remains unclear how TSLP can have a dual role in Th2-mediated disorders, such as asthma and allergy, and bleomycin-induced inflammation. Overall, the data available in the literature seem to suggest that the pro-homeostatic effects of TSLP might be tissue specific (mainly in the gastrointestinal tract) and context specific (parasitic infections and colitis), and manifest within a narrow window of concentrations50. Another plausible explanation for this paradox is that the short and long forms of TSLP (hereafter called sfTSLP and lfTSLP, respectively), which can exhibit anti-inflammatory and pro-inflammatory properties in the airways51, respectively, are differentially up-regulated during allergic inflammation and inflammation induced by cell damage. However, this possibility may only explain the dual effect of TSLP in the human system, as the sfTSLP isoform has not been yet described in mice50.
Our in vitro studies showed that TSLP can protect primary human airway epithelial cells from bleomycin-induced damage; however, the relevance of our data to human disease remains unknown. We recently reported that TSLP levels were increased in sepsis-induced ARDS4 patients, but the small number of patients tested did not allow us to statistically establish whether TSLP levels in these patients associated with clinical outcomes of the disease. We think that additional studies in this regard are imperative to establish whether patients receiving anti-TSLP therapy for the treatment of asthmatic or allergic disorders may be at higher risk of developing ARDS induced by sepsis, injury, or other causes.
Materials and methods
Mice
C57BL/6 mice were purchased from Jackson Laboratories. Tslpr−/− mice on the C57BL/6 background were previously described1. Mice with transgenic expression of Lys-Cre on the C57BL/6 background were purchased from Jackson laboratories and crossed with mice containing loxP-flanked Tslpr alleles52. Mice were bred and maintained at the Seattle Children’s Research Institute Animal Facility. Unless specified otherwise, all experiments were performed using male or female mice that were 12 weeks old at the beginning of the experiment. All animal care and experimentation was conducted in accordance with the current National Institutes of Health guidelines and with the approval of the Seattle Children’s Research Institute Institutional Animal Care and Use Committee.
Bone marrow chimeras
Tslpr+/+ and Tslpr−/− mice received total body irradiation with two exposures of 550 cGy separated by 3–4 h prior to receiving 5 × 106 bone marrow cells from either Tslpr+/+ or Tslpr−/− mice by i.v. within 4 h of the second irradiation. Mice were placed on Baytril antibiotic water (0.5 mg/ml). Baytril treatment began up to 3 days prior to irradiation and continued for 3 weeks post-irradiation. Experiments were performed with the mouse chimeras 7–8 weeks after adoptive transfer of bone marrow cells.
Peripheral blood chimerism was examined to assess the robustness of hematopoietic reconstitution in Tslpr+/+ and Tslpr−/− mice. For this purpose, Tslpr+/+ (CD45.1 genotype) and Tslpr−/− (CD45.2 genotype) recipient mice were lethally irradiated as described above and received bone marrow cells from Tslpr+/+ mice of CD45.2 or CD45.1 genotype, respectively. A separate group of Tslpr+/+ mice (CD45.2 genotype) was reconstituted with Tslpr−/− bone marrow cells (CD45.1 genotype). Peripheral blood was obtained at 7–8 weeks after irradiation and white blood cells were then stained using antibodies to CD45.1 and CD45.2 and analyzed by flow cytometry. Levels of blood chimerism were calculated as the proportion of CD45 labeled white blood cells that expressed the donor CD45.1 or CD45.2 genotypes.
Bleomycin-induced airway inflammation
Briefly, mice were anesthetized via isoflurane inhalation and then oropharyngeal (o.p) administered 100ug of bleomycin sulfate (catalog number BML-AP302, Enzo Life Sciences, Farmingdale, NY) in 25ul sterile saline (pyrogen-free 0.9% NaCl) on days 1, 3, and 5. For morbidity assessment, the mice were observed at least three times daily for the first day, and then twice a day for up to fourteen days. Additionally, the mice were weighed daily. Mice were euthanized if weight loss was above 20% from initial body weight or they were clearly moribund. Data from mice that exhibited weight loss above 20% shown in Figs. 1n and S2C were obtained at the planned euthanasia time (7 days after first bleomycin administration). After 1 or 7 days, the mice were euthanized and the lungs were flushed through an incision in the trachea with 1 mL PBS for BALF collection. The lungs were then flushed an addition four times with PBS for BALF cell collection. Then, portions of the right lungs were collected for RNA, protein, and flow cytometry evaluations.
For histological assessment, the trachea, thymus, heart, and left lungs were collected, a ligation was made between the heart and left lungs, and the lungs were inflated with 10% saline-buffered formaldehyde. Fixed tissues were paraffin embedded, cut into ~4 µm sections, and stained with hematoxylin and eosin (H&E). Pulmonary changes were scored semi-quantitatively by a board-certified veterinary pathologist, not blinded to genotype. Briefly, numerical scores were assigned based on the maximum degree of severity of inflammation in the alveolar space; bronchiolar lumen; interstitium; and perivascular space, with 0 = normal; 1 = few inflammatory cells; 2 = larger foci of inflammatory cells, with preservation of underlying architecture; 3 = larger foci of inflammatory cells with mild changes of underlying vessel, alveolar wall or bronchiolar epithelium; 4 = larger foci of inflammatory cells with disruption or loss of underlying architecture including necrosis and/or hemorrhage. Each compartment was scored individually, and a total inflammatory score was also assigned for global lung inflammation based on similar criteria. A percent lung inflammation involvement score was assigned with 0 = normal; 1 = ≤5%; 2 = 6–10%; 3 = 11–20%; and 4 = >20% pulmonary parenchymal involvement. Bronchiolar epithelium was graded for hyperplasia and inflammation. A cumulative score was calculated, with a maximum possible score of 28.
In a separate experiment, mice were euthanized at 14 days for Masson’s Trichrome staining of lung tissues and fibrosis evaluation. Lung fibrosis scoring was performed using slight modifications of a previously described scoring system53. The pathologist scoring the slides was blinded to the genotype and treatment. Briefly, lungs were assigned a severity score from 0 (normal) to 4 (marked) and an extent score from 1 (occasional alveolar duct and bronchioles) to 4, with the total score representing the severity score times the extent score.
Images of representative lesions were acquired using NIS-Elements BIR 3.2 64-bit or directly from digitally scanned images and plated in Adobe Photoshop Elements. Image brightness and contrast was adjusted using AutoSmart Fix and/or Auto levels (White Balance) manipulations applied to the entire image.
For treatment with the anti-TSLP antibody, mice were i.p. injected once with 200 μg anti-TSLP antibody (clone M702, Amgen) or control IgG 24 h prior to bleomycin administration.
Statistical analyses
All statistics were performed using Prism software (GraphPad Software, Inc., La Jolla, CA). Mann–Whitney U-test, Wilcoxon Signed Rank Test and one-way ANOVA followed by Tukey’s multiple comparisons test were performed as noted in the respective figure legends. P < 0.05 was considered statistically significant. Unless otherwise specified, all data are presented as mean + SEM.
References
Carpino, N. et al. Absence of an essential role for thymic stromal lymphopoietin receptor in murine B-cell development. Mol. Cell Biol. 24, 2584–2592 (2004).
Han, H., Roan, F. & Ziegler, S. F. The atopic march: current insights into skin barrier dysfunction and epithelial cell-derived cytokines. Immunol. Rev. 278, 116–130 (2017).
Reardon, C. et al. Thymic stromal lymphopoetin-induced expression of the endogenous inhibitory enzyme SLPI mediates recovery from colonic inflammation. Immunity 35, 223–235 (2011).
Piliponsky, A. M. et al. Thymic Stromal Lymphopoietin Improves Survival and Reduces Inflammation in Sepsis. Am. J. Respir. Cell Mol. Biol. 55, 264–274 (2016).
Sharp, C., Millar, A. B. & Medford, A. R. Advances in understanding of the pathogenesis of acute respiratory distress syndrome. Respiration 89, 420–434 (2015).
Takai, T. TSLP expression: cellular sources, triggers, and regulatory mechanisms. Allergol. Int. 61, 3–17 (2012).
Snider, G. L., Hayes, J. A. & Korthy, A. L. Chronic interstitial pulmonary fibrosis produced in hamsters by endotracheal bleomycin: pathology and stereology. Am. Rev. Respir. Dis. 117, 1099–1108 (1978).
Nagai, A. et al. Administration of alpha 1-proteinase inhibitor ameliorates bleomycin-induced pulmonary fibrosis in hamsters. Am. Rev. Respir. Dis. 145, 651–656 (1992).
Matute-Bello, G., Frevert, C. W. & Martin, T. R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L379–L399 (2008).
Allakhverdi, Z. et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 204, 253–258 (2007).
Taylor, B. C. et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J. Exp. Med. 206, 655–667 (2009).
Semlali, A., Jacques, E., Koussih, L., Gounni, A. S. & Chakir, J. Thymic stromal lymphopoietin-induced human asthmatic airway epithelial cell proliferation through an IL-13-dependent pathway. J. Allergy Clin. Immunol. 125, 844–850 (2010).
Tatsuno, K., Fujiyama, T., Yamaguchi, H., Waki, M. & Tokura, Y. TSLP directly interacts with skin-homing Th2 cells highly expressing its receptor to enhance IL-4 production in atopic dermatitis. J. Invest. Dermatol. 135, 3017–3024 (2015).
Karo-Atar, D. et al. A protective role for IL-13 receptor alpha 1 in bleomycin-induced pulmonary injury and repair. Mucosal. Immunol. 1, 240–253 (2015).
Huaux, F., Liu, T., McGarry, B., Ullenbruch, M. & Phan, S. H. Dual roles of IL-4 in lung injury and fibrosis. J. Immunol. 170, 2083–2092 (2003).
Rochman, Y. & Leonard, W. J. The role of thymic stromal lymphopoietin in CD8+ T cell homeostasis. J. Immunol. 181, 7699–7705 (2008).
Kabata, H. et al. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat. Commun. 4, 2675 (2013).
Galuppo, M. et al. GW0742, a high affinity PPAR-beta/delta agonist reduces lung inflammation induced by bleomycin instillation in mice. Int. J. Immunopathol. Pharmacol. 23, 1033–1046 (2010).
Qiu, X. et al. Hydrogen inhalation ameliorates lipopolysaccharide-induced acute lung injury in mice. Int. Immunopharmacol. 11, 2130–2137 (2011).
Ben, D. F. et al. TLR4 mediates lung injury and inflammation in intestinal ischemia-reperfusion. J. Surg. Res. 174, 326–333 (2012).
Li, T. et al. Protective effect of polydatin against burn-induced lung injury in rats. Respir. Care 59, 1412–1421 (2014).
Tao, W. et al. Platycodin D attenuates acute lung injury by suppressing apoptosis and inflammation in vivo and in vitro. Int. Immunopharmacol. 27, 138–147 (2015).
Lessene, G. et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat. Chem. Biol. 9, 390–397 (2013).
Bruey, J. M. et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129, 45–56 (2007).
Faustin, B. et al. Mechanism of Bcl-2 and Bcl-X(L) inhibition of NLRP1 inflammasome: loop domain-dependent suppression of ATP binding and oligomerization. Proc. Natl Acad. Sci. USA 106, 3935–3940 (2009).
Hoshino, T. et al. Role of proinflammatory cytokines IL-18 and IL-1beta in bleomycin-induced lung injury in humans and mice. Am. J. Respir. Cell Mol. Biol. 41, 661–670 (2009).
Li, Y. H., Wei, X., Ji, S., Gui, S. Y. & Zhang, S. M. In vivo effects of the NLRP1/NLRP3 inflammasome pathway on latent respiratory virus infection. Int. J. Mol. Med. 41, 3620–3628 (2018).
Riteau, N. et al. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 182, 774–783 (2010).
Gasse, P. et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Invest. 117, 3786–3799 (2007).
Roan, F. et al. The multiple facets of thymic stromal lymphopoietin (TSLP) during allergic inflammation and beyond. J. Leukoc. Biol. 91, 877–886 (2012).
Ware, L. B. & Matthay, M. A. The acute respiratory distress syndrome. N. Engl. J. Med. 342, 1334–1349 (2000).
Hrusch, C. L. et al. ICOS protects against mortality from acute lung injury through activation of IL-5(+) ILC2s. Mucosal Immunol. 11, 61–70 (2018).
Herro, R., Da Silva Antunes, R., Aguilera, A. R., Tamada, K. & Croft, M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J. Allergy Clin. Immunol. 136, 757–768 (2015).
Egger, C. et al. Administration of bleomycin via the oropharyngeal aspiration route leads to sustained lung fibrosis in mice and rats as quantified by UTE-MRI and histology. PLoS ONE 8, e63432 (2013).
Elder, M. J., Webster, S. J., Williams, D. L., Gaston, J. S. & Goodall, J. C. TSLP production by dendritic cells is modulated by IL-1beta and components of the endoplasmic reticulum stress response. Eur. J. Immunol. 46, 455–463 (2016).
Bernard, M. et al. IL-1beta induces thymic stromal lymphopoietin and an atopic dermatitis-like phenotype in reconstructed healthy human epidermis. J. Pathol. 242, 234–245 (2017).
Meduri, G. U. et al. Inflammatory cytokines in the BAL of patients with ARDS. Persistent elevation over time predicts poor outcome. Chest 108, 1303–1314 (1995).
Park, W. Y. et al. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 164, 1896–1903 (2001).
Dolinay, T. et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am. J. Respir. Crit. Care Med. 185, 1225–1234 (2012).
Grailer, J. J. et al. Critical role for the NLRP3 inflammasome during acute lung injury. J. Immunol. 192, 5974–5983 (2014).
Fukumoto, J. et al. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am. J. Physiol. Cell Physiol. 305, C182–C189 (2013).
Hwang, I. et al. Non-transcriptional regulation of NLRP3 inflammasome signaling by IL-4. Immunol. Cell Biol. 93, 591–599 (2015).
Koh, H. et al. Protective role of vascular endothelial growth factor in endotoxin-induced acute lung injury in mice. Respir. Res. 8, 60 (2007).
Liu, T. et al. NLRP3 contributes to inflammation, pyroptosis, and mucin production in human airway epithelium upon rhinovirus infection. J. Allergy Clin. Immunol. 3, 777–787 (2019).
McHugh, B. J. et al. Cathelicidin is a “fire alarm”, generating protective NLRP3-dependent airway epithelial cell inflammatory responses during infection with Pseudomonas aeruginosa. PLoS Pathog. 15, e1007694 (2019).
Li, C. et al. The nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 inflammasome regulates bronchial epithelial cell injury and proapoptosis after exposure to biomass fuel smoke. Am. J. Respir. Cell Mol. Biol. 55, 815–824 (2016).
Alves-Filho, J. C. et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 16, 708–712 (2010).
Lai, D. et al. Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis. Cell Death Dis. 9, 369 (2018).
Greenberg, J. A. et al. Distinct T-helper cell responses to Staphylococcus aureus bacteremia reflect immunologic comorbidities and correlate with mortality. Crit. Care 22, 107 (2018).
Tsilingiri, K., Fornasa, G. & Rescigno, M. Thymic stromal lymphopoietin: to cut a long story short. Cell Mol. Gastroenterol. Hepatol. 3, 174–182 (2017).
Dong, H. et al. Distinct roles of short and long thymic stromal lymphopoietin isoforms in house dust mite-induced asthmatic airway epithelial barrier disruption. Sci. Rep. 6, 39559 (2016).
Hongwei, H., Thelen, T. D., Comeau, M. R. & Ziegler, S. F. TSLP-mediated epicutaneous inflammation promotes acute diarrhea and anaphylaxis. J. Clin. Invest. 12, 5442–5452 (2014).
Kim, S. J. et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic. Biol. Med. 101, 482–490 (2016).
Acknowledgements
This work was supported by funding from the National Institutes of Health; Grants AI068731, HL098067, and AR059058 to S.F.Z. HL128361 to J.S.D., and HL113351 to A.M.P. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was supported by funding from the National Institutes of Health; Grants AI068731, HL098067, and AR059058 to S.F.Z. and HL113351 to A.M.P. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
N.J.S., M.C., K.N., V.K. and A.T. performed the experiments and analyzed the data. J.M.S performed the assessment of lung pathology. N.J.S. and A.M.P. designed the study. S.F.Z. analyzed data and provided with Tslpr−/− and Lys-Cre−; Tslprfl/fl mice. J.S.D. provided human bronchial epithelial cells. A.M.P. and N.J.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shubin, N.J., Clauson, M., Niino, K. et al. Thymic stromal lymphopoietin protects in a model of airway damage and inflammation via regulation of caspase-1 activity and apoptosis inhibition. Mucosal Immunol 13, 584–594 (2020). https://doi.org/10.1038/s41385-020-0271-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-0271-0
