
Abstract
Leukotriene B4 receptor 1 (BLT1) triggers the migration of granulocytes and activated T cells; however, its role in B-cell function remains unclear. Here we report that BLT1 is required to induce the production of antigen-specific IgA against oral vaccine through mediating innate immune signals from commensal bacteria. B cells acquire BLT1 expression during their differentiation to IgA+ B cells and plasma cells in Peyer’s patches and the small intestinal lamina propria, respectively. BLT1 KO mice exhibited impaired production of antigen-specific fecal IgA to oral vaccine despite normal IgG responses to systemically immunized antigen. Expression of MyD88 was decreased in BLT1 KO gut B cells and consequently led to diminished proliferation of commensal bacteria-dependent plasma cells. These results indicate that BLT1 enhances the proliferation of commensal bacteria-dependent IgA+ plasma cells through the induction of MyD88 and thereby plays a key role in the production of antigen-specific intestinal IgA.
Similar content being viewed by others


Introduction
Leukotriene B4 receptor 1 (BLT1) is a G-protein-coupled high-affinity receptor for leukotriene B4 (LTB4), a metabolite of arachidonic acid.1 BLT1 is expressed on various types of immune cells, including activated T cells such as Th1 and Th2 cells,2 CD8+ effector T cells,3,4 and Th17 cells;5 mast cell progenitor cells;6 macrophages;7 dendritic cells;8 eosinophils;9 and neutrophils.10,11 The activation of BLT1 by LTB4 primarily induces the Gαi-mediated signaling pathway, which induces cell migration to inflamed tissues where LTB4 is highly concentrated.12,13 In addition, BLT1-mediated signals enhance the FcγR-dependent phagocytic activity of macrophages,7 and IL-12 production by dendritic cells.8 Accumulating evidence has demonstrated that the LTB4–BLT1 axis plays a key role in the development of allergic, inflammatory, and immune diseases, including asthma,14,15 dermatitis,8,11 peritonitis,16 rheumatoid arthritis,17 and experimental autoimmune encephalomyelitis.18 Compared with the intensive analysis regarding the role of the LTB4–BLT1 axis in other immune cells, few studies address B cells. BLT1 is expressed on B cells in human peripheral blood and bone marrow,19,20 and LTB4 enhances B-cell proliferation and IgG and IgM production in vitro.21,22 However, little information is available regarding the role of LTB4–BLT1 signaling in B cells in vivo.
Intestinal tissues uniquely contain numerous B cells that spontaneously differentiate into IgA-producing plasma cells.23,24 Peyer’s patches (PPs) are the inductive site for the generation of antigen-specific IgA-committed B cells, and the intestinal lamina propria (iLP) is the effector site for the production of antigen-specific secretory IgA in the gut.23,24 In PPs, B cells are divided into four distinct subsets, namely B220+IgM+IgA– naive B cells, B220+IgM+IgA+ Ig-class-switching B cells, B220+IgM–IgA+ Ig-class-switched B cells, and B220–IgM–IgA+ plasmablasts.25 During their differentiation from naïve to IgA+ plasmablast cells in the germinal center, B cells start to express CCR9 and α4β7 integrin.26 These proteins enable the IgA+ plasmablasts to traffic into the iLP, where plasmablasts further differentiate into B220–IgM–IgA+ plasma cells that produce copious secretory IgA.25
Lipid is a critical factor in the regulation of gut IgA. Our group previously reported that sphingoshin-1-phosphate modulates the egress of IgA+ plasmablast cells from PPs.25 The expression of type I sphingoshin-1-phosphate receptor is downregulated during the differentiation of B220+IgM+IgA– naive B cells to mature B220+IgA+ B cells but is restored on B220–IgM–IgA+ plasmablast cells, where it is essential for emigration from PPs and trafficking to the iLP.25 In addition, we recently found that dietary palmitic acid enhances the production of intestinal IgA against orally immunized antigen by promoting the recruitment of plasmablast cells to the iLP and stimulating plasma cells to secrete IgA.27 Furthermore, short-chain fatty acid acetate increases gut IgA production through GPR43-dependent induction of retinoic acid synthesis by dendritic cells.28 Therefore, because it controls Ig class-switching, B-cell trafficking, and IgA secretion, fatty acid is an important regulator in the generation of IgA. However, the role of the LTB4–BLT1 axis in the regulation of gut IgA production has not been elucidated.
Understanding the mechanisms driving the induction and production of intestinal IgA is key to developing effective oral vaccines. Here we report that BLT1 expressed on intestinal IgA+ cells mediates innate immune signals from commensal bacteria in the gut to promote the proliferation of antigen-specific IgA in response to oral vaccines.
Results
BLT1 was expressed on intestinal IgA+ B cells
We performed flow cytometry to analyze BLT1 expression on the surfaces of intestinal B cells. The gating strategy for identifying each intestinal B-cell subset was based on the expression pattern of B220, IgM, and IgA, as described previously (Supplementary Fig. S1).25 In the PPs, BLT1 was negligible on B220+IgM+IgA– naïve B cells but was readily apparent on IgA+ B cells, including B220+IgM+IgA+ Ig-class-switching B cells, B220+IgM–IgA+ Ig-class-switched B cells, and B220–IgM–IgA+ plasmablast cells (Fig. 1a). In addition, B220–IgM–IgA+ plasma cells in the iLP showed cell surface expression of BLT1 (Fig. 1a). We then performed quantitative RT-PCR analysis of Blt1 gene expression in each of these B-cell populations (Supplementary Fig. S2 shows representative purity data). Consistent with our findings from flow cytometry, the amount of Blt1 mRNA transcripts was increased in IgA+ B cells when compared with IgM+ naïve B cells in the PPs and remained high in IgA+ plasma cells in the iLP (Fig. 1b). However, the expression of Blt1 mRNA in splenic B cells was scant to nonexistent (Supplementary Fig. S3). These findings suggest that the cell surface expression of BLT1 in intestinal IgA+ B cells is induced at the transcriptional level.

BLT1 was expressed on intestinal IgA+ B cells. a Cell surface expression of BLT1 in 8-week-old BLT1 WT male mice was examined by using flow cytometry. Data are representative of two independent experiments with n = 3 mice per group per experiment. b RNA was purified from the indicated B-cell populations, and quantitative RT-PCR assays were performed to measure Blt1 mRNA expression. The relative amount of Blt1 mRNA is expressed as the ratio to Actinb. Data are expressed as mean ± SEM (duplicate measurements) and are representative of three independent experiments
BLT1 KO mice failed to produce antigen-specific fecal IgA against oral vaccine
The finding regarding BLT1 expression on IgA+ B cells prompted us to examine the role of BLT1 in the induction of intestinal IgA production. To address this issue, we orally immunized BLT1 WT and KO mice by using OVA combined with cholera toxin (CT), a mucosal adjuvant, and measured antigen-specific fecal IgA production by ELISA. The levels of both CT-specific and OVA-specific fecal IgAs were lower in BLT1 KO mice than in WT mice (Fig. 2a). Concurrent ELISPOT assays revealed that the iLP of BLT1 KO mice had fewer antigen-specific IgA-producing cells than did that of their WT counterparts (Fig. 2b). In contrast to the antigen-specific IgA response to oral vaccine, the total amount of IgA in the feces and the number of IgA+ plasma cells in the iLP were not decreased in BLT1 KO mice in the steady state (Supplementary Fig. S4), suggesting that the effects of LTB4−BLT1 signals become evident in the acute phase of immune responses. Unlike oral immunization, intraperitoneal immunization by using OVA with alum as a systemic adjuvant yielded comparable concentrations of OVA-specific serum IgG between BLT1 WT and KO mice (Supplementary Fig. S5).

Oral immunization failed to induce antigen-specific IgA production in BLT1 KO mice. BLT1 WT and KO male mice (age, 8 weeks) were orally immunized by using OVA plus CT on days 0, 7, and 14. a One week after the final immunization, fecal extracts were prepared, and the amount of antigen-specific IgA was analyzed by ELISA. Data are combined from five independent experiments and expressed as mean ± SEM (WT, n = 13; KO, n = 11). Statistical significance was analyzed by using the Mann–Whitney test; **P < 0.01. b One week after the final immunization, cells were isolated from the iLP, and antigen-specific IgA+ plasma cells were counted by using ELISPOT assays. Data are combined from five independent experiments, and each data point represents an individual mouse; bars indicate median values. Statistical significance was analyzed by using the Mann–Whitney test; **P < 0.01 and ***P < 0.001. Representative ELISPOT plates from each group are shown
We next examined whether the impaired fecal IgA antibody responses in BLT1 KO mice led to faulty immunosurveillance. To this end, we orally challenged immunized mice with high-dose CT. Whereas nonvaccinated naïve WT mice developed severe watery diarrhea, orally vaccinated WT mice were protected against CT-induced diarrhea (Fig. 3a). In sharp contrast and despite oral vaccination with CT, BLT1 KO mice developed severe diarrhea to the same extent as nonvaccinated naïve WT mice (Fig. 3a). Indeed, the intestinal water volume of orally vaccinated BLT1 KO mice was comparable to that of nonvaccinated WT mice (Fig. 3b). These results indicate that BLT1 is important for inducing the production of antigen-specific fecal IgA against oral vaccines and thus plays a key role in the development of protective immunity against intestinal pathogens.

Oral vaccination failed to protect BLT1 KO mice from CT-induced diarrhea. BLT1 WT and KO male mice (age, 8 weeks) were orally immunized by using OVA with CT on days 0, 7, and 14. a One week after the final immunization, mice were orally challenged with a high dose of CT. Representative photos of intestines from two independent experiments with n = 2 or 3 per group are shown; arrows indicate the cecum, where water accumulates when diarrhea occurs. b Intestinal water volume was measured. Data are combined from two independent experiments and are expressed as mean ± SEM (WT without vaccination, n = 4; WT with vaccination, n = 5; and KO with vaccination, n = 5)
Signaling through BLT1 promoted the proliferation of IgA+ plasma cells
We wondered why BLT1 KO mice had decreased numbers of antigen-specific IgA+ plasma cells in the iLP and formulated three hypotheses: (i) decreased cell migration from PPs to iLP; (ii) decreased cell survival or increased apoptosis; and (iii) reduced cell proliferation in the iLP. Contrary to our hypothesis of decreased migration, cell surface levels of α4β7 integrin, CCR9, and CCR10—molecules important in cell migration—were comparable between WT and BLT1 KO B cells (Supplementary Fig. S6). In addition, an in vitro migration assay revealed that, unlike BLT1-expressing neutrophils,10,11 IgA+ B cells isolated from PPs did not respond to LTB4 (Supplementary Fig. S7). Refuting our hypothesis of decreased cell survival or increased apoptosis, the ratio of apoptotic to total IgA+ plasma cells did not differ significantly between WT and BLT1 KO mice (Supplementary Fig. S8). Together, these results indicate that neither cell migration nor cell survival or apoptosis is the target of the impaired IgA production in BLT1 KO mice.
We then treated mice with BrdU to assess the proliferation of IgA+ plasma cells in iLP. Flow cytometry revealed that BrdU+IgA+ plasma cells were decreased in BLT1 KO mice when compared with WT mice (Fig. 4a). Consistently, staining for Ki67 protein, a marker for cell proliferation, confirmed decreased IgA+ plasma cell proliferation in BLT1 KO mice (Fig. 4b).29 Immunohistologic analysis showed that WT mice had many IgA+ plasma cells, the nuclei of which contained plentiful BrdU and Ki67, whereas BLT1 KO mice had few BrdU+Ki67+IgA+ plasma cells (Fig. 4c). These data suggest that the impaired IgA production in BLT1 KO mice is likely due to abnormal proliferation of intestinal plasma cells.

BLT1 signaling promoted the proliferation of IgA+ plasma cells. BLT1 WT and KO male mice (age, 8 weeks) were orally immunized by using OVA combined with CT on days 0, 7, and 14. Mice were treated with BrdU on day 20 and euthanized on day 21. a BrdU uptake by IgA+ plasma cells in iLP was analyzed by flow cytometry to examine cell proliferation. Data are combined from three independent experiments, and each data point represents an individual mouse; bars indicate median values. Statistical significance was analyzed by using the Mann–Whitney test; **P < 0.01. b IgA+ plasma cell fraction from the iLP was examined for the expression of Ki67 by flow cytometry. Data are representative of three independent experiments with n = 3 per group. MFI mean fluorescence intensity. c Small intestine was examined by immunohistochemistry. Frozen tissue sections were stained with the indicated antibodies and reagents. Arrows indicate IgA+BrdU+Ki67+ cells. Data are representative of three independent experiments with n = 3 per group. Scale bars, 100 μm
To confirm that a BLT1-mediated signal is required for IgA+ plasma cell proliferation in the gut, we treated orally immunized mice with cyclophosphamide, which inhibits cell proliferation; we then used ELISAs to measure the amount of antigen-specific fecal IgA produced, and we performed ELISPOT assays to count IgA+ plasma cells. Whereas cyclophosphamide decreased antigen-specific IgA responses in WT mice (Fig. 5a, b, see also Fig. 2a, b for comparison data as cyclophosphamide nontreated groups),30 we noted no treatment-associated change in BLT1 KO mice (Fig. 5a, b). Therefore, the amount of antigen-specific intestinal IgA production did not differ between WT and KO mice (Fig. 5a, b). Given that decreased cell proliferation in IgA+ plasma cells in iLP is the causative factor in the decreased IgA response in BLT1 KO mice against oral vaccine, we next sought to evaluate whether disrupting germinal centers in PPs by treating WT mice with anti-CD40L Ab alters the IgA response. As expected, anti-CD40L treatment disrupted germinal centers in the PPs of WT mice (Supplementary Fig. S9a). However, despite the defective germinal center formation in PPs, IgA+ plasma cells in the iLP compartment of anti-CD40L-treated mice proliferated at a comparable level to those in the PBS-treated control group (Supplementary Fig. S9b and c). Moreover, we found that antigen-specific fecal IgA responses were not diminished by treatment with anti-CD40L Ab (Supplementary Fig. S9d). These results collectively demonstrate that cell proliferation is a prerequisite factor in the BLT1-mediated enhancement of antigen-specific intestinal IgA responses.

BLT1-mediated cell proliferation was required for fecal antigen-specific IgA after oral vaccination. BLT1 WT and KO male mice (age, 8 weeks) were orally immunized by using OVA with CT on days 0, 7, and 14 and then were treated with cyclophosphamide on days 18, 19, and 20 to inhibit cell proliferation. a Fecal extracts were prepared on day 20, and the amount of antigen-specific IgA was analyzed by ELISA. Data are combined from three independent experiments, and expressed as mean ± SEM (WT, n = 7; KO, n = 5). Statistical significance was analyzed by using the Mann–Whitney test; N.S. not significant. b On day 21, cells were isolated from iLP, and antigen-specific IgA+ plasma cells were counted by using ELISPOT assays. Data are combined from three independent experiments, and each point represents an individual mouse; bars indicate median values. Statistical significance was analyzed by using the Mann–Whitney test; N.S. not significant. Representative photos of ELISPOT plates from each group are shown
BLT1 KO B cells produced less IgA than did WT cells
Because BLT1 occurs in various immune cells other than B cells, including T cells and granulocytes, we next asked whether intrinsic expression of BLT1 on B cells is crucial for IgA production. To address this issue, we isolated B cells from PPs and incubated them in vitro with or without the IgA-inducing cytokines IL-4, IL-5, IL-6, and TGF-β.31,32,33,34,35,36,37,38 We revealed that the IgA content in the supernatant from BLT1 KO B cells was less than that of WT B cells (Fig. 6a and Supplementary Fig. S10). In addition, the number of BLT1 KO IgM–IgA+ B cells was decreased after in vitro culture, whereas counts of IgM+IgA– naïve B cells were comparable between WT and BLT1 KO samples (Fig. 6b). The selective reduction of IgA+ B cells in BLT1 KO samples was consistent with our in vivo finding that IgA+ B cells—but not IgM+ naïve B cells—expressed BLT1 (Fig. 1a). The cellular source of the LTB4 in these in vitro experiments is unclear; however, we found that IgA+ plasma cells—but not IgM+ naïve B cells—expressed all necessary enzymes responsible for LTB4 production (i.e., Alox-5, Flap, and LTA4 hydrolase) (Supplementary Fig. S11). Collectively, these results showed that BLT1 intrinsically expressed on IgA+ B cells plays a key role in enhancing IgA production by inducing cell proliferation.

BLT1-deficient B cells produced less IgA. B220+ cells were isolated from the PPs of 8-week-old BLT1 WT and KO male mice. Isolated B cells were cultured with lipopolysaccharide (LPS) in the presence or absence of the IgA-inducing cytokines IL-4, IL-5, IL-6, and TGFβ. a After culturing, the supernatant was collected, and the amount of total IgA was quantitated by ELISA. b After culturing, cell counts were calculated by using flow cytometry. Data are combined from two independent experiments with n = 3–4 per group. Statistical significance was analyzed by one-way ANOVA; **P < 0.01; N.S. not significant
BLT1 signaling upregulated microbe-dependent proliferation of intestinal IgA+ cells through the induction of MyD88 for mucosal IgA production
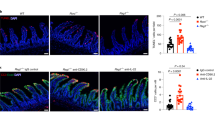
Given that BLT1 signaling promoted intestinal but not systemic antibody production, we considered it likely that environmental differences between the mucosal and systemic compartments influence BLT1-mediated IgA production. Because the intestine is exposed to the abundant microbiota, we treated mice with antibiotics and then orally immunized them with OVA plus CT.30 As expected, antibiotic-treated WT mice had decreased level of antigen-specific IgA in their feces when compared with antibiotic-nontreated WT mice (Fig. 7a). We also found that this decreased level of antigen-specific IgA in antibiotic-treated WT mice was comparable in antibiotic-treated BLT1 KO mice (Fig. 7a). Although statistically no different, WT mice with antibiotics yet tend to show certain a higher extent than BLT1 KO mice with antibiotics (Fig. 7a), indicating that microbiota-dependent and -independent pathway are involved in BLT1-mediated enhancement of IgA. These results indicate that the intestinal microbiota mainly contribute to BLT1-mediated enhancement of the antigen-specific intestinal IgA response.

BLT1 signaling enhanced microbe-dependent proliferation of intestinal IgA+ cells through the induction of MyD88 for mucosal IgA production. a Mice were provided antibiotics mixed in drinking water beginning 1 week prior to oral immunization and continuing until the end of the experiment. BLT1 WT and KO male mice (age, 8 weeks) were orally immunized by using OVA with CT on days 0, 7, and 14. One week after the final immunization, fecal extracts were prepared, and the amount of antigen-specific IgA was analyzed by ELISA. Square symbols indicate WT mice without antibiotic treatment as a control. Data are combined from three independent experiments, and expressed as mean ± SEM (n = 6 per group). Statistical significance was analyzed by using the Mann–Whitney test; N.S. not significant. b RNA was purified from the indicated B-cell populations of 8-week-old BLT1 WT naïve male mice, and quantitative RT-PCR analysis was performed to measure Myd88 mRNA expression. The relative amount of Myd88 mRNA is expressed as the ratio to Actinb. Data are expressed as mean ± SEM (duplicate measurements) and are representative of three independent experiments. c BLT1 WT, BLT1 KO, MyD88 KO, and BLT1/MyD88 double-KO (DKO) male mice (age, 8 weeks) were orally immunized by using OVA with CT on days 0, 7, and 14. One week after the final immunization, fecal extracts were collected, and the amount of antigen-specific IgA was analyzed by ELISA. Data are combined from three independent experiments, and expressed as mean ± SEM (WT, n = 19; BLT1 KO, n = 17; MyD88 KO, n = 5; BLT1/MyD88 DKO, n = 6)
Regarding a possible underlying mechanism, a previous study showed that BLT1-mediated signals induced phosphorylation of STAT1 and JAK2, which induced the expression of MyD88,39 an important TLR-mediated signaling molecule for the proliferation of IgA+ cells.40 These studies prompted us to examine whether MyD88 expression in gut B-cell populations differed between WT and BLT1 KO mice. We found that BLT1 KO IgA+ B cells had less Myd88 mRNA than WT IgA+ B cells (Fig. 7b).
We next examined whether the expression of MyD88 is involved in the production of intestinal IgA against orally administered antigen. To address this issue, we orally immunized MyD88 KO mice and BLT1/MyD88 double-KO (DKO) mice by using OVA with CT and then used an ELISA to measure antigen-specific fecal IgA. We found that antigen-specific IgA responses were diminished in MyD88 KO and BLT1/MyD88 DKO mice and were similar to those of BLT1 KO mice (Fig. 7c). These results indicate that MyD88 facilitates the BLT1-mediated promotion of IgA production. Taken together, our results suggest that BLT1 signaling enhanced the microbe-dependent proliferation of intestinal IgA+ cells through the induction of MyD88 for gut mucosal IgA production.
Discussion
In this study, we showed that BLT1 expression was required for full activation of antigen-specific IgA production against oral vaccine. Of note, BLT1 expression was synchronized with the differentiation of intestinal B cells into IgA+ cells. This process is induced by several molecules, including cytokines such as TGFβ.35,37,38 In addition to cytokines, retinoic acid reportedly induces Ig class-switching from μ to α.41 Interestingly, BLT1 initially was cloned from a human promyelocytic leukemia cell line (HL-60) that had been stimulated with retinoic acid.1 These findings suggest that retinoic acid likely is a master regulator for the induction of BLT1 and IgA transcription. Mechanistically, retinoic acid loosens the chromatin structure around the BLT1 enhancer region, thus leading to increased binding of Runx1.42 It is noteworthy that Runx proteins activate transcription of the germline Ig Cα promoter by cooperating with TGFβ–Smad signaling.43,44,45 Therefore, retinoic acid and TGFβ likely together create the appropriate environment for the simultaneous induction of BLT1 expression and IgA class-switching.
Our group previously identified a commensal bacteria-dependent and highly proliferative population of IgA+ plasma cells that secretes a large amount of IgA in the gut.30 Furthermore, this highly proliferative plasma cell population requires the expression of MyD88 for their development.30 Therefore, commensal bacteria-mediated stimuli through MyD88 likely are a key signaling cascade for the enhancement of IgA production in the gut. MyD88 primarily mediates TLR signaling, whose ligands include lipoprotein, peptidoglycan, LPS, flagellin, and bacterial and viral DNA and RNA; all of these components are abundant in the intestine, which contains tremendous numbers of commensal bacteria. In the current study, we found that MyD88 indeed was an essential molecule for the production of antigen-specific fecal IgA to oral vaccines. Precisely which ligand–TLR interactions are involved in the generation of intestinal IgA is unknown. Because in vitro stimulation with a combination of either LPS or peptidoglycan and IgA-inducing cytokines decreased the production of IgA in BLT1 KO B cells compared with that in WT B cells, we propose that the LPS–TLR4–MyD88 axis and the peptidoglycan–TLR2–MyD88 axis both contribute to the enhancement of IgA production, which was promoted by BLT1-mediated signaling.
The PPs are the induction site for the differentiation of IgA+ B cells from IgM+ naïve B cells in the gut.24 This inductive function in the generation of IgA+ B cells has been attributed to PPs mainly because of the presence of antigen-uptake M cells in the follicle-associated epithelium, retinoic acid-producing dendritic cells, helper T cells and T follicular helper cells for germinal center formation.23 In addition, PPs were recently found to be an essential site for the differentiation of commensal bacteria-dependent and highly proliferative IgA+ plasma cells.30 Furthermore, in the current study, the expression of both BLT1 and MyD88 was induced in IgA+ B cells within PPs. Together these findings suggest that PP-resident commensal bacteria influence the generation and promote the production of intestinal IgA. In this context, it merits mentioning that our group previously identified a unique PP-resident bacteria, Alcaligenes, which was absent from both the iLP and luminal compartments.46 Compared with E. coli LPS, LPS isolated from Alcaligenes only weakly activated dendritic cells to produce less IL-6, suggesting that the intensity of TLR4 signaling differs between bacterial LPS types.47 Therefore, microfloral diversity or uniqueness may influence the signaling intensity of the intestinal TLR4–MyD88 pathway, which also is regulated by the LTB4–BLT1–MyD88 axis, for the production of antigen-specific IgA during oral vaccination.
In addition to commensal bacteria, several lines of evidence suggest that dietary materials (e.g., oils and vitamins) are key factors for the regulation of intestinal immune response including IgA production.48,49 We previously showed that dietary palmitic acid enhanced fecal IgA production against orally immunized antigen by increasing the number of IgA+ plasma cells and their IgA production.27 LTB4 is synthesized from arachidonic acid, an omega-6 essential fatty acid that is not generated in mammals and therefore needs to be acquired through the diet. Because our current findings suggest that the LTB4–BLT1 axis enhances intestinal IgA production, dietary supplementation with omega-6 essential fatty acids (e.g., arachidonic acid and linoleic acid) may be an effective tool to enhance gut immune responses.
In contrast to the intestinal immune system, the LTB4–BLT1 axis was dispensable for the induction of systemic antigen-specific immune responses. Consistent with this finding, we found that the amount of BLT1 expressed in splenic B cells was scant to nonexistent. Our group previously reported that dietary palmitic acid enhanced the production of intestinal IgA but not of systemic IgG.27 In addition, supplementation with acetate in drinking water of mice promoted intestinal IgA but not systemic antibody production.28 These findings suggest that the intestinal immune system is more sensitive to dietary components than is the systemic immune system. Therefore, we propose that nutritional control is a key factor for the development of effective oral vaccines.
The LTB4–BLT1 axis induces the migration of effector T cells, macrophages, eosinophils, and neutrophils.50 In contrast, LTB4 promoted the proliferation of IgA+ intestinal B cells rather than their migration. Recent studies have indicated that the expression of receptors for advanced glycation end products on neutrophils not only promotes cell migration but also enhances LTB4–BLT1-mediated activation of ERK and inhibits the production of inflammatory cytokines.51,52 Therefore, intracellular signaling through BLT1 may influence the output following stimulation of LTB4 (i.e., cell migration, cytokine production, or cell proliferation).
CT is used as a mucosal adjuvant.53 CT binds to GM1 ganglioside, which is expressed on several types of cells, including epithelial cells and antigen-presenting cells. In its mechanism of action as a mucosal adjuvant, CT increases the permeability of intestinal epithelial cells, thereby enhancing the uptake of coadministered antigens, and activates both antigen presentation and Ig class-switching in B cells.53 In the current study, we found that oral immunization of mice with CT induced antigen-specific fecal IgA in WT but not BLT1 KO mice, suggesting that LTB4 production is another key mechanism of CT-mediated immune induction. Indeed, exposure of animals to CT induced LTB4 production in the gut.54 Therefore, CT likely acts as a mucosal adjuvant through the induction of LTB4, thus stimulating gut IgA+ plasma cells to proliferate and enhancing the production of IgA.
In summary, the results of our current study show that the expression of BLT1 on gut IgA+ B cells is important for inducing the production of antigen-specific fecal IgA after oral vaccination by promoting commensal bacteria-dependent proliferation of IgA+ plasma cells through the induction of MyD88.
Materials and methods
Mice
BLT1 KO mice14 and MyD88 KO mice55,56 were generated as previously described, and maintained in specific pathogen-free conditions at the National Institutes of Biomedical Innovation, Health and Nutrition (NIBIOHN; Osaka, Japan). For experiments, BLT1 KO mice were obtained by mating heterozygous mice, and littermate WT mice were used as controls. BLT1/MyD88 DKO mice were obtained by mating BLT1 heterozygous mice with MyD88 KO mice. Male mice were used for experiments after euthanasia by cervical dislocation under anesthesia with isoflurane (AbbVie, North Chicago, Illinois, USA). All experiments were conducted in accordance with the guidelines of the Animal Care and Use Committee and Ethics of Animal Experiments Committee of NIBIOHN (<DS25-2, DS25-3, DS27-47 and DS27-48).
Cell isolation and flow cytometry
Cell isolation and flow cytometry were performed as described previously, with modification.25 PPs were isolated and then cut into small pieces by using scissors and incubated for 30 min at 37 °C with stirring in RPMI1640 medium (Sigma-Aldrich, St. Louis, Missouri, USA) containing 2% (vol/vol) newborn calf serum (Equitech-Bio, Kerrville, Texas, USA) and 2 mg/mL collagenase (Wako Chemicals, Richmond, Virginia, USA). Cell suspensions were filtered through cell strainers (pore size, 70 μm; BD Biosciences, Franklin Lakes, New Jersey, USA).
Samples of iLP were prepared after removal of PPs. Small intestine was opened longitudinally, washed vigorously with ice-cold PBS, cut into ~2cm sections, incubated for 15 min at 37 °C in 0.5 mM EDTA (Nacalai Tesque, Kyoto, Japan) to remove intestinal epithelial cells and intestinal intraepithelial lymphocytes, and then incubated with collagenase as described earlier. Then, LP lymphocytes were obtained as the cell layer between 40 and 75% Percoll (GE Healthcare, Tokyo, Japan) after gradient centrifugation (820 × g, 20 min, 20 °C).
Samples of these cell preparations were stained with an anti-CD16/32 monoclonal antibody (mAb) (TruStain fcX; BioLegend, San Diego, California, USA) to prevent nonspecific staining and with 7-AAD (BioLegend) to detect dead cells. In addition, cells were stained with the following fluorescently labeled mAbs: FITC–anti-IgA (BD Biosciences; catalog no. 559354; dilution, 1:100), PE–Cyanin 7 (PE–Cy7)–anti-IgM (BioLegend; no. 406514; 1:100), BV421–anti-B220 (BioLegend; no. 103240; 1:100), PE–anti-Ki67 (BioLegend; no. 652404; 1:100), PE–anti-α4β7 integrin (BioLegend; no. 120605; 1:20), PE–anti-CCR9 (R&D Systems, Minneapolis, Minnesota, USA; no. FAB2160P; 1:10), Allophycocyanin (APC)–anti-CCR10 (R&D Systems; no. FAB2815A; 1:10), APC–Annexin V (BioLegend; no. 640919; 1:20), APC–anti-BrdU (BD Biosciences; no. 557892; 1:50), PE–rat IgG2a isotype control (BioLegend; no. 400508; 1:100 or 1:20), PE–rat IgG2b isotype control (BD Biosciences; no. 553989; 1:10), PE–Cy7–mouse IgG1 isotype control (BioLegend; no. 400126; 1:20), APC–rat IgG2b isotype control (BD Biosciences; no. 553991; 1:10), and APC–rat IgG1 isotype control (BD Biosciences; no. 550884; 1:100). For the staining of BLT1, cells were stained with biotinylated anti-mouse BLT1 mAb (7A8; 10 μg/mL), followed by staining with PE-Cy7–streptavidin (BD Biosciences; no. 557598; 1:100).57 Staining for Ki67 was conducted after cell surface staining and was performed by incubation in 70% ethanol for 1 h at –20 °C, according to the manufacturer’s protocol. Staining for BrdU was conducted after cell surface staining and included fixation, permeabilization, and DNase treatment according to the manufacturer’s protocol. Samples were analyzed (MACSQuant, Miltenyi Biotec, Bergisch Gladbach, Germany; or FACSAria II, BD Biosciences) and cells were isolated (FACSAria II) by using flow cytometry. Data were analyzed by using FlowJo 9.9 (Tree Star, Ashland, Oregon, USA). Dead cells were excluded from analysis.
Reverse transcription and quantitative PCR analysis
Reverse transcription (RT) and quantitative PCR analysis were performed as described previously with modification.58 Total RNA was isolated from purified cells by using Sepasol (Nacalai Tesque) and chloroform (Nacalai Tesque), precipitated with 2-propanol (Nacalai Tesque), and washed with 75% (vol/vol) ethanol (Nacalai Tesque). RNA samples were incubated with DNase I (Invitrogen, Carlsbad, California, USA) to remove contaminating genomic DNA and then reverse-transcribed into cDNA (Superscript III reverse transcriptase, VIRO cDNA Synthesis Kit; Invitrogen). Quantitative PCR analysis was performed by using a thermocycler (LightCycler 480 II, Roche, Basel, Switzerland) with FastStart Essential DNA Probes Master Mix (Roche). Primer sequences were: Blt1 sense, 5′-tctgatcagcatcagggaaa-3′; Blt1 antisense, 5′-gaaggtgctgcaggagatgt-3′; MyD88 sense, 5′-gccttgttagaccgtgaggat-3′; MyD88 antisense, 5′-ctaagtatttctggcagtcctcct-3′; Alox-5 sense, 5′-aggcacggcaaaaacagtat-3′; Alox-5 antisense, 5′-tgtggcatttggcatcaata-3′; Flap sense, 5′-catcagcgtggtccagaat-3′; Flap antisense, 5′-agagtcccggtcctctgg-3′; LTA4 hydrolase sense, 5′-tggaacacctggctctacg-3′; LTA4 hydrolase antisense, 5′-gtcagagtcacgtcgtaattgg-3′; Actinb sense, 5′-aaggccaaccgtgaaaagat-3′; and Actinb antisense, 5′-gtggtacgaccagaggcatac-3′.
Immunization
Immunization was performed as described previously with modification.30 For oral immunization, 8-week-old BLT1-deficient and WT mice were fasted for 16 h and orally immunized by using OVA (Sigma-Aldrich; 1 mg/mouse) with CT (List Biological Laboratories, Campbell, California, USA; 10 μg/mouse) dissolved in PBS (200 μL/mouse) through disposable feeding needles (Fuchigami, Kyoto, Japan) once weekly for a total of three times (days 0, 7, and 14). In some experiments, mice were intraperitoneally treated with cyclophosphamide (Sigma-Aldrich, 35 mg/kg) once daily for a total of three times (days 18, 19, and 20) as described previously.30 In some experiments, mice were intraperitoneally treated with anti-CD40L mAb (Bio X Cell, 250 μg/mouse) once daily for a total of three times (days 15, 17, and 19) to disrupt germinal center formation as described previously.59,60 In some experiments, mice were intraperitoneally injected with BrdU (BD Biosciences, 1.0 mg/mouse) once at 1 day before euthanasia to examine cell proliferation.30 For systemic immunization, mice were intraperitoneally injected with OVA (25 μg/mouse) and Imject Alum (Thermo Fisher Scientific, Tokyo, Japan; 1 mg/mouse) dissolved in PBS (200 μL/mouse) once weekly for two times, as described previously with modification.61
In vivo CT challenge
In vivo CT challenge was performed as described previously with modification.30,62 At 7 days after final immunization (day 21), mice were fasted for 8 h and orally challenged with CT (100 μg/mouse). At 15 h after CT challenge, clinical symptoms of diarrhea were recorded, and the volume of intestinal water was measured.
ELISA
ELISA was performed as described previously with modification.30 At 7 days after final immunization (day 21), feces or serum were obtained for ELISA assay. Feces were weighed, dissolved in PBS (100 mg/mL), vortexed for 30 min at 4 °C, and centrifuged (3000 × g, 10 min, 4 °C). Supernatants were used as fecal samples in the ELISAs. Serum was prepared from blood by centrifugation (5800 × g, 10 min). Supernatants were used as serum samples in the ELISAs.
For antigen-specific ELISAs, flat-bottom 96-well plates (Nunc MaxiSorp, Thermo Fisher Scientific) were coated with either CT (0.5 μg/well) or OVA (100 μg/well) and incubated overnight at 4 °C. Plates were washed three times by using 0.1% (vol/vol) Tween-20 (Nacalai Tesque) in PBS, blocked with 1% (wt/vol) bovine serum albumin (Nacalai Tesque) in PBS for 1 h at room temperature, and washed three times with 0.1% (vol/vol) Tween-20 in PBS three times. Fecal and serum samples, serially diluted with PBS containing 1% (wt/vol) bovine serum albumin, were applied to the plates and incubated for 3 h at 4 °C, and washed three times 0.1% (vol/vol) Tween-20 in PBS. Plates were incubated for 30 min at room temperature with either anti-mouse IgA-HRP (Southern Biotech, Birmingham, Alabama, USA; catalog no. SBA-1040-05-1; dilution, 1:4000) or anti-mouse IgG-HRP (Southern Biotech; SBA-1031-05-1; 1:4000) in 2% (vol/vol) bovine serum albumin and 0.1% (vol/vol) Tween-20 in PBS, followed by three washes with 0.1% (vol/vol) Tween-20 in PBS. Antigen-specific antibody titers were visualized by adding peroxidase substrate (TMB microwell peroxidase substrate, SeraCare Life Sciences, Milford, Massachusetts, USA) for 20 min at room temperature in the dark. Then, 0.5 M HCl (Nacalai Tesque) was added to the samples, and the absorbance at OD450 was measured (iMark microplate reader, Bio-Rad, Hercules, California, USA).
For the total IgA ELISA, plates were coated with goat anti-mouse Ig (H + L) (Southern Biotech; no. 1010-01; 1:1000) and detected with anti-mouse IgA-HRP (Southern Biotech; no. SBA-1040-05-1; 1:4000). Purified mouse IgA κ isotype control (BD Biosciences; no. 553476) was used as an IgA standard.
ELISPOT assay
ELISPOT assays were performed as described previously with modification.63 At 7 days after the final immunization, cell samples were prepared from iLP as described earlier. For antigen-specific ELISPOT assays, plates were coated overnight at 4 °C with either CT (0.5 μg/well) or OVA (100 μg/well). Plates were washed with PBS three times and blocked in RPMI1640 containing 10% (vol/vol) newborn calf serum for 30 min at 37 °C. Cell samples, diluted in 2% (vol/vol) newborn calf serum in PBS, were applied to wells and incubated for 4 h at 37 °C in a 5% CO2 incubator (Sanyo, Osaka, Japan). Plates were washed three times with PBS and then three times with PBS containing 0.1% (vol/vol) Tween-20, followed by incubation overnight at 4 °C with either anti-mouse IgA-HRP (Southern Biotech; 1:1000) or anti-mouse IgG–HRP (Southern Biotech; 1:1000) in PBS containing 0.1% (vol/vol) Tween-20. Plates were washed with PBS six times, and antigen-specific antibody-producing cells were visualized by adding 3-amino-9-ethylcarbazole (Tokyo Chemical Industry, Tokyo, Japan) in 0.1 M acetic acid solution plus H2O2 (Nacalai Tesque). Then, plates were washed with running water and dried at room temperature. Spots were counted (ImmunoSpot Analyzer, Cellular Technology, Cleveland, Ohio, USA).
Immunohistochemistry
Immunohistochemical analysis was performed as described previously with modification.64 Tissues samples in OCT compound (Sakura Finetek Japan, Tokyo, Japan) were frozen in liquid nitrogen. Freshly prepared cryostat sections (6 μm thick) were fixed with 95% ethanol (Nacalai Tesque) for 30 min at 4 °C, followed by incubation in 100% acetone (Nacalai Tesque) for 1 min at room temperature. After being blocked in 2% (vol/vol) newborn calf serum in PBS (Nacalai Tesque) for 30 min at room temperature, samples were incubated with antibodies for 16 h at 4 °C. Samples were washed for 5 min each with 0.1% (vol/vol) Tween-20 (Nacalai Tesque) in PBS and then PBS, followed by staining with DAPI (AAT Bioquest, Sunnyvale, California, USA) for 10 min at room temperature to visualize nuclei. Finally, samples were washed twice with PBS, mounted in Fluoromount (Diagnostic BioSystems, Pleasanton, California, USA), and examined under a fluorescence microscope (model BZ-9000, Keyence, Osaka, Japan). BrdU staining was performed as described previously with modification.65 In brief, freshly prepared cryostat sections (thickness, 6 μm) were fixed in 100% acetone for 3 min at room temperature, followed by blocking and staining with PE-anti-Ki67 mAb (BioLegend; no. 652404; 1:100) and biotin-anti-IgA mAb (Thermo Fisher Scientific; no. 13-5994-81, 1:100). Sections were fixed in 4% paraformaldehyde (Nacalai Tesque) for 20 min and, to cleave DNA, were incubated in 4 N HCl (Nacalai Tesque) for 20 min at room temperature. To neutralize acid, sections were transferred to 0.2 M sodium borate (pH 8.5) for 3 min and stained with APC-streptavidin (BioLegemd; no. 405207; 1:200) and FITC–anti-BrdU mAb (BD Biosciences, 1:50).
In vitro migration assay
In vitro migration assays were performed as described previously.25 Bone marrow neutrophils were prepared from the femur and tibia of 8-week-old WT mice and were harvested as a cloudy cell pellet after 62% Percoll gradient centrifugation (1000 × g, 30 min, 20 °C). Neutrophils were further purified as a Gr1highCD11b+CD45+ population by flow cytometry (FACSAria, BD Biosciences). IgM+IgA– B cells and IgA+ B cells were purified from PPs by using flow cytometry (FACSAria, BD Biosciences). These cells were applied to the upper chambers (1 × 104 cells/well) of Transwell plates (pore diameter, 5 μm; Corning-Costar, Corning, New York, USA). Various concentrations of LTB4 (0, 1, 10, or 100 nM; Cayman Chemical, Ann Arbor, Michigan, USA) were added to the lower wells. After a 2-h incubation, the cells that had migrated into the lower wells were counted by flow cytometry.
In vitro culture of PP B cells
In vitro culture was performed as described previously with modification.25,66 RPMI-1640 supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Gibco, Gaithersburg, Maryland, USA), 50 U/mL penicillin and 50 μg/mL streptomycin (Nacalai Tesque), 55 μM 2-mercaptoethanol (Nacalai Tesque), and 1 mM sodium pyruvate (Nacalai Tesque) was used as a complete culture medium. B220+ B cells were isolated from PPs by using autoMACS (Miltenyi Biotec). For the induction of IgA class-switching and production, cells were cultured in U-bottom, 96-well microtiter plates (1 × 105 cells/well; Thermo Scientific) containing recombinant murine IL-4 (PeproTech, New Jersey, USA; 100 ng/mL), recombinant murine IL-5 (PeproTech; 100 ng/mL), recombinant human TGF-β1 (PeproTech; 1 ng/mL), and either Escherichia coli lipopolysaccharide (LPS) 0127:B8 (Sigma-Aldrich; 10 μg/mL) or Staphylococcus aureus peptidoglycan (Sigma-Aldrich; 10 μg/mL) in complete culture medium (200 μL/well); plates were incubated for 3 days at 37 °C in a 5% CO2 incubator. After the 3-day culture period, half (100 μL) of the culture medium was replaced with complete culture medium containing recombinant murine IL-6 (PeproTech; 1 ng/mL); plates were incubated for 3 days at 37 °C in a 5% CO2 incubator. Then the supernatant and cells were collected and used in ELISAs and flow cytometric analyses, respectively.
Antibiotic treatment
Mice were provided ampicillin (1 g/L; Nacalai Tesque), vancomycin (500 mg/L; Nacalai Tesque), neomycin sulfate (1 g/L; Sigma-Aldrich), and metronidazole (1 g/L; Sigma-Aldrich) in their drinking water,67 beginning 1 week prior to oral immunization by using OVA with CT and continuing throughout the study.
Statistical analysis
Statistical significance was evaluated by one-way ANOVA for comparison of multiple groups and the Mann–Whitney test for two groups by using Prism 6 (GraphPad Software, La Jolla, California, USA). A P value less than 0.05 was considered to be significant.
References
Yokomizo, T., Izumi, T., Chang, K., Takuwa, Y. & Shimizu, T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 387, 620–624 (1997).
Tager, A. M. et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat. Immunol. 4, 982–990 (2003).
Goodarzi, K., Goodarzi, M., Tager, A. M., Luster, A. D. & von Andrian, U. H. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 4, 965–973 (2003).
Ott, V. L., Cambier, J. C., Kappler, J., Marrack, P. & Swanson, B. J. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat. Immunol. 4, 974–981 (2003).
Lee, W., Su Kim, H. & Lee, G. R. Leukotrienes induce the migration of Th17 cells. Immunol. Cell. Biol. 93, 472–479 (2015).
Weller, C. L. et al. Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J. Exp. Med. 201, 1961–1971 (2005).
Okamoto, F., Saeki, K., Sumimoto, H., Yamasaki, S. & Yokomizo, T. Leukotriene B4 augments and restores FcγRs-dependent phagocytosis in macrophages. J. Biol. Chem. 285, 41113–41121 (2010).
Toda, A. et al. Attenuated Th1 induction by dendritic cells from mice deficient in the leukotriene B4 receptor 1. Biochimie 92, 682–691 (2010).
Patnode, M. L., Bando, J. K., Krummel, M. F., Locksley, R. M. & Rosen, S. D. Leukotriene B4 amplifies eosinophil accumulation in response to nematodes. J. Exp. Med. 211, 1281–1288 (2014).
Lammermann, T. et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498, 371–375 (2013).
Oyoshi, M. K. et al. Leukotriene B4-driven neutrophil recruitment to the skin is essential for allergic skin inflammation. Immunity 37, 747–758 (2012).
Yokomizo, T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J. Biochem. 157, 65–71 (2015).
Yokomizo, T., Nakamura, M. & Shimizu, T. Leukotriene receptors as potential therapeutic targets. J. Clin. Invest. 128, 2691–2701 (2018).
Terawaki, K. et al. Absence of leukotriene B4 receptor 1 confers resistance to airway hyperresponsiveness and Th2-type immune responses. J. Immunol. 175, 4217–4225 (2005).
Miyahara, N. et al. Requirement for leukotriene B4 receptor 1 in allergen-induced airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 172, 161–167 (2005).
Haribabu, B. et al. Targeted disruption of the leukotriene B4 receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. J. Exp. Med. 192, 433–438 (2000).
Kim, N. D., Chou, R. C., Seung, E., Tager, A. M. & Luster, A. D. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. J. Exp. Med. 203, 829–835 (2006).
Kihara, Y. et al. The leukotriene B4 receptor, BLT1, is required for the induction of experimental autoimmune encephalomyelitis. Biochem. Biophys. Res. Commun. 394, 673–678 (2010).
Pettersson, A., Richter, J. & Owman, C. Flow cytometric mapping of the leukotriene B4 receptor, BLT1, in human bone marrow and peripheral blood using specific monoclonal antibodies. Int. Immunopharmacol. 3, 1467–1475 (2003).
Runarsson, G. et al. Leukotriene B4 plays a pivotal role in CD40-dependent activation of chronic B lymphocytic leukemia cells. Blood 105, 1274–1279 (2005).
Yamaoka, K. A., Claesson, H. E. & Rosen, A. Leukotriene B4 enhances activation, proliferation, and differentiation of human B lymphocytes. J. Immunol. 143, 1996–2000 (1989).
Dugas, B. et al. Leukotriene B4 potentiates the expression and release of FcεRII/CD23, and proliferation and differentiation of human B lymphocytes induced by IL-4. J. Immunol. 145, 3406–3411 (1990).
Lycke, N. Y. & Bemark, M. The regulation of gut mucosal IgA B-cell responses: recent developments. Mucosal Immunol. 10, 1361–1374 (2017).
Kunisawa, J., Nochi, T. & Kiyono, H. Immunological commonalities and distinctions between airway and digestive immunity. Trends Immunol. 29, 505–513 (2008).
Gohda, M. et al. Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer’s patches for intestinal IgA responses. J. Immunol. 180, 5335–5343 (2008).
Kunkel, E. J. & Butcher, E. C. Plasma-cell homing. Nat. Rev. Immunol. 3, 822–829 (2003).
Kunisawa, J. et al. Regulation of intestinal IgA responses by dietary palmitic acid and its metabolism. J. Immunol. 193, 1666–1671 (2014).
Wu, W. et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 10, 946–956 (2017).
Bulmer, J. N., Morrison, L. & Johnson, P. M. Expression of the proliferation markers Ki67 and transferrin receptor by human trophoblast populations. J. Reprod. Immunol. 14, 291–302 (1988).
Kunisawa, J. et al. Microbe-dependent CD11b+ IgA+ plasma cells mediate robust early-phase intestinal IgA responses in mice. Nat. Commun. 4, 1772 (2013).
Coffman, R. L., Shrader, B., Carty, J., Mosmann, T. R. & Bond, M. W. A mouse T cell product that preferentially enhances IgA production. I. Biologic characterization. J. Immunol. 139, 3685–3690 (1987).
Murray, P. D., McKenzie, D. T., Swain, S. L. & Kagnoff, M. F. Interleukin 5 and interleukin 4 produced by Peyer’s patch T cells selectively enhance immunoglobulin A expression. J. Immunol. 139, 2669–2674 (1987).
Beagley, K. W. et al. Recombinant murine IL-5 induces high rate IgA synthesis in cycling IgA-positive Peyer’s patch B cells. J. Immunol. 141, 2035–2042 (1988).
Beagley, K. W. et al. Interleukins and IgA synthesis. Human and murine interleukin 6 induce high rate IgA secretion in IgA-committed B cells. J. Exp. Med. 169, 2133–2148 (1989).
Sonoda, E. et al. Transforming growth factor β induces IgA production and acts additively with interleukin 5 for IgA production. J. Exp. Med. 170, 1415–1420 (1989).
Defrance, T. et al. Interleukin 10 and transforming growth factor β cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J. Exp. Med. 175, 671–682 (1992).
Coffman, R. L., Lebman, D. A. & Shrader, B. Transforming growth factor β specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J. Exp. Med. 170, 1039–1044 (1989).
Ehrhardt, R. O., Strober, W. & Harriman, G. R. Effect of transforming growth factor (TGF)-β1 on IgA isotype expression. TGF-β1 induces a small increase in sIgA+ B cells regardless of the method of B cell activation. J. Immunol. 148, 3830–3836 (1992).
Serezani, C. H., Lewis, C., Jancar, S. & Peters-Golden, M. Leukotriene B4 amplifies NF-κB activation in mouse macrophages by reducing SOCS1 inhibition of MyD88 expression. J. Clin. Invest. 121, 671–682 (2011).
Yanagibashi, T. et al. Differential requirements of MyD88 and TRIF pathways in TLR4-mediated immune responses in murine B cells. Immunol. Lett. 163, 22–31 (2015).
Seo, G. Y. et al. Retinoic acid, acting as a highly specific IgA isotype switch factor, cooperates with TGF-β1 to enhance the overall IgA response. J. Leukoc. Biol. 94, 325–335 (2013).
Hashidate, T. et al. AML1 enhances the expression of leukotriene B4 type-1 receptor in leukocytes. FASEB J. 24, 3500–3510 (2010).
Hanai, J. et al. Interaction and functional cooperation of PEBP2/CBF with Smads. Synergistic induction of the immunoglobulin germline Calpha promoter. J. Biol. Chem. 274, 31577–31582 (1999).
Pardali, E. et al. Smad and AML proteins synergistically confer transforming growth factor β1 responsiveness to human germ-line IgA genes. J. Biol. Chem. 275, 3552–3560 (2000).
Watanabe, K. et al. Requirement for Runx proteins in IgA class switching acting downstream of TGF-β1 and retinoic acid signaling. J. Immunol. 184, 2785–2792 (2010).
Obata, T. et al. Indigenous opportunistic bacteria inhabit mammalian gut-associated lymphoid tissues and share a mucosal antibody-mediated symbiosis. Proc. Natl. Acad. Sci. USA 107, 7419–7424 (2010).
Shibata, N. et al. Lymphoid tissue-resident Alcaligenes LPS induces IgA production without excessive inflammatory responses via weak TLR4 agonist activity. Mucosal Immunol. 11, 693–702 (2018).
Kunisawa, J. & Kiyono, H. Vitamin-mediated regulation of intestinal immunity. Front. Immunol. 4, 189 (2013).
Lamichhane, A., Kiyono, H. & Kunisawa, J. Nutritional components regulate the gut immune system and its association with intestinal immune disease development. J. Gastroenterol. Hepatol. 28(Suppl 4), 18–24 (2013).
Saeki, K. & Yokomizo, T. Identification, signaling, and functions of LTB4 receptors. Semin. Immunol. 33, 30–36 (2017).
Ichiki, T. et al. Modulation of leukotriene B4 receptor 1 signaling by receptor for advanced glycation end products (RAGE). FASEB J. 30, 1811–1822 (2016).
Ichiki, T., Koga, T. & Yokomizo, T. Receptor for advanced gglycation end products regulates leukotriene B4 receptor 1 signaling. DNA Cell Biol. 35, 747–750 (2016).
Sanchez, J. & Holmgren, J. Cholera toxin—a foe & a friend. Indian J. Med. Res. 133, 153–163 (2011).
Autore, G., Capasso, F., Di Carlo, G. & Mascolo, N. Effect of cholera toxin on the production of eicosanoids by rat jejunum. Br. J. Pharm. 92, 149–152 (1987).
Ishii, K. J. et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7, 40–48 (2006).
Ishii, K. J. et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729 (2008).
Sasaki, F. et al. Biochemical and immunological characterization of a novel monoclonal antibody against mouse leukotriene B4 receptor 1. PLoS ONE 12, e0185133 (2017).
Nagatake, T., Fujita, H., Minato, N. & Hamazaki, Y. Enteroendocrine cells are specifically marked by cell surface expression of claudin-4 in mouse small intestine. PLoS ONE 9, e90638 (2014).
Natarajan, P. et al. CD4 Depletion or CD40L blockade results in antigen-specific tolerance in a red blood cell alloimmunization model. Front. Immunol. 8, 907 (2017).
Foy, T. M. et al. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J. Exp. Med. 180, 157–163 (1994).
Takamura, K. et al. Regulatory role of lymphoid chemokine CCL19 and CCL21 in the control of allergic rhinitis. J. Immunol. 179, 5897–5906 (2007).
Nochi, T. et al. Rice-based mucosal vaccine as a global strategy for cold-chain- and needle-free vaccination. Proc. Natl. Acad. Sci. USA 104, 10986–10991 (2007).
Nagatake, T. et al. Id2-, RORγt-, and LTβR-independent initiation of lymphoid organogenesis in ocular immunity. J. Exp. Med. 206, 2351–2364 (2009).
Nagatake, T. et al. Central role of core binding factor β2 in mucosa-associated lymphoid tissue organogenesis in mouse. PLoS ONE 10, e0127460 (2015).
Gillard, G. O. & Farr, A. G. Features of medullary thymic epithelium implicate postnatal development in maintaining epithelial heterogeneity and tissue-restricted antigen expression. J. Immunol. 176, 5815–5824 (2006).
Kotani, Y. et al. Role of Lactobacillus pentosus Strain b240 and the Toll-like receptor 2 axis in Peyer’s patch dendritic cell-mediated immunoglobulin A enhancement. PLoS ONE 9, e91857 (2014).
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004).
Acknowledgements
We thank our laboratory members for helpful discussion. This work was supported by Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT)/Japanese Society for the Promotion of Science KAKENHI (grant numbers JP15K19142 to T.N.; JP18J00556 and JP18K17997 to K.H.; JP15H05904, JP15H04708, and JP18H02627 to T.Y.; and JP15H05790, JP18H02150, JP18H02674, JP17K09604, JP26670241, and JP26293111 to J.K.); the Japan Agency for Medical Research and Development (grant numbers JP17ek0410032s0102, JP17ek0210078h0002, JP17ak0101068h0001, JP17gm1010006s0101, and JP18ck0106243h0003 to J.K.); the Ministry of Health and Welfare of Japan (to J.K.); the Ministry of Health, Labour, and Welfare of Japan (JP19KA3001 to K.H.); the Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries, and Food Industry (to J.K.); a grant-in-aid for Scientific Research on Innovative Areas from MEXT (JP23116506, JP16H01373, and JP25116706 to J.K.); the Grant for Joint Research Project of the Institute of Medical Science, the University of Tokyo (to J.K.); the Ono Medical Research Foundation (to J.K.); the Canon Foundation (to J.K.); and the Takeda Science Foundation (to T.Y.). This study was supported in part by a Grant-in-Aid (S1311011) from the Foundation of Strategic Research Projects in Private Universities from MEXT (to T.Y.), a grant from the Institute for Environmental and Gender-Specific Medicine (to T.Y.).
Author information
Authors and Affiliations
Contributions
T.N. and J.K. conceived and designed the study, performed data analysis, and wrote the manuscript; S.-i.H., H. S., K.H., N.M., Y.Y., M.S., and S.M. provided technical help for the in vitro and in vivo experiments; E.K., S.K., and K.J.I. provided materials of MyD88 KO mice and discussed the results; and T.K., F.S., and T.Y. provided materials of BLT1 KO mice and anti-BLT1 mAb, and discussed the results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagatake, T., Hirata, Si., Koga, T. et al. BLT1 mediates commensal bacteria-dependent innate immune signals to enhance antigen-specific intestinal IgA responses. Mucosal Immunol 12, 1082–1091 (2019). https://doi.org/10.1038/s41385-019-0175-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-019-0175-z
This article is cited by
-
A mouse protozoan boosts antigen-specific mucosal IgA responses in a specific lipid metabolism- and signaling-dependent manner
Nature Communications (2024)
-
12-Hydroxyeicosapentaenoic acid inhibits foam cell formation and ameliorates high-fat diet-induced pathology of atherosclerosis in mice
Scientific Reports (2021)
