
Abstract
A wealth of neuromodulatory transmitters regulate synaptic circuits in the brain. Their mode of signaling, often called volume transmission, differs from classical synaptic transmission in important ways. In synaptic transmission, vesicles rapidly fuse in response to action potentials and release their transmitter content. The transmitters are then sensed by nearby receptors on select target cells with minimal delay. Signal transmission is restricted to synaptic contacts and typically occurs within ~1 ms. Volume transmission doesn’t rely on synaptic contact sites and is the main mode of monoamines and neuropeptides, important neuromodulators in the brain. It is less precise than synaptic transmission, and the underlying molecular mechanisms and spatiotemporal scales are often not well understood. Here, we review literature on mechanisms of volume transmission and raise scientific questions that should be addressed in the years ahead. We define five domains by which volume transmission systems can differ from synaptic transmission and from one another. These domains are (1) innervation patterns and firing properties, (2) transmitter synthesis and loading into different types of vesicles, (3) architecture and distribution of release sites, (4) transmitter diffusion, degradation, and reuptake, and (5) receptor types and their positioning on target cells. We discuss these five domains for dopamine, a well-studied monoamine, and then compare the literature on dopamine with that on norepinephrine and serotonin. We include assessments of neuropeptide signaling and of central acetylcholine transmission. Through this review, we provide a molecular and cellular framework for volume transmission. This mechanistic knowledge is essential to define how neuromodulatory systems control behavior in health and disease and to understand how they are modulated by medical treatments and by drugs of abuse.
Similar content being viewed by others
Neuromodulatory transmission broadly controls neural circuits and behavior. It is exceptionally diverse and includes many different types of transmitters [1,2,3,4,5]. For example, several monoaminergic systems and many neuropeptides regulate nervous system function. Disruption or dysfunction of modulatory transmission is broadly implicated in brain disease, ranging from neurodevelopmental and neuropsychiatric disorders to neurodegeneration and brain cancer. Furthermore, many medical treatments and drugs of abuse directly act on these systems. Hence, detailed molecular, cellular and systems level understanding of neuromodulation is a central goal of current neuroscience.
The main ascending modulatory systems signal through three monoamines, dopamine, norepinephrine, and serotonin. They also include the central cholinergic system. Neuropeptides, amino acid transmitters, additional monoamines (histamine, epinephrine, and melatonin), endocannabinoids, and other transmitters can also operate through volume transmission [2,3,4, 6]. Here, we focus on the ascending modulatory systems and assess their release machineries, their transcellular organization, and their transmission scales. We then discuss volume transmission features of amino acid transmitters and neuropeptides. We identify five domains through which these modulatory systems are specified, and assess them in view of the rich, mechanistic knowledge that has been developed for synaptic transmission.
Synaptic and volume transmission
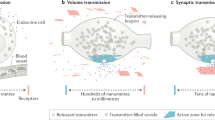
The principal difference between synaptic and volume transmission is that volume transmission is not “wired”, meaning that it does not rely on synaptic connections (Fig. 1) [3, 7, 8]. A hallmark feature of synaptic transmission is its temporal and spatial precision [9, 10]. At synapses, secretion is mediated by the SNARE proteins synaptobrevin-2/VAMP2 on vesicles, syntaxin-1 and SNAP-25 on the plasma membrane, and by Munc18. Action potentials trigger SNARE-mediated vesicle fusion via opening of voltage-gated Ca2+ channels (CaVs). Synaptotagmins sense the local Ca2+ increase to induce fusion of transmitter-laden vesicles with the plasma membrane. Synaptic vesicle exocytosis is restricted to a region at the presynaptic plasma membrane called the active zone that contains RIM, RIM-BP, Munc13, ELKS, Liprin-α, and Piccolo/Bassoon [11, 12]. Active zone protein machinery tethers vesicles to their future sites of release, primes them to render the vesicles fusion-ready, orchestrates CaV positioning, and controls the operation of SNARE proteins and Ca2+ sensors. Through this organization, action potentials trigger release without delay, and transmitters are detected postsynaptically by ionotropic receptors that are precisely apposed to presynaptic release sites. Release-receptor distances are short, tens of nanometers, and the synaptic cleft forms a contained space that limits diffusion of transmitter away from the cell-to-cell contact. This synaptic structure warrants that signal transmission occurs rapidly, within ~1 ms of a presynaptic action potential, that a vesicular fusion event reliably leads to postsynaptic receptor activation, and that intercellular signaling is restricted to the two neurons participating in the synapse.

Schematic comparison of key features of the organization of synaptic (A) and volume (B) transmission. Tight apposition is a hallmark of synapses. Volume transmission lacks this organization and release-receptor distances are often much larger than at synapses. While synaptic transmission is typically mediated by ionotropic receptors, volume transmission usually operates through G protein-coupled receptors (GPCRs). Estimated transmission distances and speeds are indicated at the bottom and discussed in the text.
Early electron microscopic studies found that neuromodulatory nerve terminals, often called varicosities, do not form synaptic appositions in most cases [13,14,15,16,17,18]. In parallel, immunostainings revealed poor colocalization between neuromodulatory axons and their corresponding receptors [19,20,21]. The asynaptic nature of varicosities and the release-receptor mismatch led to the conclusion that modulatory systems act over distance. These observations defined volume transmission as a signaling mode, yet precise mechanisms have been difficult to establish. This is in part because different modulatory systems employ distinct mechanisms, and transmission distances and speeds vary greatly (Fig. 1). Furthermore, finding an overarching and sharp definition of volume transmission has been complicated by the fact that there isn’t a morphological correlate like the synaptic structure.
In this review, we focus on the discussion of volume transmission as vesicular transmission that is not restricted to a synaptic contact. A single vesicular release event might act on multiple cells as long as their receptors are positioned close enough to be reached by transmitter diffusion. Volume transmission can be defined more broadly to include non-vesicular forms of release and many additional characteristics have been assigned to it [8, 22]. Properties like enhanced transmission delays, low specificity of coded information, and low energy cost have been proposed as defining qualities, but synaptic and volume transmission can share these features. The model of persistent ambient (“tonic”) transmitter levels, often detected with microdialysis, has been a point of focus in volume transmission. However, vesicular release generates a dynamic signal. When a vesicle fuses, transmitter originates from a point source and it is rapidly diluted in the extracellular volume. The transmitter signal is further shaped by buffering, reuptake, and degradation. Hence, “tonic” transmitter levels are highly dynamic in space and time. These dynamics matter for receptor activation as they generate massive spatiotemporal transmitter gradients. The detection of seemingly constant ambient levels arises from low sampling frequency and large collection volumes, and they are composed of highly dynamic signals even at baseline, as we recently discussed for dopamine [23].
Fundamentally, volume transmission systems can differ from classical synaptic transmission and from one another in five domains (Fig. 2): (1) the innervation patterns and firing properties of the neurons, (2) the subcellular localization of transmitter synthesis and transmitter loading into different types of vesicles, (3) the architecture of axonal release sites and release site density along an axon, (4) the balance between diffusion, reuptake and degradation of the released transmitter, and (5) the types of transmitter receptors and their relative positioning to transmitter release sites. While we focus on transmission mediated by axonal release, neurons secrete transmitter from somata and dendrites as well [24, 25]. We complement the review with important insights from the characterization of somatodendritic transmission.

Schematic display of five domains that differentiate volume transmission systems from synaptic transmission and from one another. The modulatory systems we discuss in the review are evaluated considering these domains.
We evaluate the prototypical neuromodulator dopamine considering these domains and examine how norepinephrine and serotonin might be similar or differ. We then discuss neuromodulators that also act as synaptic transmitters and neuropeptides. We focus on the transmission mode between cells without evaluating signaling pathways in target cells. While we emphasize neuron-to-neuron transmission, the principles we discuss can involve non-neuronal cells as targets or as regulators of signaling. Finally, neuromodulatory neurons often release multiple transmitters; we refer to previous reviews on the topic of co-transmission [26, 27].
Key lessons from dopamine
Dense striatal innervation and switches in firing modes of midbrain dopamine neurons
Most brain dopamine is synthesized by neurons in the substantia nigra and ventral tegmental area that send their projections to the striatum, prefrontal cortex, amygdala, hippocampus and other brain areas [23, 28, 29]. Their axonal arbors are extensively branched. In the striatum, massive arborization leads to dense innervation with an estimated ~3% of the striatal volume occupied by dopamine axons [30]. In addition to axonal release, dopamine cell bodies and dendrites also release dopamine through somatodendritic exocytosis [24].
Dopamine neurons exhibit three modes of action potential firing [31, 32]. Tonic (or pacemaker) firing is cell-autonomous at frequencies of 0.2–10 Hz [33]. Phasic (or burst) firing entails 3 to 10 action potentials at >10 Hz, and shared somatodendritic inputs synchronize this mode across dopamine neurons [34, 35]. Action potentials can also be induced in distal dopamine axons by cholinergic interneurons via the activation of nicotinic acetylcholine receptors (nAChRs) on dopamine axons [32, 36].
Changes in firing rates regulate striatal dopamine dynamics and behavior [23, 31, 37,38,39,40]. One recent model, the domain-overlap model [23], proposed that baseline activity leads to isolated and local vesicular release events with activation of nearby receptors. Shared inputs induce burst firing in groups of dopamine neurons, and this leads to activation of receptors that are only reached through overlapping dopamine domains from multiple release sites. The third mechanism, firing induced in distal axons by cholinergic interneurons, was only recently discovered [32]. It remains uncertain how it contributes to striatal regulation in vivo [41, 42], how it intersects with ascending action potentials, and how it influences somatic dopamine neuron activities. It might selectively modulate striatal dopamine or regulate somata and dendrites in the midbrain through backpropagation [32, 43].
Synthesis and vesicular loading of dopamine
Dopamine synthesis is cytosolic and occurs in two steps that are in part shared with other monoamines (Fig. 3). The enzyme tyrosine hydroxylase (TH), often used as a marker, converts the amino acid l-tyrosine to l-dihydroxyphenylalanine (l-DOPA), a precursor for dopamine and norepinephrine. Aromatic l-amino acid decarboxylase (AADC) then catalyzes the generation of dopamine from l-DOPA, and dopamine is loaded into vesicles or degraded via monoamine oxidase (MAO) [43,44,45,46,47] (Fig. 3A).

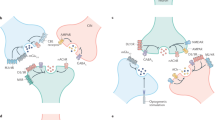
Schematic of the metabolism of dopamine (A), serotonin (B) and norepinephrine (C) with shared (pink) and distinct (blue) steps highlighted. Monoamines are synthesized from amino acids through enzymatic reactions. The vesicular transporter VMAT2 is shared across the three central monoamines, while reuptake is mediated by a specific transporter for each monoamine. Intracellular degradation is mediated by MAO for all three discussed monoamines; 5-HT (5-hydroxytryptamine, serotonin), 5-HTP (5-hydroxytryptophan), AADC (aromatic l-amino acid decarboxylase), DA (dopamine), DAT (dopamine transporter), DBH (dopamine-β-hydroxylase), l-DOPA (l-dihydroxyphenylalanine, levodopa), MAO (monoamine oxidase), NE (norepinephrine), NET (norepinephrine transporter), SERT (serotonin transporter), TH (tyrosine hydroxylase); TpH2 (tryptophan hydroxylase 2), VMAT2 (vesicular monoamine transporter 2).
In the brain, vesicular loading of dopamine is mediated by the vesicular monoamine transporter 2 (VMAT2) [48, 49]. Neurons load neurotransmitters into multiple types of vesicles [50]. Synaptic vesicles are ~50 nm in diameter, have a clear core, and are filled locally with neurotransmitter. Large dense core vesicles (LDCVs), as their name infers, are larger (typically 80–120 nm diameter), have a protein-dense core, and are supplied through long-range transport. Dopamine can in principle be loaded into both types of vesicles. In the adrenal gland, dopamine and other catecholamines are stored in LDCVs and released from these vesicles upon stimulation [51,52,53]. In striatal dopamine axons, dopamine is mostly loaded into small, clear vesicles [54, 55]. First, the axon is filled with clear vesicles and LDCVs are overall sparse [17, 56,57,58], although some varicosities contain clear vesicles that are larger than synaptic vesicles [56]. Second, in dopamine neurons, VMAT2 is preferentially present on small, clear vesicles [59, 60]. Third, dopamine release is strongly dependent on proteins that are typical for synaptic vesicles, for example synaptotagmin-1 (Syt-1) and synaptobrevin-2/VAMP2 [61,62,63,64]. Finally, as discussed in the next section, the release properties of dopamine are similar to those of classical neurotransmitters stored in synaptic vesicles. Hence, most striatal dopamine is released from synaptic vesicles.
The intracellular compartment for somatodendritic dopamine release remains unclear. Early work suggested release from tubulovesicular structures rather than vesicles [65,66,67]. A recent study has found that the vesicular Ca2+ sensor Syt-1 is important for somatodendritic exocytosis [68], perhaps suggesting the involvement of synaptic vesicles.
Sparse dopamine release sites confer a high vesicular release probability
Action potentials trigger vesicular dopamine release, which relies on SNAREs [61, 62] and on extracellular Ca2+ [64, 69, 70]. At classical synapses, Ca2+ triggering is fully mediated by CaV2 channels with major roles for CaV2.1 (P/Q-type) and CaV2.2 (N-type), and contributions of CaV2.3 (R-type) are limited [9, 10]. In contrast, the CaV2-dependence of dopamine release is partial, and CaV1 (L-type) and CaV3 (T-type) channels also contribute [32, 71]. Dopamine release occurs with synchrony to increase local dopamine concentration within milliseconds of axonal depolarization, and dopamine secretion has a high vesicular release probability [63, 72]. This suggests the presence of molecular machinery to organize these processes.
Recent work indeed established that active zone-like protein architecture is essential for evoked dopamine release (Fig. 4). The scaffold RIM is present in small clusters in dopamine axons, and its ablation from dopamine neurons disrupts evoked release [63]. Moreover, these axons contain Bassoon, ELKS2 and Munc13-1 clustered in release site-like structures, and Liprin-α also contributes to evoked dopamine secretion [63, 73,74,75]. At synapses, RIM primes vesicles through recruitment and activation of Munc13 [76, 77], it links primed vesicles to Ca2+ channels [78, 79], and it couples these processes to the membrane phospholipid PIP2 [80, 81]. In dopamine axons, RIM mediates fusion through Munc13 as well [75, 82]. RIM C2B domains, which bind to PIP2 and Liprin-α, also participate in dopamine release [75, 80, 83]. Consistent with only partial reliance on CaV2s, and with the dispensability of RIM-BP and ELKS for dopamine release, the RIM sequences that bind to these proteins are not essential for dopamine release [32, 63, 71, 75].

Schematics of the proteins that mediate the release of dopamine (A), serotonin (B) and norepinephrine (C). Evidence for the importance of active zone proteins, Ca2+ sources and Ca2+ sensors stems from conditional mouse gene knockouts; evidence for the importance of SNARE proteins stems from cleavage with bacterial neurotoxins; evidence for the importance of voltage-gated Ca2+ channels (CaVs) is further supported by pharmacological blockade. Properties (D) of the release of dopamine, serotonin, and norepinephrine as assessed in brain slices and in vivo by a variety of techniques. These monoamines show a range of properties with depression detected in brain slices for dopamine, and facilitation for norepinephrine in some cases.
Altogether, these analyses led to the model of active zone-like release sites for rapid, synchronous dopamine secretion with a high vesicular release probability (Fig. 4). Proximity-based proteomic approaches identified additional release site proteins in dopamine axons [84, 85]. One remarkable morphological feature of dopamine axons is that active zone protein clusters are sparse, and ~75% of vesicle-containing varicosities do not have detectable levels of active zone proteins [63, 75]. This matches with functional experiments in which only a subset of dopamine varicosities appears to release dopamine upon stimulation [86].
The synchronous nature of evoked dopamine release indicates the need for Ca2+ sensors that rapidly respond to Ca2+ entry. At conventional synapses, synchronous release is mediated by Syt-1, -2, or -9, Ca2+ sensors with a low Ca2+ affinity and fast kinetics [10, 87]. In dopamine neurons, axonal and somatodendritic dopamine release evoked by action potentials depend on Syt-1 [64, 68]. When Syt-1 is ablated, a form of dopamine release that can be induced with strong depolarizations or with action potential trains persists and is mediated by Ca2+ sensors that have not been identified yet. Early work revealed a different extracellular Ca2+ dependence and suggested reliance on distinct Ca2+ sensors for somatodendritic release [69, 88, 89], but it is mediated by RIM and Syt-1 similar to axonal release [68, 90].
Diffusion and reuptake are key determinants of dopamine transmission
After dopamine is released, local dopamine concentrations are shaped by diffusion and reuptake. Diffusion rapidly dilutes dopamine in the extracellular volume and the dopamine transporter (DAT) mediates reuptake into dopamine neurons [23, 91]. It has remained challenging to measure local dopamine concentrations and dynamics, which chiefly determine dopamine receptor activation. A single dopamine vesicle contains a few thousands to tens of thousands of dopamine molecules, and dopamine concentration inside a vesicle might be as high as ~1 M [91,92,93,94]. In consequence, the dopamine concentration at the mouth of a fusing vesicle must be hundreds of millimolar. Dopamine concentration from this point source likely drops to a nanomolar range within a few milliseconds and within micrometers of a fusing vesicle [23, 91, 95]. In most cases, dopamine diffusion is not limited by the constraints of a synaptic cleft, but tortuosity in the extracellular space may limit or direct diffusion [17, 56, 91]. While diffusion dominates within a few micrometers of the fusion event, DAT further shapes the signal through reuptake at longer distances [91, 96, 97]. Currently available detection methods, including electrochemistry, microdialysis and fluorescent sensors, do not allow for measuring these dopamine dynamics at the relevant spatiotemporal nanoscales. Instead, they often average over extended space and/or time.
Models of dopamine release-receptor appositions
Dopamine is sensed by G protein-coupled receptors (GPCRs) classified into D1-like (D1 and D5; mostly Gαs) and D2-like (D2, D3 and D4; mostly Gαi) receptors [98]. In the striatum, D1 and D2 receptors together account for most striatal dopamine receptors [95, 99,100,101]. They are on medium spiny neurons (MSNs) and their cell-type specific expression defines D1- and D2-MSNs, which form the direct (D1-MSNs) and indirect (D2-MSNs) pathways [28, 102]. D1 receptors are also expressed on glial cells and D2 receptors on acetylcholine and dopamine axons (reviewed in [23]).
The relative positioning of release sites and receptors is a critical determinant of receptor activation. Because experimental resources including appropriate microscopy and antibodies to define dopamine release hotspots and dopamine receptor distributions are not readily available, the spatial organization of release and receptors has not been conclusively determined. Hence, models of dopamine transmission have been inferred from properties other than measured release-receptor distances. One important finding is that dopamine release sites are sparse, with only every ~4th varicosity containing active zone-like machinery and with frequent occurrence of varicosities with a limited ability to release [63, 73, 75, 86]. One possibility is that the varicosities which form synaptic appositions are the ones that release dopamine in response to stimulation. Initial electron microscopic studies have found that roughly one quarter of varicosities have synaptic appositions [17], similar to the sparsity of release sites [63, 86]. At these varicosities, dopamine receptors are not found in the apposed postsynaptic densities, but ~100 nm lateral, suggesting that the receptors are not as precisely aligned as ionotropic receptors at synapses [9, 57]. A recent study, however, established that synaptic appositions in dopamine axons are much sparser: only ~1% of the dopamine varicosities have them [56]. This finding makes it highly unlikely that these are the relevant signaling units and support the model that dopamine signaling does not rely on synaptic organization.
Dopamine receptor affinities have been used to develop models on receptor activation and release-receptor organization. Initial studies found that D1 receptors have a lower dopamine affinity (Kd ~1 μM) than D2 receptors (Kd ~25 nM) [100]. These differences formed the basis for models in which dopamine activates D1 receptors during burst firing close to release sites and D2 receptors sense steady-state tonic dopamine further away. These affinity-based models need to be updated [23, 95, 103, 104]. First, tonic dopamine release is vesicular and generates small, transient “sparks” that have a high dopamine concentration; the “stable” tonic dopamine level of ~2 nM is likely an artifact of sampling that averages over a large space and long times [23]. Second, the affinity measurements may not reflect in vivo receptor affinities, and both types of receptors may have a low affinity [72, 105,106,107]. Third, receptor positioning cannot be inferred from available data; it has to be measured in relationship to dopamine sources.
Studies of the properties of D2 receptor-induced inhibitory postsynaptic currents (D2-IPSCs) support that D2 receptors are rapidly activated by high, local dopamine concentrations. D2-IPSCs are mediated via coupling of D2 receptors to endogenous (midbrain) or exogenously expressed (striatum) G protein-gated inwardly rectifying potassium (GIRK) channels [72, 108]. In brain slices of both brain areas, D2-IPSCs are readily elicited by single stimuli. D2 receptor activation necessitates ~100 μM dopamine within a short time window after stimulation and D2-IPSCs have fast kinetics with a response onset of tens of milliseconds that is limited by GPCR signaling speeds [72, 108,109,110]. These studies establish that dopamine needs to rise quickly to a high concentration for D2 receptor activation [23].
In conclusion, the working model is that dopamine is secreted effectively via vesicular exocytosis at sparse sites with major contributions of RIM, Munc13, and Syt-1. Diffusion chiefly determines local dopamine levels and reuptake shapes the signal. Receptor activation strongly depends on the relative positioning of dopamine receptors to release sites, but these spatial relationships remain unknown.
Are dopamine concepts applicable to central serotonin and norepinephrine transmission?
Innervation patterns and firing properties
Innervation and firing patterns of norepinephrine and serotonin neurons have similarities with the dopamine system. Most norepinephrine and serotonin neuron somata are in the locus coeruleus and the raphe nuclei, respectively, and their axons project throughout the brain [111,112,113,114]. Compared with the exceptionally high striatal dopamine innervation, norepinephrine and serotonin axons are less dense in their target areas [114, 115]. Similar to dopamine, norepinephrine and serotonin are released from axonal varicosities and synaptic appositions are largely absent [13,14,15, 17, 116,117,118].
Firing properties are shared across monoaminergic systems. Serotonin neurons fire tonically at 0.03–3 Hz and in bursts up to 17 Hz, and these firing modes can encode distinct information [119,120,121,122]. Norepinephrine neurons also switch between tonic and phasic firing to modulate behavioral states [123,124,125]. Repetitive stimulation of serotonin and norepinephrine neurons increases extracellular transmitter levels [126,127,128], and how firing shapes release and signaling is discussed further below.
Synthesis and vesicular loading
Most norepinephrine and serotonin are released from vesicles in an action potential- and Ca2+-dependent manner. This is supported by the sensitivity of release to Ca2+ removal, to toxins that cleave SNARE proteins, and to blockade of vesicular loading [127, 129,130,131,132,133,134,135,136,137]. VMAT2 is the shared vesicular transporter for dopamine, norepinephrine, and serotonin (Fig. 3). Constitutive VMAT2 ablation in mice, like that of the synthesis enzymes TH and dopamine β-hydroxylase (DBH), is lethal [138,139,140], while conditional VMAT2 knockout in serotonin or norepinephrine neurons depletes the corresponding transmitters but does not induce lethality [141,142,143].
Norepinephrine is synthesized from l-tyrosine via dopamine (Fig. 3C). After the first two enzymatic steps with TH and AADC, dopamine is loaded into vesicles by VMAT2. The intravesicular enzyme DBH then converts dopamine to norepinephrine and DBH is partially co-released with the transmitter [144, 145]. Because norepinephrine is synthesized from dopamine, the commonly used marker TH labels both types of neurons. Serotonin is synthesized in the cytosol from its precursor l-tryptophan (Fig. 3B) via two steps involving the brain-specific tryptophan hydroxylase 2 (TpH2), and AADC [146].
It remains uncertain which vesicle types mediate norepinephrine and serotonin release in the vertebrate CNS. Retzius cells of the leech are mechanosensory neurons that use serotonin as the main transmitter. There, serotonin is released from small, clear vesicles and from LDCVs [147, 148]. These vesicles are estimated to discharge ~5000 or ~80,000 serotonin molecules, respectively. Rat brain serotonin and norepinephrine neurons also contain both vesicle types [13], and pharmacological studies supported the presence of multiple compartments for serotonin storage and release [149]. VMAT2 is present on both synaptic vesicles and LDCVs, and its levels may be high on LDCVs of serotonin axons while being mostly on synaptic vesicles in dopamine axons [59, 60, 150]. Together, these studies suggest that multiple vesicle types contribute to norepinephrine and serotonin release, and signaling with different kinetics and functions might be a result of this organization [151].
Release machinery and release site distribution
While norepinephrine and serotonin release are vesicular, the machineries for their secretion (Fig. 4B, C) and the distribution of release sites within an axon are largely unknown. Botulinum neurotoxin A and tetanus neurotoxin, which cleave SNAREs, inhibit the release of 3[H]-labeled norepinephrine and serotonin from synaptosomes or in brain slices [130, 131, 134]. Despite the similar SNARE-dependence of norepinephrine, serotonin and dopamine release, studies using electrochemistry, fluorescent sensors, and mouse genetics suggest differences (Fig. 4D). When imaged with GPCR-based sensors, dopamine and serotonin only moderately build-up during stimulus trains [152, 153], consistent with the depression observed for dopamine with amperometry or D2-IPSCs in brain slices [63, 72] (Fig. 4D). While dopamine release in response to stimulus trains is enhanced upon knockout of the vesicle-associated protein Synapsin, serotonin release appears to be unaffected [154]. In hippocampal interneurons, serotonin elicits ionotropic, 5HT3-receptor-mediated responses within a few milliseconds after a single stimulus [155]. Norepinephrine release often necessitates stimulus trains for effective detection with voltammetry or fluorescent sensors, suggesting that release might facilitate and might have slower kinetics (Fig. 4D) [156,157,158]. In contrast, whole-cell recordings effectively detect single stimulus-evoked IPSCs mediated by somatodendritic release that activates adrenergic receptors [109]. Overall, these studies indicate that the release machineries for these monoamines might differ between transmitters and cellular compartments.
The strong extracellular Ca2+ dependence of evoked serotonin and norepinephrine release suggests that voltage-gated Ca2+ channels mediate their release [127, 129, 135, 137, 159]. Indeed, blockers of CaV2.1 and CaV2.2 impair norepinephrine and serotonin release detected by high-performance liquid chromatography, microdialysis, or as 3[H]-labeled transmitter [160,161,162,163,164]. Because of the low spatiotemporal resolution of these methods, they do not establish that Ca2+ entry through these channels directly triggers vesicular exocytosis. Different from dopamine release, both norepinephrine and serotonin release appear to be resistant to CaV1 blockade [137, 159, 165]. Finally, conditional ablation of CaV2.1 in serotonin neurons increased the firing rate of these neurons and impacted mouse behavior, but it was not assessed whether CaV2.1 is involved in serotonin release [166].
Optical tracer experiments suggested that norepinephrine axons have many varicosities that are not release-competent, and morphological studies revealed that the density of varicosities in norepinephrine axons differs across brain areas [167,168,169]. Hence, heterogeneity in axonal organization might contribute to variable signaling properties within an axon and across brain areas.
Future studies should assess the composition and distribution of release machinery in serotonin and norepinephrine axons and determine the resulting release and signaling properties. Recent advances in the development of fluorescent sensors for detecting serotonin and norepinephrine provide tools for answering these questions [153, 156, 170,171,172,173].
Diffusion and reuptake
Once released, the balance between diffusion and reuptake determines neurotransmitter concentration at the corresponding receptors. Extracellular serotonin levels measured by voltammetry reach micromolar concentrations upon stimulation [127]. The local serotonin concentration is likely much higher for a brief amount of time in the vicinity of a fusing vesicle given the high intravesicular serotonin concentration [147, 148]. Norepinephrine transporter (NET) and serotonin transporter (SERT) move the corresponding transmitters back into cells. Transporter expression is often limited to the neurons that release the specific transmitter [174, 175], and cross-talk between transmitters and transporters can occur [176]. After reuptake, monoamines are either loaded back into vesicles via VMAT2 or degraded by MAO (Fig. 3) [44,45,46]. For some neuromodulators, degradation in the extracellular space controls transmitter concentration [177,178,179]. In contrast, monoamine degradation is intracellular and does not directly contribute to clearing the extracellular space. Monoamine reuptake and degradation mechanisms are targeted by tricyclic antidepressants (TCAs), by selective serotonin reuptake inhibitors (SSRIs), by cocaine (which inhibits DAT, SERT, and NET), and by MAO inhibitors [175, 180, 181].
For striatal dopamine, modeling of the competition between diffusion and reuptake indicates that DAT shapes signaling by limiting dopamine spread [55, 91]. A key factor is the high density of DAT-containing dopamine axons in the striatum. Because serotonin and norepinephrine axons are much less dense, the corresponding transporters do not cover a large fraction of the extracellular volume and the transmitters are less likely to encounter transporters as they diffuse away from the release site. Hence, reuptake might be less powerful in shaping the signaling in sparse modulatory systems [127, 136, 158].
Receptors and release-receptor appositions
Serotonin and norepinephrine exert diverse functions via large receptor families expressed in brain-area and cell-type specific manners. Most receptors are GPCRs that are widely expressed on neurons and glial cells, and they can help excite or inhibit their targets [182,183,184,185,186,187]. One exception is the ionotropic 5HT3 receptor, through which serotonin acts as a fast excitatory transmitter [155, 188, 189].
Serotonin receptors are organized into seven classes (5HT1–7) with 14 subtypes based on ligand affinities, sequence homology, and intracellular transduction mechanisms: 5HT1 (mostly Gαi; subtypes 5HT1A, 5HT1B, 5HT1D, 5HT1E, 5HT1F), 5HT2 (Gαq; subtypes 5HT2A, 5HT2B, 5HT2C), 5HT3 (ionotropic), 5HT4 (Gαs and Gαi), 5HT5 (Gαi; subtypes 5HT5A, 5HT5B), 5HT6 (Gαs), and 5HT7 (Gαs) [186, 187]. In many brain areas, the density of serotonin innervation corresponds to receptor density, but mismatches exist [19]. Receptor autoradiography, immunostainings, and electron microscopy found associations between serotonin axons and 5HT2A receptors, but synaptic appositions are overall rare [13, 15, 20, 21, 190]. In the parietal cortex, for example, the mean varicosity to receptor distance is estimated to be ~8 μm, and in the hippocampus, dense receptor staining is present despite sparse serotonin axonal innervation [21].
Two classes of norepinephrine receptors, α- and β-adrenergic receptors, are divided into 9 subtypes: α1 (Gαq; subtypes α1A, α1B and α1D), α2 (Gαi; subtypes α2A, α2B and α2C), and β1 to β3 (Gαs and Gαi) [185, 191]. Norepinephrine receptors show non-uniform expression, and there are mismatches with broad receptor expression but low norepinephrine fiber density, or vice versa [19, 183, 184, 192, 193].
Overall, synaptic appositions are sparse and discrepancy between axon densities and receptor patterns exist [19]. Because the distribution of release sites is uncertain, exact release-receptor associations remain unknown. For striatal dopamine, we proposed the domain-overlap model in which multiple release sites contribute to receptor activation during phasic firing [23]. A similar model might not operate for neuromodulators with a low innervation density because overlap of release domains is less likely to occur. Precise assessment of release site distribution and of the relative positioning of receptors will be essential for understanding functional properties of serotonin and norepinephrine signaling.
From fast transmitters to neuropeptides: expanding scales of volume transmission
Fast transmitters
Amino acids are the main synaptic transmitters, but they also act as volume transmitters. For example, extrasynaptic GABA and glutamate receptors detect spillover transmitter from synapses [194,195,196]. In this case, innervation, vesicle types, and release mechanisms are defined by the synaptic properties, but neurotransmitters escape the synaptic cleft to activate distant receptors for volume transmission. Spillover can operate on ionotropic receptors or on GPCRs. The transmitter can also enter neighboring synapses and activate distant synaptic receptors [197]. It is noteworthy that volume transmission of synaptic neurotransmitters is amplified in states with enhanced activity, for example, during epilepsy. Amino acid transmitters can also be co-released from neurons that use a neuromodulator as their primary transmitter. For example, GABA is co-released with dopamine in the retina and in the striatum [198,199,200], and this GABA co-transmission shares many properties of dopamine. These examples illustrate that volume transmission is a broad concept, and important forms of signaling can arise as a secondary process from synaptic transmission.
Acetylcholine acts as a synaptic transmitter at the neuromuscular junction. In contrast, acetylcholine is considered a volume transmitter in the central nervous system, although the mode of action remains debated [14, 22, 32, 36, 177, 201,202,203]. Cholinergic neurons are found in multiple brain regions [204, 205]. Neurons located in the pedunculo-pontine and lateral dorsal tegmental nuclei and in the basal forebrain send ascending projections throughout the brain. In the striatum, local cholinergic interneurons form a dense network, and they account for ~2% of striatal neurons [206, 207]. Their high density, their firing properties and recent work on these interneurons render them well-suited for a discussion of central acetylcholine transmission mechanisms.
In the striatum, cholinergic interneurons activate metabotropic muscarinic acetylcholine receptors expressed on MSNs and on local interneurons. In addition, β2-containing nAChRs are present at high levels on dopamine axons [208]. These ionotropic nAChRs allow for a rapid readout of released acetylcholine and robustly trigger dopamine release [32, 36, 63, 209,210,211,212,213]. The axons of the cholinergic interneurons spread over a large striatal area [214], and the neurons have tonic firing activity, pause-rebound responses, and phasic firing similar to monoamine neurons [215,216,217]. Ultrastructural characterization suggested that synaptic contacts of striatal cholinergic varicosities are very sparse if existent at all [16, 18], and recent superresolution microscopic studies found that the contact frequency between cholinergic and dopaminergic axons is not higher than a predicted random association given their local densities [32]. Despite this sparse contact frequency, stimulation of cholinergic interneurons powerfully and rapidly, within a few milliseconds, depolarizes the dopamine axonal plasma membrane to induce axonal action potential firing (Fig. 5) [32]. Furthermore, individual cholinergic events that are resistant to action potential blockade can be recorded from dopamine axons as subthreshold depolarizations, and even spontaneous events can lead to dopamine axon action potential firing [36]. Released acetylcholine is rapidly degraded by acetylcholine esterase which is present at very high levels in the striatum [218]. Inhibiting acetylcholine esterase slows the decay kinetics of spontaneous events and leads to a transient increase in event frequency recorded from dopamine axons [36], suggesting that degradation limits acetylcholine spread.

Model of axo-axonal volume transmission through which acetylcholine triggers action potential firing in distal dopamine axons. Acetylcholine is released from local cholinergic interneurons and activates nicotinic acetylcholine receptors (nAChRs) on dopamine axons. This leads to local initiation of firing in dopamine axons. The delay from the time point of cholinergic axon stimulation to detection of dopamine release triggered by an axonal action potential is ~10 ms. Inhibitory dopamine feedback is provided to acetylcholine and dopamine axons via D2 receptors.
Further insight on acetylcholine transmission comes from electrophysiological recordings of D1-MSNs after expression of GIRK channels. This leads to M4 muscarinic receptor-coupled IPSCs (M4-IPSCs) [203], similar to D2-IPSCs [72, 108], and provides an electrophysiological readout of acetylcholine transmission. Despite the low synaptic connectivity [16], spontaneous unitary M4-IPSCs occur, and M4-IPSCs are sensitive to action potential blockade. M4-IPSCs have a lag of ~40 ms and a rise time of ~80 ms, consistent with the time course of GPCR signaling, and they are rapidly depressing during repetitive stimulation. Acetylcholine esterase inhibition slows M4-IPSC kinetics, suggesting that receptor activation upon diffusion is limited by acetylcholine degradation. While M4 receptors can be positioned on MSN spines [219], it is unclear whether M4 receptor clusters are apposed to acetylcholine releasing nerve terminals. These findings argue against the importance of tonic ambient levels of acetylcholine, but point toward rapid, metabotropic transmission.
In summary, current data indicate that central cholinergic signaling is likely asynaptic and mediated by volume transmission. Like dopamine [23, 72, 108], it has functional features of fast transmission including the induction of spontaneous events and rapid action in the target cell. Striatal acetylcholine secretion might therefore rely on protein machinery that supports high release synchrony and probability. The frequency and architecture of release sites, and their organization relative to various types of acetylcholine receptors remain major open questions. Altogether, work on striatal acetylcholine suggests that volume transmission may modulate target cells rapidly.
Neuropeptides
Neuropeptide signaling is widespread and diverse with more than 100 neuropeptides, and it regulates behavior with broad spatiotemporal characteristics [4, 5, 220]. These modulators are typically released from LDCVs, and their transmission is mediated by GPCRs. A small number of neurons located in the hypothalamus use neuropeptides as their primary transmitter, while most central neurons release one or multiple neuropeptides from LDCVs in addition to their synaptic transmitter. Hence, for most neuropeptide signaling, innervation patterns and activities reflect the broad range of properties of the neurons that release fast transmitters and co-release neuropeptides.
LDCVs are heterogeneous and are located in somata, dendrites and axons. In contrast to the local production of monoamines and amino acid transmitters, neuropeptides are produced in the soma and supplied via long-range transport [50, 221]. Most synapses contain a very low number of LDCVs, making it challenging to study LDCV exocytosis. Their release generally necessitates strong stimulation, depends on Ca2+ and voltage-gated Ca2+ channels, and occurs at synapses and extrasynaptically [222,223,224,225,226].
In vertebrate neurons, neuropeptide release machinery has been studied with loss-of-function approaches combined with various release measurements in cultured cells and in brain slices (Table 1). The fundamental message of this body of work is that proteins for synaptic vesicle release also mediate fusion of neuropeptide-containing LDCVs, although the exact properties differ qualitatively and quantitatively. One specific finding is that, different from synaptic vesicle exocytosis, Rab3 is essential for neuropeptide release and operates via its effector RIM [227]. Interestingly, distinct secretory pathways for multiple neuropeptides can exist within a neuron. In olfactory mitral cells, IGF-1 release is mediated by Syt-10, while release of other LDCVs and synaptic vesicles depends on Syt-1 [228, 229]. This heterogeneity of release pathways within a neuron might amplify the signaling capacity greatly.
Neuropeptide receptors are mostly metabotropic GPCRs, and they are expressed throughout the brain and on most neurons [230, 231]. Mismatches at the scale of millimeters between neuropeptide-releasing axons and corresponding receptors are common and have defined neuropeptides as volume transmitters [19, 232]. Signaling is terminated through neuropeptide cleavage by extracellular peptidases, and by receptor desensitization and internalization [178, 179, 221, 233]. Despite important advances on release mechanisms, major knowledge gaps on neuropeptide signaling persist. The transcellular organization of release and receptors and the signaling distances are not well understood.
Conclusions and outlook
We here assessed current knowledge on volume transmission. We focused on ascending monoamine systems and provided a comparative discussion of amino acid transmitters, acetylcholine, and neuropeptides. The central conclusion is that volume transmission mechanisms are heterogeneous, and that analyses of each system are required. Detailed, transmitter-specific knowledge on release site structure and function, on receptors, and on the relative positioning of these elements is essential for understanding neuromodulatory systems and for generating accurate models on how each modulator might control circuit function and behavior.
Major knowledge gaps persist. For dopamine, recent advances on release mechanisms now allow for generating models on signaling [23, 54]. These models need to be challenged and further developed with assessment of cell-type specific positioning of dopamine receptors. For serotonin and norepinephrine, as well as neuropeptides, more groundwork on the five domains (Fig. 2) that guide this review is necessary to build initial biophysical models of their signaling. Tool development, including of fluorescent sensors, has started delivering means for a systematic assessment of neuromodulatory transmission mechanisms [220, 234,235,236]. It offers new approaches to study additional modulators. For example, recent work dissected mechanisms of endocannabinoids, a family of lipophilic modulators, and surprisingly found a dependence on membrane trafficking machinery [6, 237, 238]. Ultimately, these new tools will help bridge knowledge gaps between mechanistic features of volume transmission and in vivo neuromodulatory dynamics. A molecular and cellular understanding of volume transmission will help define how these systems regulate brain function and how they can be targeted for treating disease.
References
Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–30.
Avery MC, Krichmar JL. Neuromodulatory systems and their interactions: a review of models, theories, and experiments. Front Neural Circuits. 2017;11:108.
Agnati LF, Zoli M, Strömberg I, Fuxe K. Intercellular communication in the brain: wiring versus volume transmission. Neuroscience. 1995;69:711–26.
van den Pol AN. Neuropeptide transmission in brain circuits. Neuron. 2012;76:98–115.
Russo AF. Overview of neuropeptides: awakening the senses? Headache. 2017;57:37–46.
Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81.
Agnati LF, Fuxe K, Zoli M, Ozini I, Toffano G, Ferraguti F. A correlation analysis of the regional distribution of central enkephalin and beta-endorphin immunoreactive terminals and of opiate receptors in adult and old male rats. Evidence for the existence of two main types of communication in the central nervous system: the volume transmission and the wiring transmission.Acta Physiol Scand. 1986;128:201–7.
Agnati LF, Leo G, Zanardi A, Genedani S, Rivera A, Fuxe K, et al. Volume transmission and wiring transmission from cellular to molecular networks: history and perspectives. Acta Physiol. 2006;187:329–44.
Biederer T, Kaeser PS, Blanpied TA. Transcellular nanoalignment of synaptic function. Neuron. 2017;96:680–96.
Kaeser PS, Regehr WG. Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol. 2014;76:333–63.
Südhof TC. The presynaptic active zone. Neuron. 2012;75:11–25.
Emperador-Melero J, Kaeser PS. Assembly of the presynaptic active zone. Curr Opin Neurobiol. 2020;63:95–103.
Beaudet A, Descarries L. The monoamine innervation of rat cerebral cortex: synaptic and nonsynaptic axon terminals. Neuroscience. 1978;3:851–60.
Descarries L, Mechawar N. Ultrastructural evidence for diffuse transmission by monoamine and acetylcholine neurons of the central nervous system. Prog Brain Res. 2000;125:27–47.
Descarries L, Beaudet A, Watkins KC. Serotonin nerve terminals in adult rat neocortex. Brain Res. 1975;100:563–88.
Contant C, Umbriaco D, Garcia S, Watkins KC, Descarries L. Ultrastructural characterization of the acetylcholine innervation in adult rat neostriatum. Neuroscience. 1996;71:937–47.
Descarries L, Watkins KC, Garcia S, Bosler O, Doucet G. Dual character, asynaptic and synaptic, of the dopamine innervation in adult rat neostriatum: a quantitative autoradiographic and immunocytochemical analysis. J Comp Neurol. 1996;375:167–86.
Chang HT. Dopamine-acetylcholine interaction in the rat striatum: a dual-labeling immunocytochemical study. Brain Res Bull. 1988;21:295–304.
Herkenham M. Mismatches between neurotransmitter and receptor localizations in brain: observations and implications. Neuroscience. 1987;23:1–38.
Blue ME, Yagaloff KA, Mamounas LA, Hartig PR, Molliver ME. Correspondence between 5-HT2 receptors and serotonergic axons in rat neocortex. Brain Res. 1988;453:315–28.
Jansson A, Tinner B, Bancila M, Vergé D, Steinbusch HWM, Agnati LF, et al. Relationships of 5-hydroxytryptamine immunoreactive terminal-like varicosities to 5-hydroxytryptamine-2A receptor-immunoreactive neuronal processes in the rat forebrain. J Chem Neuroanat. 2001;22:185–203.
Sarter M, Parikh V, Howe WM. Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nat Rev Neurosci. 2009;10:383–90.
Liu C, Goel P, Kaeser PS. Spatial and temporal scales of dopamine transmission. Nat Rev Neurosci. 2021;22:345–58.
Rice ME, Patel JC. Somatodendritic dopamine release: recent mechanistic insights. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140185.
Ludwig M, Apps D, Menzies J, Patel JC, Rice ME. Dendritic release of neurotransmitters. Compr Physiol. 2016;7:235–52.
Hnasko TS, Edwards RH. Neurotransmitter corelease: mechanism and physiological role. Annu Rev Physiol. 2012;74:225–43.
Wallace ML, Sabatini BL. Synaptic and circuit functions of multitransmitter neurons in the mammalian brain. Neuron. 2023;111:2969–83.
Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–66.
Garritsen O, van Battum EY, Grossouw LM, Pasterkamp RJ. Development, wiring and function of dopamine neuron subtypes. Nat Rev Neurosci. 2023;24:134–52.
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–53.
Grace AA. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci. 2016;17:524–32.
Liu C, Cai X, Ritzau-Jost A, Kramer PF, Li Y, Khaliq ZM, et al. An action potential initiation mechanism in distal axons for the control of dopamine release. Science. 2022;375:1378–85.
Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–76.
Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984;4:2877–90.
Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, et al. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci USA. 2009;106:7281–8.
Kramer PF, Brill-Weil SG, Cummins AC, Zhang R, Camacho-Hernandez GA, Newman AH, et al. Synaptic-like axo-axonal transmission from striatal cholinergic interneurons onto dopaminergic fibers. Neuron. 2022;110:2949–60.e4.
Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–73.
Venton BJ, Zhang H, Garris PA, Phillips PEM, Sulzer D, Wightman RM. Real-time decoding of dopamine concentration changes in the caudate-putamen during tonic and phasic firing. J Neurochem. 2003;87:1284–95.
Berke JD. What does dopamine mean? Nat Neurosci. 2018;21:787–93.
Niv Y. Cost, benefit, tonic, phasic: what do response rates tell us about dopamine and motivation? Ann N Y Acad Sci. 2007;1104:357–76.
Krok AC, Maltese M, Mistry P, Miao X, Li Y, Tritsch NX. Intrinsic dopamine and acetylcholine dynamics in the striatum of mice. Nature. 2023;621:543–9.
Chantranupong L, Beron CC, Zimmer JA, Wen MJ, Wang W, Sabatini BL. Dopamine and glutamate regulate striatal acetylcholine in decision-making. Nature. 2023;621:577–85.
Grace AA, Bunney BS. Nigral dopamine neurons: intracellular recording and identification with l-dopa injection and histofluorescence. Science. 1980;210:654–6.
Meiser J, Weindl D, Hiller K. Complexity of dopamine metabolism. Cell Commun Signal. 2013;11:34.
Henry JP, Sagné C, Bedet C, Gasnier B. The vesicular monoamine transporter: from chromaffin granule to brain. Neurochem Int. 1998;32:227–46.
Jain M, Sands F, Von Korff RW. Monoamine oxidase activity measurements using radioactive substrates. Anal Biochem. 1973;52:542–54.
McGeer PL, McGeer EG. Neurotransmitter synthetic enzymes. Prog Neurobiol. 1973;2:69–117.
Erickson JD, Eiden LE, Hoffman BJ. Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proc Natl Acad Sci USA. 1992;89:10993–7.
Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, et al. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell. 1992;70:539–51.
De Camilli P, Jahn R. Pathways to regulated exocytosis in neurons. Annu Rev Physiol. 1990;52:625–45.
Wightman RM, Jankowski JA, Kennedy RT, Kawagoe KT, Schroeder TJ, Leszczyszyn DJ, et al. Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc Natl Acad Sci USA. 1991;88:10754–8.
Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, et al. Munc18-1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–91.
Li H, Waites CL, Staal RG, Dobryy Y, Park J, Sulzer DL, et al. Sorting of vesicular monoamine transporter 2 to the regulated secretory pathway confers the somatodendritic exocytosis of monoamines. Neuron. 2005;48:619–33.
Liu C, Kaeser PS. Mechanisms and regulation of dopamine release. Curr Opin Neurobiol. 2019;57:46–53.
Sulzer D, Cragg SJ, Rice ME. Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia. 2016;6:123–48.
Wildenberg G, Sorokina A, Koranda J, Monical A, Heer C, Sheffield M, et al. Partial connectomes of labeled dopaminergic circuits reveal non-synaptic communication and axonal remodeling after exposure to cocaine. Elife. 2021;10:e71981.
Uchigashima M, Ohtsuka T, Kobayashi K, Watanabe M. Dopamine synapse is a neuroligin-2-mediated contact between dopaminergic presynaptic and GABAergic postsynaptic structures. Proc Natl Acad Sci USA. 2016;113:201514074.
Yung KK, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience. 1995;65:709–30.
Zhang S, Qi J, Li X, Wang H-L, Britt JP, Hoffman AF, et al. Dopaminergic and glutamatergic microdomains in a subset of rodent mesoaccumbens axons. Nat Neurosci. 2015;18:386–92.
Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Vesicular monoamine transporter-2: immunogold localization in striatal axons and terminals. Synapse. 1997;26:194–8.
Fortin GD, Desrosiers CC, Yamaguchi N, Trudeau LE. Basal somatodendritic dopamine release requires snare proteins. J Neurochem. 2006;96:1740–9.
Bergquist F, Niazi HS, Nissbrandt H. Evidence for different exocytosis pathways in dendritic and terminal dopamine release in vivo. Brain Res. 2002;950:245–53.
Liu C, Kershberg L, Wang J, Schneeberger S, Kaeser PS. Dopamine secretion is mediated by sparse active zone-like release sites. Cell. 2018;172:706–18.e15.
Banerjee A, Lee J, Nemcova P, Liu C, Kaeser PS. Synaptotagmin-1 is the Ca2+ sensor for fast striatal dopamine release. Elife. 2020;9:e58359.
Nirenberg MJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. J Neurosci. 1996;16:4135–45.
Pickel VM, Chan J, Nirenberg MJ. Region-specific targeting of dopamine D2-receptors and somatodendritic vesicular monoamine transporter 2 (VMAT2) within ventral tegmental area subdivisions. Synapse. 2002;45:113–24.
Mercer L, del Fiacco M, Cuello AC. The smooth endoplasmic reticulum as a possible storage site for dendritic dopamine in substantia nigra neurones. Experientia. 1979;35:101–3.
Lebowitz JJ, Banerjee A, Qiao C, Bunzow JR, Williams JT, Kaeser PS. Synaptotagmin-1 is a Ca2+ sensor for somatodendritic dopamine release. Cell Rep. 2023;42:111915.
Chen BT, Patel JC, Moran KA, Rice ME. Differential calcium dependence of axonal versus somatodendritic dopamine release, with characteristics of both in the ventral tegmental area. Front Syst Neurosci. 2011;5:39
Ford CP, Gantz SC, Phillips PEM, Williams JT. Control of extracellular dopamine at dendrite and axon terminals. J Neurosci. 2010;30:6975–83.
Brimblecombe KR, Gracie CJ, Platt NJ, Cragg SJ. Gating of dopamine transmission by calcium and axonal N-, Q-, T- and L-type voltage-gated calcium channels differs between striatal domains. J Physiol. 2015;593:929–46.
Marcott PF, Mamaligas AA, Ford CP. Phasic dopamine release drives rapid activation of striatal D2-receptors. Neuron. 2014;84:164–76.
Ducrot C, Bourque M-J, Delmas CVL, Racine A-S, Guadarrama Bello D, Delignat-Lavaud B, et al. Dopaminergic neurons establish a distinctive axonal arbor with a majority of non-synaptic terminals. FASEB J. 2021;35:e21791.
Daniel JA, Galbraith S, Iacovitti L, Abdipranoto A, Vissel B. Functional heterogeneity at dopamine release sites. J Neurosci. 2009;29:14670–80.
Banerjee A, Imig C, Balakrishnan K, Kershberg L, Lipstein N, Uronen R-L, et al. Molecular and functional architecture of striatal dopamine release sites. Neuron. 2022;110:248–65.e9.
Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, et al. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–96.
Deng L, Kaeser PS, Xu W, Südhof TC. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron. 2011;69:317–31.
Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, et al. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–95.
Held RG, Liu C, Ma K, Ramsey AM, Tarr TB, De Nola G, et al. Synapse and active zone assembly in the absence of presynaptic Ca2+ channels and Ca2+ entry. Neuron. 2020;107:667–83.e9.
de Jong APH, Roggero CM, Ho M-R, Wong MY, Brautigam CA, Rizo J, et al. RIM C2B domains target presynaptic active zone functions to PIP2-containing membranes. Neuron. 2018;98:335–49.e7.
Di PaoloG, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, et al. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431:415–22.
Elizarova S, Chouaib AA, Shaib A, Hill B, Mann F, Brose N, et al. A fluorescent nanosensor paint detects dopamine release at axonal varicosities with high spatiotemporal resolution. Proc Natl Acad Sci USA. 2022;119:e2202842119.
Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, et al. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–6.
Kershberg L, Banerjee A, Kaeser PS. Protein composition of axonal dopamine release sites in the striatum. Elife. 2022;11:e83018.
Hobson BD, Choi SJ, Mosharov E V, Soni RK, Sulzer D, Sims PA. Subcellular proteomics of dopamine neurons in the mouse brain. Elife. 2022;11:e70921.
Pereira DB, Schmitz Y, Mészáros J, Merchant P, Hu G, Li S, et al. Fluorescent false neurotransmitter reveals functionally silent dopamine vesicle clusters in the striatum. Nat Neurosci. 2016;19:578–86.
Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–81.
Mendez JA, Bourque M-J, Fasano C, Kortleven C, Trudeau L-E. Somatodendritic dopamine release requires synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J Biol Chem. 2011;286:23928–37.
Delignat-Lavaud B, Ducrot C, Kouwenhoven W, Feller N, Trudeau L-É. Implication of synaptotagmins 4 and 7 in activity-dependent somatodendritic dopamine release in the ventral midbrain. Open Biol. 2022;12:210339.
Robinson BG, Cai X, Wang J, Bunzow JR, Williams JT, Kaeser PS. RIM is essential for stimulated but not spontaneous somatodendritic dopamine release in the midbrain. Elife. 2019;8:e47972.
Cragg SJ, Rice ME. DAncing past the DAT at a DA synapse. Trends Neurosci. 2004;27:270–7.
Jaffe EH, Marty A, Schulte A, Chow RH. Extrasynaptic vesicular transmitter release from the somata of substantia nigra neurons in rat midbrain slices. J Neurosci. 1998;18:3548–53.
Pothos EN, Davila V, Sulzer D. Presynaptic recording of quanta from midbrain dopamine neurons and modulation of the quantal size. J Neurosci. 1998;18:4106–18.
Omiatek DM, Bressler AJ, Cans A-S, Andrews AM, Heien ML, Ewing AG. The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Sci Rep. 2013;3:1447.
Hunger L, Kumar A, Schmidt R. Abundance compensates kinetics: similar effect of dopamine signals on D1 and D2 receptor populations. J Neurosci. 2020;40:2868–81.
Gonon F. Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo. J Neurosci. 1997;17:5972–8.
Garris P, Ciolkowski E, Pastore P, Wightman R. Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci. 1994;14:6084–93.
Beaulieu J-M, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217.
Callier S, Snapyan M, Le Crom S, Prou D, Vincent JD, Vernier P. Evolution and cell biology of dopamine receptors in vertebrates. Biol Cell. 2003;95:489–502.
Richfield EK, Penney JB, Young AB. Anatomical and affinity state comparisons between dopamine D1 and D2 receptors in the rat central nervous system. Neuroscience. 1989;30:767–77.
Caillé I, Dumartin B, Bloch B. Ultrastructural localization of D1 dopamine receptor immunoreactivity in rat striatonigral neurons and its relation with dopaminergic innervation. Brain Res. 1996;730:17–31.
Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, et al. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–32.
Skinbjerg M, Sibley DR, Javitch JA, Abi-Dargham A. Imaging the high-affinity state of the dopamine D2 receptor in vivo: fact or fiction? Biochem Pharmacol. 2012;83:193–8.
Goto Y, Grace AA. Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci. 2008;31:552–8.
Yapo C, Nair AG, Clement L, Castro LR, Hellgren Kotaleski J, Vincent P. Detection of phasic dopamine by D1 and D2 striatal medium spiny neurons. J Physiol. 2017;595:7451–75.
Bowery B, Rothwell LA, Seabrook GR. Comparison between the pharmacology of dopamine receptors mediating the inhibition of cell firing in rat brain slices through the substantia nigra pars compacta and ventral tegmental area. Br J Pharmacol. 1994;112:873–80.
Nair AG, Gutierrez-Arenas O, Eriksson O, Vincent P, Hellgren Kotaleski J. Sensing positive versus negative reward signals through adenylyl cyclase-coupled GPCRs in direct and indirect pathway striatal medium spiny neurons. J Neurosci. 2015;35:14017–30.
Beckstead MJ, Grandy DK, Wickman K, Williams JT. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron. 2004;42:939–46.
Courtney NA, Ford CP. The timing of dopamine- and noradrenaline-mediated transmission reflects underlying differences in the extent of spillover and pooling. J Neurosci. 2014;34:7645–56.
Condon AF, Robinson BG, Asad N, Dore TM, Tian L, Williams JT. The residence of synaptically released dopamine on D2 autoreceptors. Cell Rep. 2021;36:109465.
Dahlström A, Fuxe K. Localization of monoamines in the lower brain stem. Experientia. 1964;20:398–9.
Schwarz LA, Luo L. Organization of the locus coeruleus-norepinephrine system. Curr Biol. 2015;25:R1051–56.
Okaty BW, Commons KG, Dymecki SM. Embracing diversity in the 5-HT neuronal system. Nat Rev Neurosci. 2019;20:397–424.
Moore RY, Bloom FE. Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci. 1979;2:113–68.
Maddaloni G, Bertero A, Pratelli M, Barsotti N, Boonstra A, Giorgi A, et al. Development of serotonergic fibers in the post-natal mouse brain. Front Cell Neurosci. 2017;11:202.
Fuxe K, Hamberger B, Hökfelt T. Distribution of noradrenaline nerve terminals in cortical areas of the rat. Brain Res. 1968;8:125–31.
Soghomonian JJ, Descarries L, Watkins KC. Serotonin innervation in adult rat neostriatum. II. Ultrastructural features: a radioautographic and immunocytochemical study. Brain Res. 1989;481:67–86.
Séguéla P, Watkins KC, Geffard M, Descarries L. Noradrenaline axon terminals in adult rat neocortex: an immunocytochemical analysis in serial thin sections. Neuroscience. 1990;35:249–64.
Kocsis B, Varga V, Dahan L, Sik A. Serotonergic neuron diversity: identification of raphe neurons with discharges time-locked to the hippocampal theta rhythm. Proc Natl Acad Sci USA. 2006;103:1059–64.
Allers KA, Sharp T. Neurochemical and anatomical identification of fast- and slow-firing neurones in the rat dorsal raphe nucleus using juxtacellular labelling methods in vivo. Neuroscience. 2003;122:193–204.
Hajós M, Gartside SE, Villa AEP, Sharp T. Evidence for a repetitive (burst) firing pattern in a sub-population of 5-hydroxytryptamine neurons in the dorsal and median raphe nuclei of the rat. Neuroscience. 1995;69:189–97.
Cohen JY, Amoroso MW, Uchida N. Serotonergic neurons signal reward and punishment on multiple timescales. Elife. 2015;4:1–25.
Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–50.
Foote SL, Aston-Jones G, Bloom FE. Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc Natl Acad Sci USA. 1980;77:3033–7.
Hobson JA, Mccarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–8.
Berridge CW, Abercrombie ED. Relationship between locus coeruleus discharge rates and rates of norepinephrine release within neocortex as assessed by in vivo microdialysis. Neuroscience. 1999;93:1263–70.
Bunin MA, Wightman RM. Quantitative evaluation of 5-hydroxytryptamine (serotonin) neuronal release and uptake: an investigation of extrasynaptic transmission. J Neurosci. 1998;18:4854–60.
Florin-Lechner SM, Druhan JP, Aston-Jones G, Valentino RJ. Enhanced norepinephrine release in prefrontal cortex with burst stimulation of the locus coeruleus. Brain Res. 1996;742:89–97.
O’Connor JJ, Kruk ZL. Fast cyclic voltammetry can be used to measure stimulated endogenous 5-hydroxytryptamine release in untreated rat brain slices. J Neurosci Methods. 1991;38:25–33.
Nakov R, Habermann E, Hertting G, Wurster S, Allgaier C. Effects of botulinum A toxin on presynaptic modulation of evoked transmitter release. Eur J Pharmacol. 1989;164:45–53.
Ashton AC, Dolly JO. Characterization of the inhibitory action of botulinum neurotoxin type A on the release of several transmitters from rat cerebrocortical synaptosomes. J Neurochem. 1988;50:1808–16.
Kataoka M, Yamamori S, Suzuki E, Watanabe S, Sato T, Miyaoka H, et al. A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS ONE. 2011;6:e25158.
Kim JC, Cook MN, Carey MR, Shen C, Regehr WG, Dymecki SM. Linking genetically defined neurons to behavior through a broadly applicable silencing allele. Neuron. 2009;63:305–15.
Najib A, Pelliccioni P, Gil C, Aguilera J. Clostridium neurotoxins influence serotonin uptake and release differently in rat brain synaptosomes. J Neurochem. 1999;72:1991–8.
Palij P, Stamford JA. Real-time monitoring of endogenous noradrenaline release in rat brain slices using fast cyclic voltammetry: 1. Characterisation of evoked noradrenaline efflux and uptake from nerve terminals in the bed nucleus of stria terminalis, pars ventralis. Brain Res. 1992;587:137–46.
Bunin MA, Prioleau C, Mailman RB, Wightman RM. Release and uptake rates of 5-hydroxytryptamine in the dorsal raphe and substantia nigra reticulata of the rat brain. J Neurochem. 1998;70:1077–87.
Sharp T, Bramwell SR, Grahame-Smith DG. Release of endogenous 5-hydroxytryptamine in rat ventral hippocampus evoked by electrical stimulation of the dorsal raphe nucleus as detected by microdialysis: sensitivity to tetrodotoxin, calcium and calcium antagonists. Neuroscience. 1990;39:629–37.
Wang YM, Gainetdinov RR, Fumagalli F, Xu F, Jones SR, Bock CB, et al. Knockout of the vesicular monoamine transporter 2 gene results in neonatal death and supersensitivity to cocaine and amphetamine. Neuron. 1997;19:1285–96.
Zhou QY, Quaife CJ, Palmiter RD. Targeted disruption of the tyrosine hydroxylase gene reveals that catecholamines are required for mouse fetal development. Nature. 1995;374:640–3.
Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374:643–6.
Narboux-Nême N, Sagné C, Doly S, Diaz SL, Martin CBP, Angenard G, et al. Severe serotonin depletion after conditional deletion of the vesicular monoamine transporter 2 gene in serotonin neurons: neural and behavioral consequences. Neuropsychopharmacology. 2011;36:2538–50.
Isingrini E, Perret L, Rainer Q, Sagueby S, Moquin L, Gratton A, et al. Selective genetic disruption of dopaminergic, serotonergic and noradrenergic neurotransmission: insights into motor, emotional and addictive behaviour. J Psychiatry Neurosci. 2016;41:169–81.
Isingrini E, Perret L, Rainer Q, Amilhon B, Guma E, Tanti A, et al. Resilience to chronic stress is mediated by noradrenergic regulation of dopamine neurons. Nat Neurosci. 2016;19:560–3.
Stewart LC, Klinman JP. Dopamine beta-hydroxylase of adrenal chromaffin granules: structure and function. Annu Rev Biochem. 1988;57:551–90.
Weinshilboum RM, Thoa NB, Johnson DG, Kopin IJ, Axelrod J. Proportional release of norepinephrine and dopamine-beta-hydroxylase from sympathetic nerves. Science. 1971;174:1349–51.
Pratelli M, Pasqualetti M. Serotonergic neurotransmission manipulation for the understanding of brain development and function: Learning from Tph2 genetic models. Biochimie. 2019;161:3–14.
Bruns D, Jahn R. Real-time measurement of transmitter release from single synaptic vesicles. Nature. 1995;377:62–5.
Bruns D, Riedel D, Klingauf J, Jahn R. Quantal release of serotonin. Neuron. 2000;28:205–20.
Shields PJ, Eccleston D. Evidence for the synthesis and storage of 5-hydroxytryptamine in two separate pools in the brain. J Neurochem. 1973;20:881–8.
Nirenberg MJ, Liu Y, Peter D, Edwards RH, Pickel VM. The vesicular monoamine transporter 2 is present in small synaptic vesicles and preferentially localizes to large dense core vesicles in rat solitary tract nuclei. Proc Natl Acad Sci USA. 1995;92:8773–7.
Trueta C, De-Miguel FF. Extrasynaptic exocytosis and its mechanisms: a source of molecules mediating volume transmission in the nervous system. Front Physiol. 2012;3:319.
Sun F, Zeng J, Jing M, Zhou J, Feng J, Owen SF, et al. A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell. 2018;174:481–96.e19.
Wan J, Peng W, Li X, Qian T, Song K, Zeng J, et al. A genetically encoded sensor for measuring serotonin dynamics. Nat Neurosci. 2021;24:746–52.
Kile BM, Guillot TS, Venton BJ, Wetsel WC, Augustine GJ, Wightman RM. Synapsins differentially control dopamine and serotonin release. J Neurosci. 2010;30:9762-70.
Varga V, Losonczy A, Zemelman BV, Borhegyi Z, Nyiri G, Domonkos A, et al. Fast synaptic subcortical control of hippocampal circuits. Science. 2009;326:449–53.
Feng J, Zhang C, Lischinsky JE, Jing M, Zhou J, Wang H, et al. A genetically encoded fluorescent sensor for rapid and specific in vivo detection of norepinephrine. Neuron. 2019;102:745–61.e8.
Park J, Kile BM, Wightman MR. In vivo voltammetric monitoring of norepinephrine release in the rat ventral bed nucleus of the stria terminalis and anteroventral thalamic nucleus. Eur J Neurosci. 2009;30:2121–33.
Park J, Takmakov P, Wightman RM. In vivo comparison of norepinephrine and dopamine release in rat brain by simultaneous measurements with fast-scan cyclic voltammetry. J Neurochem. 2011;119:932–44.
Okada M, Mine K, Fujiwara M. Differential calcium dependence between the release of endogenous dopamine and noradrenaline from rat brain synaptosomes. J Neurochem. 1990;54:1947–52.
Hirning LD, Fox AP, McCleskey EW, Olivera BM, Thayer SA, Miller RJ, et al. Dominant role of N-type Ca2+ channels in evoked release of norepinephrine from sympathetic neurons. Science. 1988;239:57–61.
Feuerstein TJ, Dooley DJ, Seeger W. Inhibition of norepinephrine and acetylcholine release from human neocortex by omega-conotoxin GVIA. J Pharmacol Exp Ther. 1990;252:778–85.
Friedman DJ, Duckles SP. Effect of calcium channel blockers on norepinephrine release and modulation by prejunctional D2 dopamine receptors. Life Sci. 1994;54:1545–57.
Kimura M, Yamanishi Y, Hanada T, Kagaya T, Kuwada M, Watanabe T, et al. Involvement of P-type calcium channels in high potassium-elicited release of neurotransmitters from rat brain slices. Neuroscience. 1995;66:609–15.
Harvey J, Wedley S, Findlay JD, Sidell MR, Pullar IA. omega-Agatoxin IVA identifies a single calcium channel subtype which contributes to the potassium-induced release of acetylcholine, 5-hydroxytryptamine, dopamine, gamma-aminobutyric acid and glutamate from rat brain slices. Neuropharmacology. 1996;35:385–92.
Mitchell K, Adams RN. Comparison of the effects of voltage-sensitive calcium channel antagonism on the electrically stimulated release of dopamine and norepinephrine in vivo. Brain Res. 1993;604:349–53.
Bohne P, Volkmann A, Schwarz MK, Mark MD. Deletion of the P/Q-type calcium channel from serotonergic neurons drives male aggression in mice. J Neurosci. 2022;42:6637–53.
Agster KL, Mejias-Aponte CA, Clark BD, Waterhouse BD. Evidence for a regional specificity in the density and distribution of noradrenergic varicosities in rat cortex. J Comp Neurol. 2013;521:2195–207.
Plummer NW, Chandler DJ, Powell JM, Scappini EL, Waterhouse BD, Jensen P. An intersectional viral-genetic method for fluorescent tracing of axon collaterals reveals details of noradrenergic locus coeruleus structure. ENeuro. 2020;7:ENEURO.0010-20.2020.
Dunn M, Henke A, Clark S, Kovalyova Y, Kempadoo KA, Karpowicz RJ, et al. Designing a norepinephrine optical tracer for imaging individual noradrenergic synapses and their activity in vivo. Nat Commun. 2018;9:2838.
Unger EK, Keller JP, Altermatt M, Liang R, Matsui A, Dong C, et al. Directed evolution of a selective and sensitive serotonin sensor via machine learning. Cell. 2020;183:1986–2002.e26.
Kagiampaki Z, Rohner V, Kiss C, Curreli S, Dieter A, Wilhelm M, et al. Sensitive multicolor indicators for monitoring norepinephrine in vivo. Nat Methods. 2023;20:1426–36.
Deng F, Wan J, Li G, Dong H, Xia X, Wang Y, et al. Improved green and red GRAB sensors for monitoring spatiotemporal serotonin release in vivo. Nat Methods. 2024;21:692–702.
Feng J, Dong H, Lischinsky JE, Zhou J, Deng F, Zhuang C, et. al. Monitoring norepinephrine release in vivo using next-generation GRABNE sensors. Neuron. 2024:S0896-6273(24)00155-7.
Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, et al. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev. 2011;63:585–640.
Aggarwal S, Mortensen OV. Overview of monoamine transporters. Curr Protoc Pharmacol. 2017;79:12.16.1–12.16.17.
Morón JA, Brockington A, Wise RA, Rocha BA, Hope BT. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci. 2002;22:389.
Nosaka D, Wickens JR. Striatal cholinergic signaling in time and space. Molecules. 2022;27:1202.
Littlewood GM, Iversen LL, Turner AJ. Neuropeptides and their peptidases: functional considerations. Neurochem Int. 1988;12:383–9.
Malfroy B, Swerts JP, Guyon A, Roques BP, Schwartz JC. High-affinity enkephalin-degrading peptidase in brain is increased after morphine. Nature. 1978;276:523–6.
Otte C, Gold SM, Penninx BW, Pariante CM, Etkin A, Fava M, et al. Major depressive disorder. Nat Rev Dis Primers. 2016;2:16065.
Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4:13–25.
Huang J, Spier AD, Pickel VM. 5-HT3A receptor subunits in the rat medial nucleus of the solitary tract: subcellular distribution and relation to the serotonin transporter. Brain Res. 2004;1028:156–69.
Aoki C. Beta-adrenergic receptors: astrocytic localization in the adult visual cortex and their relation to catecholamine axon terminals as revealed by electron microscopic immunocytochemistry. J Neurosci. 1992;12:781–92.
Aoki C, Pickel VM. Ultrastructural relations between beta-adrenergic receptors and catecholaminergic neurons. Brain Res Bull. 1992;29:257–63.
Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci. 2011;32:213–8.
McCorvy JD, Roth BL. Structure and function of serotonin G protein-coupled receptors. Pharmacol Ther. 2015;150:129–42.
Nichols DE, Nichols CD. Serotonin receptors. Chem Rev. 2008;108:1614–41.
Sugita S, Shen KZ, North RA. 5-hydroxytryptamine is a fast excitatory transmitter at 5-HT3 receptors in rat amygdala. Neuron. 1992;8:199–203.
Roerig B, Nelson DA, Katz LC. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci. 1997;17:8353–62.
Pazos A, Probst A, Palacios JM. Serotonin receptors in the human brain-IV. Autoradiographic mapping of serotonin-2 receptors. Neuroscience. 1987;21:123–39.
Wu Y, Zeng L, Zhao S. Ligands of adrenergic receptors: a structural point of view. Biomolecules. 2021;11:936.
Young WS, Kuhar MJ. Noradrenergic alpha 1 and alpha 2 receptors: light microscopic autoradiographic localization. Proc Natl Acad Sci USA. 1980;77:1696–1700.
Sutin J, Minneman KP. Adrenergic beta receptors are not uniformly distributed in the cerebellar cortex. J Comp Neurol. 1985;236:547–54.
Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of delta subunit-containing GABA(A) receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–61.
Rossi DJ, Hamann M. Spillover-mediated transmission at inhibitory synapses promoted by high affinity alpha6 subunit GABA(A) receptors and glomerular geometry. Neuron. 1998;20:783–95.
Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–93.
Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–31.
Tritsch NX, Granger AJ, Sabatini BL. Mechanisms and functions of GABA co-release. Nat Rev Neurosci. 2016;17:139–45.
Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature. 2012;490:262–6.
Hirasawa H, Betensky RA, Raviola E. Corelease of Dopamine and GABA by a Retinal Dopaminergic Neuron. J Neurosci. 2012;32:13281–91.
Disney AA, Higley MJ. Diverse spatiotemporal scales of cholinergic signaling in the neocortex. J Neurosci. 2020;40:720–5.
Sarter M, Lustig C. Forebrain cholinergic signaling: wired and phasic, not tonic, and causing behavior. J Neurosci. 2020;40:712–9.
Mamaligas AA, Ford CP. Spontaneous synaptic activation of muscarinic receptors by striatal cholinergic neuron firing. Neuron. 2016;91:574–86.
Ballinger EC, Ananth M, Talmage DA, Role LW. Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron. 2016;91:1199–218.
Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524.
Gonzales KK, Smith Y. Cholinergic interneurons in the dorsal and ventral striatum: anatomical and functional considerations in normal and diseased conditions. Ann N Y Acad Sci. 2015;1349:1–45.
Zhou F, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. 2002;53:590–605.
Jones IW, Bolam JP, Wonnacott S. Presynaptic localisation of the nicotinic acetylcholine receptor beta2 subunit immunoreactivity in rat nigrostriatal dopaminergic neurones. J Comp Neurol. 2001;439:235–47.
Zhou F-M, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–9.
Cachope R, Mateo Y, Mathur BN, Irving J, Wang H-L, Morales M, et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41.
Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron.2012;75:58–64.
Soliakov L, Wonnacott S. Voltage-sensitive Ca2+ channels involved in nicotinic receptor-mediated [3H]dopamine release from rat striatal synaptosomes. J Neurochem. 1996;67:163–70.
Giorguieff MF, Le Floc’h ML, Westfall TC, Glowinski J, Besson MJ. Nicotinic effect of acetylcholine on the release of newly synthesized [3H]dopamine in rat striatal slices and cat caudate nucleus. Brain Res. 1976;106:117–31.
Bolam JP, Wainer BH, Smith AD. Characterization of cholinergic neurons in the rat neostriatum. A combination of choline acetyltransferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience. 1984;12:711–8.
Schulz JM, Oswald MJ, Reynolds JNJ. Visual-induced excitation leads to firing pauses in striatal cholinergic interneurons. J Neurosci. 2011;31:11133–43.
Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–43.
Cai Y, Ford CP. Dopamine cells differentially regulate striatal cholinergic transmission across regions through corelease of dopamine and glutamate. Cell Rep. 2018;25:3148–57.e3.
Hoover DB, Muth EA, Jacobowitz DM. A mapping of the distribution of acetycholine, choline acetyltransferase and acetylcholinesterase in discrete areas of rat brain. Brain Res. 1978;153:295–306.
Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–63.
Girven KS, Mangieri L, Bruchas MR. Emerging approaches for decoding neuropeptide transmission. Trends Neurosci. 2022;45:899–912.
Zhang X, Bao L, Ma GQ. Sorting of neuropeptides and neuropeptide receptors into secretory pathways. Prog Neurobiol. 2010;90:276–83.
van de Bospoort R, Farina M, Schmitz SK, de Jong A, de Wit H, Verhage M, et al. Munc13 controls the location and efficiency of dense-core vesicle release in neurons. J Cell Biol. 2012;199:883–91.
Dean C, Liu H, Mark Dunning F, Chang PY, Jackson MB, Chapman ER. Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci. 2009;12:767–76.
Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, et al. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci. 2009;29:14185–98.
Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–97.
Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–407.
Persoon CM, Hoogstraaten RI, Nassal JP, van Weering JRT, Kaeser PS, Toonen RF, et al. The RAB3-RIM pathway is essential for the release of neuromodulators. Neuron. 2019;104:1065–80.e12.
Cao P, Maximov A, Südhof TC. Activity-dependent IGF-1 exocytosis is controlled by the Ca(2+)-sensor synaptotagmin-10. Cell. 2011;145:300–11.
Cao P, Yang X, Südhof TC. Complexin activates exocytosis of distinct secretory vesicles controlled by different synaptotagmins. J Neurosci. 2013;33:1714–27.
Darlison MG, Richter D. Multiple genes for neuropeptides and their receptors: co-evolution and physiology. Trends Neurosci. 1999;22:81–8.
Brain SD, Cox HM. Neuropeptides and their receptors: innovative science providing novel therapeutic targets. Br J Pharmacol. 2006;147 Suppl 1(Suppl 1):S202–11.
Chini B, Verhage M, Grinevich V. The action radius of oxytocin release in the mammalian cns: from single vesicles to behavior. Trends Pharmacol Sci. 2017;38:982–91.
Gicquiaux H, Lecat S, Gaire M, Dieterlen A, Mély Y, Takeda K, et al. Rapid internalization and recycling of the human neuropeptide Y Y(1) receptor. J Biol Chem. 2002;277:6645–55.
Qian T, Wang H, Xia X, Li Y. Current and emerging methods for probing neuropeptide transmission. Curr Opin Neurobiol. 2023;81:102751.
Wu Z, Lin D, Li Y. Pushing the frontiers: tools for monitoring neurotransmitters and neuromodulators. Nat Rev Neurosci. 2022;23:257–74.
Sabatini BL, Tian L. Imaging neurotransmitter and neuromodulator dynamics in vivo with genetically encoded indicators. Neuron. 2020;108:17–32.
Dong A, He K, Dudok B, Farrell JS, Guan W, Liput DJ, et al. A fluorescent sensor for spatiotemporally resolved imaging of endocannabinoid dynamics in vivo. Nat Biotechnol. 2022;40:787–98.
Albarran E, Sun Y, Liu Y, Raju K, Dong A, Li Y, et al. Postsynaptic synucleins mediate endocannabinoid signaling. Nat Neurosci. 2023;26:997–1007.
van Keimpema L, Kooistra R, Toonen RF, Verhage M. CAPS-1 requires its C2, PH, MHD1 and DCV domains for dense core vesicle exocytosis in mammalian CNS neurons. Sci Rep. 2017;7:10817.
Sadakata T, Kakegawa W, Mizoguchi A, Washida M, Katoh-Semba R, Shutoh F, et al. Impaired cerebellar development and function in mice lacking CAPS2, a protein involved in neurotrophin release. J Neurosci. 2007;27:2472–82.
Shinoda Y, Sadakata T, Nakao K, Katoh-Semba R, Kinameri E, Furuya A, et al. Calcium-dependent activator protein for secretion 2 (CAPS2) promotes BDNF secretion and is critical for the development of GABAergic interneuron network. Proc Natl Acad Sci USA. 2011;108:373–8.
Farina M, van de Bospoort R, He E, Persoon CM, van Weering JRT, Broeke JH, et al. CAPS-1 promotes fusion competence of stationary dense-core vesicles in presynaptic terminals of mammalian neurons. Elife. 2015;4:1–22.
Wong YH, Lee CM, Xie W, Cui B, Poo MM. Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc Natl Acad Sci USA. 2015;112:E4475–84.
Moro A, van Nifterick A, Toonen RF, Verhage M. Dynamin controls neuropeptide secretion by organizing dense-core vesicle fusion sites. Sci Adv. 2021;7:eabf0659.
Puntman DC, Arora S, Farina M, Toonen RF, Verhage M. Munc18-1 is essential for neuropeptide secretion in neurons. J Neurosci. 2021;41:5980–93.
Shimojo M, Courchet J, Pieraut S, Torabi-Rander N, Sando R, Polleux F, et al. SNAREs controlling vesicular release of BDNF and development of callosal axons. Cell Rep. 2015;11:1054-66.
Arora S, Saarloos I, Kooistra R, van de Bospoort R, Verhage M, Toonen RF. SNAP-25 gene family members differentially support secretory vesicle fusion. J Cell Sci. 2017;130:1877–89.
van Westen R, Poppinga J, Arazola RD, Toonen RF, Verhage M. Neuromodulator release in neurons requires two functionally redundant calcium sensors. Proc Natl Acad Sci USA. 2021;118:e2012137118.
Seibert MJ, Evans CS, Stanley KS, Wu Z, Chapman ER. Synaptotagmin 9 modulates spontaneous neurotransmitter release in striatal neurons by regulating substance P secretion. J Neurosci. 2023;43:1475–91.
Emperador-Melero J, Huson V, van Weering J, Bollmann C, Fischer von Mollard G, Toonen RF, et al. Vti1a/b regulate synaptic vesicle and dense core vesicle secretion via protein sorting at the Golgi. Nat Commun. 2018;9:3421.
Acknowledgements
Our work on synaptic and neuromodulatory transmission is supported by NIH grants (R01NS103484, R01DA056109, R01MH113349, R01NS083898 to PSK), by a Harvard Medical School Neurobiology Spark Grant (to PSK) and by a Harvard Brain Initiative Bipolar Disorder Seed Grant (to PSK). AB is supported by a Brain and Behavior Research Foundation young investigator grant, and ODO is a Human Frontier Science Program fellow. We thank R Lopez, L Silenzi, K Beeson, K Nozawa and J Emperador Melero for feedback on the manuscript, and all members of our laboratory for insightful discussions.
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing of the original draft and to review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Özçete, Ö.D., Banerjee, A. & Kaeser, P.S. Mechanisms of neuromodulatory volume transmission. Mol Psychiatry (2024). https://doi.org/10.1038/s41380-024-02608-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41380-024-02608-3
