
Abstract
Drug addiction, one of the major health problems worldwide, is characterized by the loss of control in drug intake, craving, and withdrawal. At the individual level, drugs of abuse produce serious consequences on health and have a negative impact on the family environment and on interpersonal and work relationships. At a wider scale, they have significant socio-economic and public health consequences and they cause delinquency and citizen insecurity. Cocaine, a psychostimulant substance, is one of the most used illicit drugs, especially in America, Western Europe, and Australia. Cocaine use disorders (CUD) are complex multifactorial conditions driven by both genetic and environmental influences. Importantly, not all people who use cocaine develop CUD, and this is due, at least in part, to biological factors that are encoded in the genome of individuals. Acute and repeated use of cocaine induces epigenetic and gene expression changes responsible for the neuronal adaptations and the remodeling of brain circuits that lead to the transition from use to abuse or dependence. The purpose of this review is to delineate such factors, which should eventually help to understand the inter-individual variability in the susceptibility to cocaine addiction. Heritability estimates for CUD are high and genetic risk factors for cocaine addiction have been investigated by candidate gene association studies (CGAS) and genome-wide association studies (GWAS), reviewed here. Also, the high comorbidity that exists between CUD and several other psychiatric disorders is well known and includes phenotypes like schizophrenia, aggression, antisocial or risk-taking behaviors. Such comorbidities are associated with a worse lifetime trajectory, and here we report shared genetic factors that may contribute to them. Gene expression changes and epigenetic modifications induced by cocaine use and chronic abuse in humans are addressed by reviewing transcriptomic studies performed on neuronal cells and on postmortem brains. We report some genes which expression is altered by cocaine that also bear genetic risk variants for the disorder. Finally, we have a glance to the pharmacogenetics of CUD treatments, still in early stages. A better understanding of the genetic underpinnings of CUD will foster the search of effective treatments and help to move forward to personalized medicine.
Similar content being viewed by others


Clinical definition
The term substance use disorders (SUD), including cocaine use disorders (CUD), refers to different types of behaviors that range from sporadic use to abuse, dependence or addiction. The differential clinical diagnosis is based on the Diagnostic and Statistical Manual of Mental Disorders (DSM), which current version is DSM-5 [1], although many studies still use the previous DSM-IV-TR [2]. The main change of DSM-5 with respect to the previous version of the manual is the unification of abuse and dependence into a unidimensional category, SUD, that is qualified on a severity scale (i.e. mild, moderate and severe addiction) based on the number of symptoms endorsed, over a total of 10 (Table 1). In addition, DSM-5 drops one of the diagnostic criteria (legal problems) due to infrequent endorsement and poor discriminant validity [3], and adds a new one: craving. The World Health Organization developed another diagnostic interview, the International Classification of Diseases (ICD-11; https://icd.who.int/en), that keeps substance dependence as the main diagnostic criterion. Several authors have attempted to establish more homogeneous subgroups of cocaine-related phenotypes that may be useful for subsequent genetic analyses. Thus, five subtypes of cocaine abusers have been defined on the basis of clinical presentation, family history, and response to treatment [4]. More recently, cluster analysis was not only used to classify individuals on different groups according to cocaine-related measures but also to demographic features and prevalence rates of comorbid substance use and psychiatric disorders. Interestingly, those clusters characterized by a more severe phenotype yielded higher heritability estimates [5, 6].
Epidemiology
According to the United Nations Office on Drugs and Crime, the number of people aged 15–64 years that use illegal drugs increased from ~210 to ~269 million over the period 2009–2018 (more than 28%, partly as a result of the global population growth) and the prevalence raised from 4.8 to 5.4% (12% increase) [7]. From these, over 35 million people suffer from SUD. Cocaine, together with metamphetamines, dominates the psychostimulants share. Some 19 million people used cocaine in 2018 (0.4% of the global population aged 15–64), fueled by the drug’s popularity in North and South America, Western Europe, and Australia. In Europe, cocaine is the most used illegal psychostimulant, around 5.4% of adults have tried cocaine [8], but at most 20% of them will develop addiction [9, 10]. From these data, the prevalence of cocaine dependence in the European population can be estimated around 1.1%, a value similar to that observed in American populations [11]. The prevalence of CUD is higher in males than in females, with 85% of cocaine users that initiate a treatment being males in contrast to 15% of females [8]. Ethnicity is also relevant, for example with rates of cocaine overdose deaths being much higher in African–American (AA) individuals in the United States [12]. For different reasons, research studies, and clinical trials have underrepresented women and ethnic minorities. This may lead to gaps in the discovery of risk factors for addiction, as several factors that influence the origin and development of the disorder differ across gender and ethnic groups. These factors include psychiatric comorbidities, sociodemographic status, frequency and severity of cocaine use, and also biological determinants [13, 14].
Neurobiology
At the molecular level, cocaine binds the monoaminergic transporters (DAT, NET, and SERT) blocking the reuptake of these neurotransmitters by the presynaptic neuron, increasing the levels of dopamine, serotonin, and noradrenaline at the synaptic cleft [15]. Cocaine pleasurable and rewarding effects are mediated mainly by the increase of dopamine activity in the limbic system. Chronic cocaine use induces alterations and adaptations in several neurotransmitter systems and affects the function of several circuits and areas such as the mesocorticolimbic system (including the nucleus accumbens (NAc) and ventral tegmental area as well as prefrontal cortex). Serotonin neurotransmission is also key to cocaine addiction since it contributes to relapse by modulating impulsivity and responsivity to cocaine-associated stimuli [16].
Preventing relapse is the main challenge for treating CUD and SUD in general. Learning and memory processes associate memories between cocaine’s reinforcing and rewarding effects and environmental stimuli, which then will trigger cocaine craving during abstinence. This vulnerability to relapse, even after a long period of abstinence, involves stable gene expression changes and epigenetic modifications, especially in the corticostriatolimbic circuitry (hippocampus, prefrontal cortex, NAc, dorsal striatum, and amygdala) [17]. Stress plays an important role in relapse since increases drug craving, involving mainly the HPA axis and corticotropin-releasing factor [18].
Cocaine-induced changes and adaptations in the brain through repeated use will depend on the genetic background of each individual. Also, these functional modifications are modulated by environmental factors and the interplay between them and genetic risk factors. Thus, some individuals are more genetically susceptible to develop CUD and addiction than others, being only around 16–20% of cocaine users. In this review, we focus on the genetics of CUD.
Genetic predisposition to cocaine dependence
Heritability: family and twin studies
SUD, in contrast to other psychiatric disorders, are often seen as preventable and attributed to individual’s choice, but many studies have shown that they are heritable, highlighting the relevance of genetic risk factors [19]. Familial aggregation of alcoholism and addiction to illicit drugs is well described, and relatives of probands with SUD have an eightfold increased risk of drug use disorders, being 4.4-fold in the case of cocaine [20]. Familiar, adoption, and especially twin studies have brought consistent evidence that both environmental and genetic risk factors contribute to initiating the use of drugs of abuse, the transition to abuse, and the development of dependence. Heritability estimates described by different twin studies have shown that the genetic contribution to SUD and addictions is in general high, and also variable depending on the drug of abuse [21].
Heritability estimates for CUD are summarized in Table 2. In general, heritability is lower for drug use than for dependence for all drugs of abuse [19]. In the case of cocaine, the contribution of genetic factors for cocaine use is estimated ~0.39–0.44 [22,23,24,25], although higher estimates, 0.61–0.7, were found in other studies [26, 27] (Table 2). For cocaine abuse, heritability estimates are also highly variable, ranging from 0.32 to 0.79 [22, 24, 27,28,29]. Finally, genetic risk factors explaining the variance for cocaine dependence have been estimated to be consistently high across studies 0.65–0.79 [24, 27, 30] (Table 2), being one of the most heritable psychiatric disorders. Some of these studies estimated the contribution of common genetic risk factors for several substances of abuse, and the ones that are specific to cocaine, reporting a high percentage of 92–93% of common factors with other drugs [28, 29].
Genetic architecture of cocaine addiction
Although the contribution of genetic risk factors to cocaine addiction is very high, as described before, identifying the specific factors that underlie addiction is challenging due to the high polygenicity and complexity of this disorder. The contribution of common variants through association studies, as well as rare variants, has been investigated for cocaine dependence. Some relevant variants and genes have been highlighted, but most genetic risk factors are still largely unknown.
Common variants
Candidate gene association studies (CGAS) have focused on specific sets of genes based on a priori assumptions about their role in the mechanism of action of cocaine and in the development of addiction, including dopamine and serotonin neurotransmission (Supplementary Table S1). Although transgenic mice for some of these genes showed alterations in cocaine-seeking behavior, particularly those encoding the monoamine transporters [31], no robust associations have been found in CGAS. These association studies are typically underpowered and the significant associations identified so far have not been successfully replicated, except for the SNPs rs806368 in the CNR1 gene [32,33,34] and rs16969968 in the CHRNA5-CHRNA3-CHRNB4 gene cluster [35,36,37], found significantly associated with cocaine dependence in several studies with limited sample sizes (Supplementary Table S1).
So far, only one copy number variant (CNV) has been found associated with cocaine dependence, a large polymorphic CNV that partially spans the NSF gene, involved in synaptic vesicle turnover [38]. In this study, individuals with a low number of copies showed a quicker transition from cocaine use to dependence than individuals with a high number of copies.
The first genome-wide association study (GWAS) on cocaine dependence with positive findings was published in 2014 [39]. The authors developed a model (Sympcountadj) to remove the effect attributable to other substances (alcohol, opioids and nicotine), thus facilitating the identification of genetic risk variants for cocaine addiction. In addition, they used cocaine-exposed controls that did not develop addiction. In the European–American (EA) discovery sample (1809 cases and 292 controls), a genome-wide significant (GWS) association was found between cocaine dependence and rs150954431 (Table 3), located in the NCOR2 gene (nuclear receptor corepressor 2) that regulates gene expression by activating histone deacetylase 3, and has been involved in memory [40]. However, this finding could not be replicated in two follow-up samples (including 4063 and 2549 subjects, respectively). The metanalysis of AA–EA samples revealed a different GWS hit on the FAM53B gene (family with sequence similarity 53 member B), rs2629540 (Table 3), that was almost GWS in the discovery sample. Although genetic variants in this gene have been related to changes in brain volumes [41], very little is known about FAM53B function and, more importantly, about its contribution to cocaine addiction susceptibility. Nevertheless, this association could not be replicated in a Spanish cohort of cocaine dependence (1011 cases and 1719 controls) [42]. Recently, a transcriptome-wide association study (TWAS) was performed using these data in several brain regions, although no significant associations were found in any tissue for cocaine dependence [43].
A GWAS metanalysis of four samples of subjects with European ancestry (2085 cocaine-dependent patients and 4293 controls) found suggestive associations (P < 1E−05) of several SNPs with cocaine dependence, being rs3075660 in the NRG3 gene the most significant one (Table 3) [44]. This gene, encoding a signaling protein involved in neurodevelopmental processes, had previously been associated with several psychiatric conditions, including schizophrenia (SCZ) [45]. In addition, at gene level, HIST1H2BD, encoding a histone protein, was found significantly associated with cocaine dependence [44]. Interestingly, this study estimated a SNP-based heritability (h2SNP) for cocaine dependence around 0.27–0.30 using two different approaches, and very similar results were obtained using GWAS data from Gelernter et al. (h2SNP = 0.28; Table 3) [46].
Two GWAS on CUD have been recently published, one accounting for gene–environment interactions [6], and another one evaluating time to develop dependence [47] (Table 3). In the first one, two GWS hits were found associated with DSM-5 diagnostic criterion counts in AA individuals (N = 2998) [6], only when the interactive effect of childhood environmental risk factors were considered: change in residence for rs10188036 in TRAK2 and household drinking and illicit drug use for del-13:61274071 in LINC00378 (Table 3). In addition, a cluster analysis was performed to divide the sample in five CUD groups. For the subtypes 4 (N = 3258) and 5 (N = 1916; the highest heritable and heavy-cocaine-use clusters) they identified 11 additional GWS loci for which the effect on CUD was moderated by environmental factors (Table 3), highlighting the importance of considering environmental interactions in the statistical models [6]. In addition, this study supports the idea that CUD subjects can be decomposed into different subgroups, as previously mentioned, and the study of more homogeneous subgroups may result in more powerful genetic analyses [5]. The second one identified two GWS hits associated with time from cocaine use to dependence onset, rs61835088 identified in the meta-analysis of EA–AA individuals, and located in the gene FAM78B, and rs2825295 in AA individuals [47]. This approach highlights the importance of investigating addiction-related phenotypes to uncover genetic risk factors involved in the development of dependence.
Today, the most important limitation for the association studies on cocaine addiction is the sample size. Hypothesis-free association studies, in the form of GWAS, have taken over from the old CGAS, allowing identification of new, unexpected associations that would have never been explored in hypothesis-driven studies. Nonetheless, a GWAS involves testing millions of genetic variants, which imposes a highly astringent statistical price. This, together with the fact that psychiatric disorders are highly polygenic, with each variant contributing individually to a small amount of the risk (odds ratios typically < 1.3), makes it necessary to gather tens of thousands of patients and controls to achieve appropriate statistical power [48]. Compared to other substances misuse, the sample sizes of GWASs of CUD are substantially smaller. For example, GWAS on problematic alcohol use (about 435 K participants) [49], opioid use disorder (~80 K) [50], cannabis lifetime use (~180 K) [51], and several stages of tobacco and alcohol use (~1.2 M individuals) [52], have been performed so far. When we look at other psychiatric conditions, we find that the most recent GWAS on attention-deficit/hyperactivity disorder (ADHD, 20 K cases) and autism spectrum disorders (ASD, 19 K cases) yielded only 12 and 5 independent GWS hits, respectively [53, 54]. Currently available GWAS for cocaine phenotypes are all below 11 K samples, and therefore larger sample sizes are needed for robust findings.
Due to the difficulty in recruiting patients that are dependent only on cocaine, some authors have studied the general genetic liability of several illicit drugs of abuse (e.g., cocaine, cannabis, and opioids) by combining individuals addicted to any of them [55,56,57,58,59]. These studies identified several SNPs that show a significant association with the phenotype, with subsequent replication in independent samples. However, the sample size of these studies is still limited and further studies are needed.
Another important and controversial consideration in association studies for SUD is the selection of the control sample. Some studies use control individuals that have been exposed to the drug at least once in their life [39], hence excluding the genetic risk factors related to impulsivity or risk-taking behavior, which are very important in this disorder, as they facilitate the first contact with the drug. Indeed, genetic correlation analyses have recently shown the existence of shared genetic factors between cocaine addiction and risk-taking behavior [44]. For that reason, other studies used screened controls that do not meet the DSM criteria for addiction or unselected controls from the general population [44, 57, 60]. The last approach could eventually dilute positive findings due to the presence of some cases among the controls; however, based on the prevalence of cocaine dependence in the general population (about 1.1%), the occurrence of false-negative results due to this effect is likely to be neglectable.
As previously mentioned, the prevalence of CUD varies among ethnicities. Two of the three GWAS performed on CUD [6, 39] included both AA and EA individuals, and their results, as well as those from other SUD [57], suggest that some genetic risk variants can be associated with the phenotype only in a particular population. Although this could be due to limited sample size, further studies in larger samples and considering different ethnicities are warranted.
While GWAS have begun to identify genes that underlie several traits relevant to drug abuse, they still explain only a small fraction of the heritability of this disorder. One of the most important limitations of these studies is the intrinsic difficulty in obtaining large and homogeneous human samples. This has fostered the emergence of a new project (www.ratgenes.org) that will perform GWAS on different behavioral traits relevant to drug abuse using thousands of male and female outbred rats (heterogeneous stock rats). A similar approach has already been taken on obesity‐relevant traits [61]. In addition, expression data of several brain regions relevant to the addiction process will be analyzed to identify expression QTLs (eQTLs). This approach can help to identify new genes that influence drug abuse-related behaviors in rats, providing potential candidates for the genetic susceptibility to drug addiction in humans.
Rare variants
Only two studies have explored the contribution of rare (Minor Allele Frequency, MAF<1%) and low-frequency variants (1%<MAF<5%) to cocaine addiction. The first one, with focus on the family of acetylcholine receptors, found increased DSM-IV cocaine dependence symptoms among carriers of rare missense variants in CHRNB3, coding for the β3 nicotinic acetylcholine receptor [62]. The other one, focusing on the gene encoding the μ-opioid receptor (OPRM1), revealed an association between rs62638690 and cocaine addiction [63]. Further studies with larger samples sizes that use methodological approaches other than GWAS are needed to dissect the role of rare genetic risk variants on cocaine dependence.
Shared genetics of CUD with other psychiatric conditions and traits
Recognition of co-occurring psychiatric disorders among people with SUD has been growing in recent years. Several studies have shown that SUD is highly comorbid with other psychiatric disorders such as SCZ, major depressive disorder (MDD), or ADHD [64,65,66,67], and traits like aggressive, antisocial, or risk-taking behaviors [68, 69]. For example, about 81% of SUD patients have at least one comorbid mental disorder: 33% MDD, 11% SCZ, and 9% personality disorders [64]. Conversely, the occurrence of lifetime SUD in patients with SCZ is 70–80%, 39.2% for ADHD, and 16.1% for MDD [65,66,67]. The pattern is similar with cocaine: 73.4% of cocaine abuse/dependence patients have comorbid mental disorders (49.7% personality disorders, 23.4% MDD, and 20.5% anxiety), and 94.9% have other SUD [70]. Such comorbidities determine a worse lifetime trajectory in patients, lower rates of treatment success, and a higher prevalence of suicide. Several studies have started to investigate whether shared genetics is behind these comorbid patterns using different statistical tools, such as the estimation of genetic correlation between phenotypes, polygenic risk score (PRS) analysis to quantify the fraction of the genetics of a given phenotype that predicts a second condition, and Mendelian Randomization or Latent Causal Variable model to infer causal relationships. These analyses support the genetic overlap between different SUD and other mental disorders, including SCZ, ADHD, MDD, bipolar disorder (BD), eating disorders, or insomnia, among others [71,72,73,74,75,76,77,78,79,80]. However, studies focusing particularly on cocaine are still scarce due to the limited availability of properly sized GWAS.
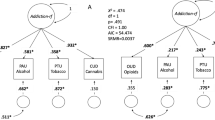
A recent GWAS metanalysis on cocaine dependence [44] explored genetic correlations between this phenotype and six previously described comorbid conditions and found significant results for SCZ (rg = 0.2 ± 0.05), ADHD (0.5 ± 0.08), MDD (0.4 ± 0.08), and risk taking (0.35 ± 0.06). Testing 690 traits from LDHub [44] yielded 109 significant findings, including negative correlations with cognitive phenotypes (e.g., college completion) or reproductive traits (e.g., age at first child) and positive correlations with several psychological or psychiatric phenotypes like neuroticism, depressive symptoms, or loneliness. Also, PRS for SCZ, ADHD, antisocial behavior, MDD, risk-taking and children’s aggressive behavior were found associated with cocaine dependence (pseudo-R2 = 2.28%, 1.39%, 1.33%, 1.21%, 0.60%, and 0.30%, respectively) [44]. Several other PRS studies with focus on SUD included also analyses of a cocaine sub-sample: PRS for SCZ (but not for BIP) associated with CUD in a study of Icelandic subjects (R2 = 0.62%) [75], and PRS for SCZ associated with stimulant use disorders in subjects from the Family Study of Cocaine Dependence (FSCD) (pseudo-R2 = 1.7–3.5%) [73]. Finally, PRS for five psychiatric disorders (SCZ, ASD, ADHD, MDD, and BD) were tested in different cocaine phenotypes from the FSCD, the Collaborative Study of the Genetics of Nicotine Dependence (COGEND), and the Collaborative Study of the Genetics of Alcohol Dependence (COGA) samples. Most associations found were driven by general substance liability, but some substance-specific associations were also identified between MDD, BD, and SCZ PRSs and severe cocaine dependence [71].
Pleiotropy has also been reported at the level of single genes: For example, a missense polymorphic variant (rs16969968:G>A, p.Asp398Asn) at CHRNA5, encoding the α5 nicotinic acetylcholine receptor, leads to hypofunction of the protein and is likely the most robust and replicated association with risk for nicotine addiction [81]. This SNP has also been found associated with cocaine addiction in several studies [35,36,37], but the direction of the effect is opposite, with allele A being a risk factor for nicotine addiction but protective for cocaine addiction [35]. The molecular basis of this distinction is not clear but seems to be related with the localization of this receptor in the mesolimbic dopamine system (both in excitatory dopaminergic and in inhibitory GABAergic neurons) and the mechanism of action of nicotine and cocaine [35]. Interestingly, antagonists of the receptor decrease the reinforcing effects of cocaine, whereas Chrna5 knock-out mice have an increased intake of nicotine [82, 83].
To summarize, we know that CUD is highly comorbid with other psychiatric disorders and cognitive or personality traits, but the role of shared genetics on these co-occurrences has been poorly studied. Thus, we do not know whether risk alleles are acting independently on CUD and on its comorbid phenotypes (i.e., horizontal pleiotropy, also known as biological pleiotropy) or whether one phenotype is causally related to the other one, so that the variants associated with one condition are indirectly associated with the second one (i.e., vertical pleiotropy, also known as mediated pleiotropy). The latter may be exemplified by genetic risk factors contributing to ADHD, which in turn would lead to the onset of CUD under the hypothesis that subjects with ADHD use cocaine to reduce symptomatology (“self-medication” model of comorbidity) [84]. Using analytical tools that investigate causal relationships is needed to clarify this issue.
Another relevant issue is whether the different SUD share genetic risk factors or if substance-specific factors are predominant. In this regard, the fact that heritability estimates for substance-related phenotypes significantly differ depending on the drug of abuse supports the view that at least certain risk factors may be substance-specific [85]. However, there is evidence from twin studies that a large proportion of genetic liability is shared across substances [29, 86], and molecular studies show that increased cross-disorder polygenic risk (e.g. from psychopathological conditions) would be associated with greater general substance involvement [71].
Changes in gene expression induced by cocaine
Cocaine use induces changes in the structure and function of the brain, such as neuronal connectivity and synaptic plasticity [87,88,89,90]. Some of these changes may become stable, contributing to addiction and relapse in cocaine use. Epigenetic and gene expression changes underlie these neuroadaptations induced by the drug and help to explain functional alterations [91, 92].
Transcriptomic studies
Several studies have assessed gene expression alterations in postmortem brain samples from cocaine abusers or in neuronal cells in vitro, the vast majority using microarrays (Table 4). It should be mentioned that these studies are based on few individuals due to the inherent difficulty in obtaining these samples. Across the different studies, some functions are recurrently identified among the genes that are differentially expressed, such as transcription regulation [93,94,95,96,97,98,99] and signal transduction [93,94,95,96, 99,100,101], but certain functions and pathways have been identified only in some particular studies.
Alteration of gene expression in the prefrontal cortex of cocaine abusers was assessed in two studies, using postmortem samples of dorsolateral and anterior prefrontal cortex (dlPFC and aPFC, respectively). The first study focused on expression alterations in aPFC shared in cocaine, cannabis, and phencyclidine abuse, highlighting genes related to calmodulin signaling, Golgi and endoplasmic reticulum [100]. In dlPFC, expression changes identified in cocaine abusers involved genes implicated in mitochondrial and oligodendrocyte function, among others [93]. Interestingly, in the NAc of cocaine abusers, myelin-related genes and genes involved in glial function were also found altered, consistent with a loss of MBP-positive oligodendrocytes [94], and alterations in the expression of PLP1, encoding a major constituent of myelin, were identified [93,94,95].
Furthermore, expression changes in neurotransmission-related genes were identified in dlPFC and NAc [93,94,95], in particular genes involved in synaptic function or cell adhesion [94, 95]. In hippocampus of cocaine abusers, two different studies detected alterations in the expression of genes involved in the regulation of the extracellular matrix [96, 97], and also in cell adhesion, neurogenesis and axon guidance [96], or mitochondrial function, oxidative phosphorylation and long-term potentiation [97]. Another study using postmortem brain samples assessed the expression in midbrain of dopamine cell-enriched regions identifying alterations in expression related to dopamine metabolic process and neuronal differentiation [98] (Table 4). Remarkably, the first study also assessed the substance-specific and shared gene expression changes between cocaine and alcohol in hippocampus, observing that cocaine induced many more transcriptomic alterations than alcohol, but also identifying common changes in the same direction for both drugs that were related to neuroadaptations [97].
The effects of cocaine on human gene expression have also been evaluated in human cells in vitro. Human neuronal progenitor cells showed alterations in the expression of genes mainly involved in immune and inflammatory processes and cell death when exposed to cocaine [101], in line with several studies mentioned above [94,95,96, 98]. In a dopaminergic neuron-like model, changes in gene expression involved chromatin modification [99], also occurring in dopamine cell-enriched regions and hippocampus [97, 98]. Other functions were also identified such as cell cycle, adhesion, cell projection and neuroadaptations [99], and epigenetic regulation by several miRNAs could explain, at least partially, the expression changes induced by cocaine [102] (Table 4).
Gene expression changes induced by cocaine have been widely investigated in animal models, adding valuable information to human studies and contributing to the understanding of the molecular changes involved in the development of CUD. Convergent gene expression changes between animal and human studies have highlighted pathways and biological processes that may be relevant for CUD, including the ERK/MAPK signaling pathway, long-term potentiation, synaptic plasticity (synucleins), and mitochondrial function [103]. Other studies have used the data generated by the above-mentioned studies in humans to assess convergence of gene expression mechanisms with rodents, highlighting genes involved in dopamine and serotonin function such as SLC1A2, CALM3, ALDOA, ALDOC, and ENO2 [104] and in brain plasticity like APP, GRIN2A, GRIN2B, KCNA2, MAP4, PCDH10, PPP3CA, SNCB, and SV2C [105].
It should be mentioned that several of the studies previously performed in humans did not apply multiple testing corrections, a relevant statistical filter when thousands of transcripts are interrogated. Nevertheless, all of them validated expression differences of selected genes by quantitative real-time PCR (Table 4). It is important to note that biological samples from cocaine-addict individuals are difficult to obtain and involve a wide range of variables that cannot be controlled and add heterogeneity, such as dose and frequency of use, time of last exposure, use of other drugs or cause of death. In contrast, in vitro experiments evaluating cocaine effect on cell lines allow to control for variables such as cocaine concentration and time of exposure, but they cannot mimic the effect of chronic cocaine use in the brain of an addicted individual and the effect of the crosstalk with other cell types and remodeling of circuits. By the use of novel techniques, such as single-cell RNA-sequencing, postmortem brain regions could be used to dissect which cell types show more relevant expression changes in each area and, then, study in vitro the effect of cocaine in a controlled environment using specific iPSCs-derived models. Moreover, the information that can be obtained from a few human samples is limited, making it difficult to fully understand the changes in gene expression and in the biological processes that occur in the brain of cocaine users. Larger biorepositories are needed that gather postmortem brain samples for CUD (and other drugs of abuse) with properly phenotyped individuals, similar to other initiatives, such as PsychEncode or GTEx (http://resource.psychencode.org/ and https://gtexportal.org, respectively). Due to the intrinsic difficulties in obtaining expression data from the brain of patients, several tools have been developed to use transcriptomic imputation to integrate genotype and expression data from large consortia, like GTEx, through machine learning. These TWAS approaches allow identification of regulatory variants associated with a given disorder, such as CUD.
Epigenetics
As mentioned above, cocaine addiction is a maladaptive neural plasticity process that occurs in response to repeated drug exposure in vulnerable individuals, depending on the genetic and environmental risk factors, and their interaction. Epigenetics is a vehicle through which environment, including the effects of drugs of abuse, interacts with an individual’s genome to reversibly regulate gene expression independently of the DNA sequence, and determine aspects of function, in health and disease, including addiction. Several studies have demonstrated cocaine-induced changes in epigenetic mechanisms like histone modifications, DNA methylation, and microRNAs, all of them recently reviewed [92, 106,107,108,109,110,111]. However, only a few studies have been conducted in human samples. Regarding histone posttranslational modifications, we found only one study that inspected genome-wide changes in H3K4Me3 in postmortem hippocampus of individuals with chronic exposure to cocaine, revealing changes in promoters of 1115 genes, although only five of them overcame multiple testing corrections [97]. On the other hand, a recent study examined DNA methylation profiles in the peripheral blood of CUD patients and found 186 differentially methylated positions, proposing these regions as potential biomarkers [112]. Finally, two studies investigated changes in the expression of microRNAs induced by cocaine in human cultured cells [102, 113] and two others in peripheral blood and in postmortem brains of cocaine abusers [114, 115]. Interestingly, all of them found alterations in the expression of miR-124, a miRNA that has also been related to cocaine effects in rodents [116, 117]. Further studies are needed to explore the epigenetic mechanisms that underlie cocaine addiction and identify potential biomarkers and therapeutic targets.
Convergence of expression and genetic studies
Genes which expression is altered by cocaine, and that possibly mediate its effects and neuroadaptations induced in the brain, could hold genetic risk variants that contribute to the susceptibility to cocaine addiction. These risk variants may have an impact on the expression or function of these genes prior to the use of the drug and/or confer a differential response to cocaine that could be relevant for the establishment of changes in neuronal circuits, necessary for the development of addiction. Under this hypothesis, some recent studies have pinpointed genes that show altered expression and bear genetic risk variants.
Expression of NFAT5 was found increased in dopaminergic neuron-like cells upon acute exposure to cocaine and, additionally, five SNPs in this gene were associated with cocaine dependence [99]. NFAT5 (TonEBP) is a transcription factor, and previous evidence suggests that cocaine-induced activation of gene expression may be mediated, in part, by NFAT-dependent transcription [118].
Three miRNAs, miR-9, miR-153, and miR-124, were downregulated by cocaine in the above-mentioned dopaminergic model, possibly regulating expression changes observed in the previous study. Interestingly, these miRNAs were found associated with cocaine dependence in a gene-based association study [102].
PLCB1, Phospholipase C beta 1 protein, carry genetic variants associated with both cocaine dependence [119] and drug dependence [119, 120]. Increased expression of PLCB1 is found in both the NAc of human cocaine abusers and in cultured dopaminergic-like human neurons treated with cocaine [119]. Increased expression has been also found in the NAc of mice self-administering cocaine and during withdrawal [121], and a recent study in mice suggests that this gene may play an important role in relapse to cocaine consumption [122].
KCTD20, a regulator of AKT signaling, was identified in a recent study that combined genomic and transcriptomic data to detect candidates for cocaine addiction. This gene was one of the three GWS findings in a GWAS of cocaine dependence [39], and altered expression was found in hippocampus of cocaine abusers, being a key node in a gene network associated with human cocaine use [46]. This gene is a member of the KCTD family, which is involved in a wide range of processes, including proteasome function, GABA signaling, and regulation of transcription responses.
These studies seem to support the hypothesis that genes mediating cocaine’s effect can also participate in the vulnerability to addiction. But the number of works is still limited and further studies are needed to investigate the convergence between expression and genetic studies. Research in the field would benefit from large GWAS and transcriptomic studies and the use of integrative analyses (e.g., TWAS) to get more insight on the genetic basis of this disorder.
Treatment and pharmacogenetics
Currently, there is no government-approved medication available to treat CUD. The medications tested in clinical trials target multiple systems altered by cocaine, including (1) dopamine agonists, as releasers or uptake inhibitors; (2) antagonists/blockers, as antipsychotics, cocaine vaccine, GABA modulators, noradrenergic agents (e.g., disulfiram, doxazosin); (3) other approaches (e.g., cholinesterase inhibitor and N‐methyl‐D‐aspartate receptor antagonist); and (4) combination of medications (reviewed by [123, 124]). However, the efficacy of these treatments requires further exploration [125].
Pharmacogenetic approaches investigate the genetic factors responsible for the inter-individual medication response variability. The final aim of pharmacogenetics is to identify the most effective “personal therapy” based on the genetic individual background, minimizing medical side effects, ensuring compliance, and adjusting dosage [86, 126]. This core element of personalized medicine is a reality in the oncology field. Patient’s specific cancer driver genes can be used as biomarkers to select the best-suited therapy in terms of response to treatment and prognosis [127]. The identification of these correlations has been possible thanks to huge consortiums, such as the Pan-Cancer Atlas, studying thousands of samples in depth [128].
The influence of genetic variants in treatment response in SUD is still at an early stage. An illustrative example is the study of the interaction genotype-treatment in smokers [129]. In African Americans, smokers with the GG genotype at the missense SNP variant rs16969968 in CHRNA5 responded better to a combination nicotine replacement therapy, whereas for A carriers the efficacy of varenicline was higher [130]. Although these findings require additional replications, selection of medication based on genotype could lead to higher rates of smoking cessation.
Early CUD pharmacogenetic studies focused on mouse models (reviewed by [131]). Now, some double-blind randomized placebo-control trials in humans have been performed to analyze cocaine drug treatment response based on the genotypes of specific functional variants on ten genes (ADRA1A, ADRA1D, ANKK1, DRD2, DBH, MTHFR, OPRK1, THP2, SLC6A4, and SLC6A3) related with cocaine physiological mechanisms [132,133,134,135,136,137,138,139,140,141,142,143]. The medications assessed include doxazosin, an α1A adrenergic antagonist that decreases noradrenergic activity; levodopa, a dopamine precursor; cocaine vaccine, succinylnorcocaine covalently linked to cholera B [137]; and disulfiram, a cooper chelator that inhibits noradrenergic synthesis. The results of these studies are summarized in Table 5. Extensive reviews of cocaine medication pharmacogenetics have been published [123, 144,145,146]. However, the knowledge in the field is still in its early stages.
Interestingly, the SNP rs161115 (-1021C>T) in the dopamine β-hydroxylase gene (DBH), responsible for up to 50% of plasma activity variation of the enzyme, has been analyzed in multiple studies. Different clinical responses for each allele (C/T) were observed when distinct treatments were used [134,135,136,137,138], suggesting that the individual’s genotype could help to make treatment choices. For instance, individuals with the CC genotype, associated with normal DBH levels, presented significant cocaine-positive urine reduction rates on disulfiram treatment in one study [136], although the results were not supported by another study [135]. Contrarily, T-allele carriers (CT and TT genotypes), associated with lower levels of circulating DBH, seem to respond better to doxazosin, levodopa, or cocaine vaccine medications [134, 137, 138].
The responses to treatments considering multiple polymorphic variants in different genes at the same time are of particular interest. These interactions were tested in two studies [139, 142]. The analysis of genotype combinations at ANKK1 rs1800497 and DRD2 rs2283265, in low linkage disequilibrium (R2 = 0.57), showed that carrying at least one minor allele in one of these SNPs is associated with better response to disulfiram [139]. In the other study, carriers of the S′ allele at the serotonin transporter (SLC6A4) 5-HTTLPR polymorphic variant and the A allele at the rs4290270 SNP of tryptophan hydroxylase (THP2), both corresponding to the low activity variants, presented significant reduction in cocaine urine levels (71–53%) on disulfiram treatment [142].
Pharmacogenetic studies published until now present some limitations. (1) The samples sizes are very limited, particularly when compared to GWAS. This is due both to the cost of the clinical trials, and to the limited adherence/abandonment of participants. (2) Multiple testing corrections are not systematically applied as each study focus on one or two functional variants. (3) Only for DBH more than one article has studied the gene/variant. Therefore, replication of the other results in independent samples is needed to confirm the participation of these polymorphisms in differential drug response. (4) The populations studied are ethnically diverse but the groups are not similarly represented. However, population stratification has been assessed and taken into consideration in statistical analyses. Larger samples representing the multiple ethnic groups are warranted to extract generalizable conclusions. (5) Incomplete knowledge on the effect of gene variants, and on the physiopathology of cocaine use dependence. (6) The drug may not be selective for a specific target, making the individual genetic variant approach insufficient. In addition, in some studies, multiple drugs were given (e.g., disulfiram and methadone). (7) Finally, individuals participating in the study present comorbidities with additional SUD (alcohol, opioids, etc.), although comorbid psychiatric disorders have been specifically excluded. Larger and more comprehensive studies are needed to be able to move toward personalized medicine in cocaine use disorder therapy.
Concluding remarks
In the last decades, GWAS have been extensively used to study complex disorders such as cocaine dependence; however, common variants identified so far by this approach only explain a fraction of its genetic liability, probably around 30% [44, 46]. To identify the molecular components that underlie the “missing heritability” of cocaine dependence we need to (1) significantly increase the sample size of GWAS, (2) consider other types of genetic variation that cannot be properly addressed in those studies, including structural variants (e.g., CNVs) and rare variants that have been poorly investigated so far, (3) explore epistasis, i.e., interaction among genes, (4) investigate epigenetic mechanisms, as well as gene-environment interactions, (5) combine GWAS results with functional data such as expression data, 3D chromatin interaction and/or regulatory histone marks to identify most likely causal variants, (6) improve the classification of cases in different subgroups (e.g., using cluster analysis rather than DSM-5 criteria) to have more homogeneous groups and increase the statistical power of genetic studies and (7) use new reference imputation panels for GWAS, such as the Haplotype Reference Consortium [147] or TOPMed [148], that improve the quality of the imputation of common genetic variants and provide a better coverage of rare variants. On the other hand, further transcriptomic studies are warranted to improve the understanding of cocaine effects on the brain and the molecular basis of the neuroadaptations that underlie compulsive drug seeking, even after long periods of abstinence. Interestingly, several studies support the idea that genes mediating cocaine’s effect can participate in the predisposition to addiction (e.g., NFAT5, PLCB1, KCTD20), highlighting potential therapeutic targets. However, we need more studies that investigate the convergence between altered expression and genetic variation in CUD. We have learned from twin studies and from molecular genetics research that there is a common genetic architecture across different SUDs, but they also reveal some genetic risk factors that are substance-specific. Understanding the weight and nature of each of these components is an important challenge in addiction research. For some drugs of abuse with considerable larger samples sizes, GWAS have started to reveal genetic risk variants in few genes that have been consistently associated and replicated, for instance SNPs in the ADH1B gene for alcohol dependence and CHRNA5 gene for nicotine dependence (recently reviewed [149]). Importantly, a SNP in CHRNA5 has been associated with differential success rate of treatments in smoking cessation [130]. Despite significant advances in the study of the molecular underpinnings of CUD during the last decade, we are still far from individual genetic prediction to aid prevention, diagnostics or to anticipate disease course and therapeutic response. However, there are promising venues in cocaine research, especially those related to GWAS data: On one hand, we know that PRSs have already been able to identify individuals with risk equivalent to monogenic mutations in several somatic conditions [150, 151] and this is currently being explored also in psychiatric conditions. On the other hand, the relevance of the supporting genetic evidence in the selection of candidates for drug development has been demonstrated across human diseases, increasing by twofold the drug success rate in clinical trials compared to non-genetically selected targets [152, 153]. However, advancing toward more comprehensive pharmacogenetic studies will only be possible once our knowledge on the genetic bases of CUD is wider and more precise.
References
Hasin DS, O’Brien CP, Auriacombe M, Borges G, Bucholz K, Budney A, et al. DSM-5 criteria for substance use disorders: recommendations and rationale. Am J Psychiatry. 2013;170:834–51.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed., Text Revision). American Psychiatric Press Inc.: Washington, DC, 2000.
Agrawal A, Heath AC, Lynskey MT. DSM-IV to DSM-5: the impact of proposed revisions on diagnosis of alcohol use disorders. Addiction. 2011;106:1935–43.
Weiss RD, Mirin SM. Subtypes of cocaine abusers. Psychiatr Clin N Am. 1986;9:491–501.
Kranzler HR, Wilcox M, Weiss RD, Brady K, Hesselbrock V, Rounsaville B, et al. The validity of cocaine dependence subtypes. Addict Behav. 2008; 33:41-53.
Sun J, Kranzler HR, Gelernter J, Bi J. A genome-wide association study of cocaine use disorder accounting for phenotypic heterogeneity and gene–environment interaction. J Psychiatry Neurosci. 2020;45:34–44.
United Nations publication, Sales No. E.20.XI.6. World Drug Report 2020. Vienna, Austria, 2020.
EMCDDA. European Drug Report Trends and Developments. Lisbon, Portugal, 2019.
Wagner FA, Anthony JC. From first drug use to drug dependence: developmental periods of risk for dependence upon marijuana, cocaine, and alcohol. Neuropsychopharmacology. 2002;26:479–88.
Reboussin BA, Anthony JC. Is there epidemiological evidence to support the idea that a cocaine dependence syndrome emerges soon after onset of cocaine use? Neuropsychopharmacology. 2006;31:2055–64.
Compton WM, Thomas YF, Stinson FS, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV drug abuse and dependence in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2007;64:566–76.
Cano M. Racial/ethnic differences in US drug overdose mortality, 2017–8. Addict Behav. 2021;112:106625.
Becker JB, McClellan ML, Reed BG. Sex differences, gender and addiction. J Neurosci Res. 2017;95:136–47.
Miguel AQC, Jordan A, Kiluk BD, Nich C, Babuscio TA, Mari JJ, et al. Sociodemographic and clinical outcome differences among individuals seeking treatment for cocaine use disorders. The intersection of gender and race. Subst Abus Treat. 2019;106:65–72.
Hall FS, Sora I, Drgonova J, Li XF, Goeb M, Uhl GR. Molecular mechanisms underlying the rewarding effects of cocaine. Ann N Y Acad Sci. 2004;1025:47–56.
Cunningham KA, Anastasio NC. Serotonin at the nexus of impulsivity and cue reactivity in cocaine addiction. Neuropharmacology. 2014;76:460–78.
Bender BN, Torregrossa MM. Molecular and circuit mechanisms regulating cocaine memory. Cell Mol Life Sci. 2020;77:3745–68.
Kreek MJ, Levran O, Reed B, Schlussman SD, Zhou Y, Butelman ER. Opiate addiction and cocaine addiction: Underlying molecular neurobiology and genetics. J Clin Investig. 2012;122:3387–91.
Goldman D, Oroszi G, Ducci F. The genetics of addictions: uncovering the genes. Nat Rev Genet. 2005;6:521–32.
Merikangas KR, Stolar M, Stevens DE, Goulet J, Preisig MA, Fenton B, et al. Familial transmission of substance use disorders. Arch Gen Psychiatry. 1998;55:973–9.
Walters RK, Polimanti R, Johnson EC, McClintick JN, Adams MJ, Adkins AE, et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat Neurosci. 2018;21:1656–69.
Van Den Bree MBM, Johnson EO, Neale MC, Pickens RW. Genetic and environmental influences on drug use and abuse/dependence in male and female twins. Drug Alcohol Depend. 1998;52:231–41.
Kendler KS, Aggen SH, Tambs K, Reichborn-Kjennerud T. Illicit psychoactive substance use, abuse and dependence in a population-based sample of Norwegian twins. Psychol Med. 2006;36:955–62.
Kendler KS, Prescott CA. Cocaine use, abuse and dependence in a population-based sample of female twins. Br J Psychiatry. 1998;173:345–50.
Beseler C, Jacobson KC, Kremen WS, Lyons MJ, Glatt SJ, Faraone SV, et al. Is there heterogeneity among syndromes of substance use disorder for illicit drugs? Addict Behav. 2006;31:929–47.
Kalayasiri R, Kranzler HR, Weiss R, Brady K, Gueorguieva R, Panhuysen C, et al. Risk factors for cocaine-induced paranoia in cocaine-dependent sibling pairs. Drug Alcohol Depend. 2006;84:77–84.
Kendler KS, Karkowski LM, Neale MC, Prescott CA. Illicit psychoactive substance use, heavy use, abuse, and dependence in a US population-based sample of male twins. Arch Gen Psychiatry. 2000;57:261–9.
Kendler KS, Ohlsson H, Maes HH, Sundquist K, Lichtenstein P, Sundquist J. A population-based Swedish Twin and Sibling Study of cannabis, stimulant and sedative abuse in men. Drug Alcohol Depend. 2015;149:49–54.
Kendler KS, Jacobson KC, Prescott CA, Neale MC. Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. Am J Psychiatry. 2003;160:687–95.
Kendler KS, Myers J, Prescott CA. Specificity of genetic and environmental risk factors for symptoms of cannabis, cocaine, alcohol, caffeine, and nicotine dependence. Arch Gen Psychiatry. 2007;64:1313–20.
Hall FS, Drgonova J, Jain S, Uhl GR. Implications of genome wide association studies for addiction: Are our a priori assumptions all wrong? Pharmacol Ther. 2013;140:267–79.
Ballon N, Leroy S, Roy C, Bourdel MC, Charles-Nicolas A, Krebs MO. et al. (AAT)n repeat in the cannabinoid receptor gene (CNR1): association with cocaine addiction in an African-Caribbean population. Pharmacogenomics J. 2006;6:126–30.
Clarke TK, Bloch PJ, Ambrose-Lanci LM, Ferraro TN, Berrettini WH, Kampman KM, et al. Further evidence for association of polymorphisms in the CNR1 gene with cocaine addiction: confirmation in an independent sample and meta-analysis. Addict Biol. 2013;18:702–8.
Zuo L, Kranzler HR, Luo X, Yang BZ, Weiss R, Brady K, et al. Interaction between two independent CNR1 variants increases risk for cocaine dependence in european americans: a replication study in family-based sample and population-based sample. Neuropsychopharmacology. 2009;34:1504–13.
Grucza RA, Wang JC, Stitzel JA, Hinrichs AL, Saccone SF, Saccone NL, et al. A risk allele for nicotine dependence in CHRNA5 Is a protective allele for cocaine dependence. Biol Psychiatry. 2008;64:922–9.
Sherva R, Kranzler HR, Yu Y, Logue MW, Poling J, Arias AJ, et al. Variation in nicotinic acetylcholine receptor genes is associated with multiple substance dependence phenotypes. Neuropsychopharmacology. 2010;35:1921–31.
Aroche AP, Rovaris DL, Grevet EH, Stolf AR, Sanvicente-Vieira B, Kessler FHP, et al. Association of CHRNA5 gene variants with crack cocaine addiction. NeuroMolecular Med. 2020;22:384–90.
Cabana-Domínguez J, Roncero C, Grau-López L, Rodríguez-Cintas L, Barral C, Abad AC, et al. A highly polymorphic copy number variant in the NSF gene is associated with cocaine dependence. Sci Rep. 2016;6:31033.
Gelernter J, Sherva R, Koesterer R, Almasy L, Zhao H, Kranzler HR, et al. Genome-wide association study of cocaine dependence and related traits: FAM53B identified as a risk gene. Mol Psychiatry. 2014;19:717–23.
Zhou W, He Y, Rehman AU, Kong Y, Hong S, Ding G, et al. Loss of function of NCOR1 and NCOR2 impairs memory through a novel GABAergic hypothalamus–CA3 projection. Nat Neurosci. 2019;22:205–17.
Zhao B, Luo T, Li T, Li Y, Zhang J, Shan Y, et al. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat Genet. 2019;51:1637–44.
Pineda-Cirera L, Cabana-Domínguez J, Roncero C, Cozar M, Grau-López L, Abad AC, et al. Evaluation of previous substance dependence genome-wide significant findings in a Spanish sample. Drug Alcohol Depend. 2018;187:358–62.
Marees AT, Gamazon ER, Gerring Z, Vorspan F, Fingal J, van den Brink W, et al. Post-GWAS analysis of six substance use traits improves the identification and functional interpretation of genetic risk loci. Drug Alcohol Depend. 2020;206:107703.
Cabana-Domínguez J, Shivalikanjli A, Fernàndez-Castillo N, Cormand B. Genome-wide association meta-analysis of cocaine dependence: Shared genetics with comorbid conditions. Prog Neuro-Psychopharmacol Biol Psychiatry. 2019;94:109667.
Avramopoulos D. Neuregulin 3 and its roles in schizophrenia risk and presentation. Am J Med Genet Part B Neuropsychiatr Genet. 2018;177:257–66.
Huggett SB, Stallings MC. Cocaine’omics: genome-wide and transcriptome-wide analyses provide biological insight into cocaine use and dependence. Addict Biol. 2020;25:e12719.
Sherva R, Zhu C, Wetherill L, Edenberg HJ, Johnson E, Degenhardt L, et al. Genome-wide association study of phenotypes measuring progression from first cocaine or opioid use to dependence reveals novel risk genes. Explor Med. 2021;2:60–73.
Hao K, Chudin E, McElwee J, Schadt EE. Accuracy of genome-wide imputation of untyped markers and impacts on statistical power for association studies. BMC Genet. 2009;10:27.
Zhou H, Sealock JM, Sanchez-Roige S, Clarke TK, Levey DF, Cheng Z, et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci. 2020;23:809-818.
Zhou H, Rentsch CT, Cheng Z, Kember RL, Nunez YZ, Sherva RM, et al. Association of OPRM1 functional coding variant with opioid use disorder: a Genome-Wide Association Study. JAMA Psychiatry. 2020;77:1072-1080.
Pasman JA, Verweij KJH, Gerring Z, Stringer S, Sanchez-Roige S, Treur JL, et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat Neurosci. 2018;21:1161-1170.
Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–244.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44.
Alblooshi H, Al Safar H, Fisher HF, Cordell HJ, El Kashef A, Al Ghaferi H, et al. A case–control genome wide association study of substance use disorder (SUD) identifies novel variants on chromosome 7p14.1 in patients from the United Arab Emirates (UAE). Am J Med Genet Part B Neuropsychiatr Genet. 2019;180:68-79.
McGue M, Zhang Y, Miller MB, Basu S, Vrieze S, Hicks B, et al. A genome-wide association study of behavioral disinhibition. Behav Genet. 2013;43:363-73.
Johnson EO, Hancock DB, Levy JL, Gaddis NC, Page GP, Glasheen C, et al. KAT2B polymorphism identified for drug abuse in African Americans with regulatory links to drug abuse pathways in human prefrontal cortex. Addict Biol. 2016;21:1217–32.
Wetherill L, Agrawal A, Kapoor M, Bertelsen S, Bierut LJ, Brooks A, et al. Association of substance dependence phenotypes in the COGA sample. Addict Biol. 2015;20:617-27.
Wetherill L, Lai D, Johnson EC, Anokhin A, Bauer L, Bucholz KK, et al. Genome-wide association study identifies loci associated with liability to alcohol and drug dependence that is associated with variability in reward-related ventral striatum activity in African- and European-Americans. Genes Brain Behav. 2019;18:e12580.
Ikeda M, Okahisa Y, Aleksic B, Won M, Kondo N, Naruse N, et al. Evidence for shared genetic risk between methamphetamine-induced psychosis and schizophrenia. Neuropsychopharmacology. 2013;38:1864–70.
Chitre AS, Polesskaya O, Holl K, Gao J, Cheng R, Bimschleger H, et al. Genome-wide association study in 3,173 outbred rats identifies multiple loci for body weight, adiposity, and fasting glucose. Obesity. 2020;28:1964-1973.
Haller G, Kapoor M, Budde J, Xuei X, Edenberg H, Nurnberger J, et al. Rare missense variants in CHRNB3 and CHRNA3 are associated with risk of alcohol and cocaine dependence. Hum Mol Genet. 2014;23:810–9.
Clarke TK, Crist RC, Kampman KM, Dackis CA, Pettinati HM, O’Brien CP, et al. Low frequency genetic variants in the μ-opioid receptor (OPRM1) affect risk for addiction to heroin and cocaine. Neurosci Lett. 2013;542:71–75.
Shantna K, Chaudhury S, Verma A, Singh A. Comorbid psychiatric disorders in substance dependence patients: a control study. Ind Psychiatry J. 2009;18:84.
Currie SR, Patten SB, Williams JVA, Wang JL, Beck CA, El-Guebaly N, et al. Comorbidity of major depression with substance use disorders. Can J Psychiatry. 2005;50:660–6.
Piñeiro-Dieguez B, Balanzá-Martínez V, García-García P, Soler-López B, Domingo MA, Labarra JDA, et al. Psychiatric comorbidity at the time of diagnosis in adults with ADHD: the CAT study. J Atten Disord. 2016;20:1066–75.
Westermeyer J. Comorbid schizophrenia and substance abuse: a review of epidemiology and course. Am J Addict. 2006;15:345–55.
Bezinović P, Malatestinić D. Perceived exposure to substance use and risk-taking behavior in early adolescence: cross-sectional study. Croat Med J. 2009;50:157–64.
Hasin D, Kilcoyne B. Comorbidity of psychiatric and substance use disorders in the United States: Current issues and findings from the NESARC. Curr Opin Psychiatry. 2012;25:165–71.
Arias F, Szerman N, Vega P, Mesias B, Basurte I, Morant C, et al. Madrid study on the prevalence and characteristics of outpatients with dual pathology in community mental health and substance misuse services. Adicciones. 2013;25:118–27.
Carey CE, Agrawal A, Bucholz KK, Hartz SM, Lynskey MT, Nelson EC, et al. Associations between polygenic risk for psychiatric disorders and substance involvement. Front Genet. 2016;7:149.
Du Rietz E, Coleman J, Glanville K, Choi SW, O’Reilly PF, Kuntsi J. Association of polygenic risk for attention-deficit/hyperactivity disorder with co-occurring traits and disorders. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:635–43.
Hartz SM, Horton AC, Oehlert M, Carey CE, Agrawal A, Bogdan R, et al. Association between substance use disorder and polygenic liability to schizophrenia. Biol Psychiatry. 2017;82:709–15.
Treur JL, Demontis D, Smith GD, Sallis H, Richardson TG, Wiers RW, et al. Investigating causality between liability to ADHD and substance use, and liability to substance use and ADHD risk, using Mendelian randomization. Addict Biol. 2021;26:e12849.
Reginsson GW, Ingason A, Euesden J, Bjornsdottir G, Olafsson S, Sigurdsson E, et al. Polygenic risk scores for schizophrenia and bipolar disorder associate with addiction. Addict Biol. 2018;23:485–92.
Soler Artigas M, Sánchez-Mora C, Rovira P, Richarte V, Garcia-Martínez I, Pagerols M, et al. Attention-deficit/hyperactivity disorder and lifetime cannabis use: genetic overlap and causality. Mol Psychiatry. 2020;25:2493–503.
Jang SK, Saunders G, Liu M, Jiang Y, Liu DJ, Vrieze S. Genetic correlation, pleiotropy, and causal associations between substance use and psychiatric disorder. Psychol Med. 2020;1–11.
Pasman JA, Smit DJA, Kingma L, Vink JM, Treur JL, Verweij KJH. Causal relationships between substance use and insomnia. Drug Alcohol Depend. 2020;214:108151.
Wimberley T, Agerbo E, Horsdal HT, Ottosen C, Brikell I, Als TD, et al. Genetic liability to ADHD and substance use disorders in individuals with ADHD. Addiction. 2020;115:1368–77.
Munn-Chernoff MA, Johnson EC, Chou YL, Coleman JRI, Thornton LM, Walters RK, et al. Shared genetic risk between eating disorder- and substance-use-related phenotypes: evidence from genome-wide association studies. Addict Biol. 2021;26:e12880.
Gorwood P, Strat YLE, Ramoz N. Genetics of addictive behavior: the example of nicotine dependence. Dialogues Clin Neurosci. 2017;19:237–45.
Hansen ST, Mark GP. The nicotinic acetylcholine receptor antagonist mecamylamine prevents escalation of cocaine self-administration in rats with extended daily access. Psychopharmacology. 2007;194:53–61.
Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular α5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601.
Wilens TE, Adamson J, Sgambati S, Whitley J, Santry A, Monuteaux MC, et al. Do individuals with ADHD self-medicate with cigarettes and substances of abuse? Results from a controlled family study of ADHD. Am J Addict. 2007;16:14–23.
Fulton SL, Maze I. Translational molecular approaches in substance abuse research. Handb Exp Pharmacol. 2020;258:31–60.
Agrawal A, Verweij KJH, Gillespie NA, Heath AC, Lessov-Schlaggar CN, Martin NG, et al. The genetics of addiction - a translational perspective. Transl Psychiatry. 2012;2:e140.
Buchta WC, Riegel AC. Chronic cocaine disrupts mesocortical learning mechanisms. Brain Res. 2015;1628:88–103.
McReynolds JR, Peña DF, Blacktop JM, Mantsch JR. Neurobiological mechanisms underlying relapse to cocaine use: Contributions of CRF and noradrenergic systems and regulation by glucocorticoids. Stress. 2014;17:22–38.
Schmidt HD, Pierce RC. Cocaine-induced neuroadaptations in glutamate transmission: Potential therapeutic targets for craving and addiction. Ann N. Y Acad Sci. 2010;1187:35–75.
Cooper S, Robison AJ, Mazei-Robison MS. Reward circuitry in addiction. Neurotherapeutics. 2017;14:687–97.
Zhou Z, Enoch MA, Goldman D. Gene expression in the addicted brain. Int Rev Neurobiol. 2014;116:251–73.
López AJ, Siciliano CA, Calipari ES. Activity-dependent epigenetic remodeling in cocaine use disorder. Handb Exp Pharmacol. 2020;258:231–63.
Lehrmann E, Oyler J, Vawter MP, Hyde TM, Kolachana B, Kleinman JE, et al. Transcriptional profiling in the human prefrontal cortex: evidence for two activational states associated with cocaine abuse. Pharmacogenomics J. 2003;3:27–40.
Albertson DN, Pruetz B, Schmidt CJ, Kuhn DM, Kapatos G, Bannon MJ. Gene expression profile of the nucleus accumbens of human cocaine abusers: Evidence for dysregulation of myelin. J Neurochem. 2004;88:1211–9.
Bannon MJ, Kapatos G, Albertson DN. Gene expression profiling in the brains of human cocaine abusers. Addict Biol. 2005;10:119–26.
Mash DC, Ffrench-Mullen J, Adi N, Qin Y, Buck A, Pablo J. Gene Expression in Human Hippocampus from Cocaine Abusers Identifies Genes which Regulate Extracellular Matrix Remodeling. PLoS One. 2007;2:e1187.
Zhou Z, Yuan Q, Mash DC, Goldman D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc Natl Acad Sci USA. 2011;108:6626–31.
Bannon MJ, Johnson MM, Michelhaugh SK, Hartley ZJ, Halter SD, David JA, et al. A molecular profile of cocaine abuse includes the differential expression of genes that regulate transcription, chromatin, and dopamine cell phenotype. Neuropsychopharmacology. 2014;39:2191–9.
Fernàndez-Castillo N, Cabana-Domínguez J, Soriano J, Sànchez-Mora C, Roncero C, Grau-López L, et al. Transcriptomic and genetic studies identify NFAT5 as a candidate gene for cocaine dependence. Transl Psychiatry. 2015;5:e667.
Lehrmann E, Colantuoni C, Deep-Soboslay A, Becker KG, Lowe R, Huestis MA, et al. Transcriptional changes common to human cocaine cannabis and phencyclidine abuse. PLoS One. 2006;1:e114.
Crawford FC, Wood ML, Wilson SE, Mathura VS, Hollen TR, Geall F, et al. Cocaine induced inflammatory response in human neuronal progenitor cells. J Neurochem. 2006;97:662–74.
Cabana-Domínguez J, Arenas C, Cormand B, Fernàndez-Castillo N. MiR-9, miR-153 and miR-124 are down-regulated by acute exposure to cocaine in a dopaminergic cell model and may contribute to cocaine dependence. Transl Psychiatry. 2018;8:173.
Lull ME, Freeman WM, Vrana KE, Mash DC. Correlating human and animal studies of cocaine abuse and gene expression. Ann N Y Acad Sci. 2008;1141:58–75.
Huggett SB, Bubier JA, Chesler EJ, Palmer RHC. Do gene expression findings from mouse models of cocaine use recapitulate human cocaine use disorder in reward circuitry? Genes Brain Behav. 2021;20:e12689.
Forero DA, González-Giraldo Y. Convergent functional genomics of cocaine misuse in humans and animal models. Am J Drug Alcohol Abus. 2020;46:22–30.
Vaillancourt K, Ernst C, Mash D, Turecki G. DNA methylation dynamics and cocaine in the brain: Progress and prospects. Genes. 2017;8:138.
Efimova OA, Koltsova AS, Krapivin MI, Tikhonov AV, Pendina AA. Environmental epigenetics and genome flexibility: focus on 5-hydroxymethylcytosine. Int J Mol Sci. 2020;21:3223.
Mahna D, Puri S, Sharma S. DNA methylation signatures: Biomarkers of drug and alcohol abuse. Mutat Res Rev Mutat Res. 2018;777:19–28.
Smith ACW, Kenny PJ. MicroRNAs regulate synaptic plasticity underlying drug addiction. Genes Brain Behav. 2018;17:e12424.
Pierce RC, Fant B, Swinford-Jackson SE, Heller EA, Berrettini WH, Wimmer ME. Environmental, genetic and epigenetic contributions to cocaine addiction. Neuropsychopharmacology. 2018;43:1471–80.
Gowen AM, Odegaard KE, Hernandez J, Chand S, Koul S, Pendyala G, et al. Role of microRNAs in the pathophysiology of addiction. Wiley Interdiscip Rev RNA. 2021;12:e1637
Camilo C, Maschietto M, Vieira HC, Tahira AC, Gouveia GR, Dos Santos ACF, et al. Genome-wide DNA methylation profile in the peripheral blood of cocaine and crack dependents. Braz J Psychiatry. 2019;41:485–93.
Rodrigues AC, Li X, Radecki L, Pan YZ, Winter JC, Huang M, et al. MicroRNA expression is differentially altered by xenobiotic drugs in different human cell lines. Biopharm Drug Dispos. 2011;32:355–67.
Sessa F, Maglietta F, Bertozzi G, Salerno M, Di Mizio G, Messina G, et al. Human brain injury and mirnas: an experimental study. Int J Mol Sci. 2019;20:1546.
Viola TW, Heberle BA, Zaparte A, Sanvicente-Vieira B, Wainer LM, Fries GR, et al. Peripheral blood microRNA levels in females with cocaine use disorder. J Psychiatr Res. 2019;114:48–54.
Chandrasekar V, Dreyer JL. microRNAs miR-124, let-7d and miR-181a regulate Cocaine-induced Plasticity. Mol Cell Neurosci. 2009;42:350–62.
Chandrasekar V, Dreyer JL. Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology 2011;36:1149–64.
Groth RD, Weick JP, Bradley KC, Luoma JI, Aravamudan B, Klug JR, et al. D1 dopamine receptor activation of NFAT-mediated striatal gene expression. Eur J Neurosci. 2008;27:31–42.
Cabana-Domínguez J, Roncero C, Pineda-Cirera L, Palma-Álvarez RF, Ros-Cucurull E, Grau-López L, et al. Association of the PLCB1 gene with drug dependence. Sci Rep. 2017;7:10110.
Drgon T, Johnson CA, Nino M, Drgonova J, Walther DM, Uhl GR. ‘Replicated’ genome wide association for dependence on illegal substances: Genomic regions identified by overlapping clusters of nominally positive SNPs. Am J Med Genet Part B Neuropsychiatr Genet. 2011;156:125–38.
Eipper-Mains JE, Kiraly DD, Duff MO, Horowitz MJ, Mcmanus CJ, Eipper BA, et al. Effects of cocaine and withdrawal on the mouse nucleus accumbens transcriptome. Genes, Brain Behav. 2013;12:21–33.
Cabana-Domínguez J, Martín-García E, Gallego-Roman A, Maldonado R, Fernàndez-Castillo N, Cormand B. Reduced cue-induced reinstatement of cocaine-seeking behavior in Plcb1+/− mice. 2021. Transl Psychiatry, in press. https://www.biorxiv.org/content/10.1101/2020.06.18.158964v3.full.
Shorter D, Domingo CB, Kosten TR. Emerging drugs for the treatment of cocaine use disorder: a review of neurobiological targets and pharmacotherapy. Expert Opin Emerg Drugs. 2015;20:15–29.
Brandt L, Chao T, Comer SD, Levin FR Pharmacotherapeutic strategies for treating cocaine use disorder—what do we have to offer? Addiction. 2021;116:694-710.
Chan B, Kondo K, Freeman M, Ayers C, Montgomery J, Kansagara D. Pharmacotherapy for cocaine use disorder—a systematic review and meta-analysis. J Gen Intern Med. 2019;34:2858–73.
Roden DM, McLeod HL, Relling MV, Williams MS, Mensah GA, Peterson JF, et al. Pharmacogenomics. Lancet. 2019;394:521–32.
Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018;10:25.
Rubio-Perez C, Tamborero D, Schroeder MP, Antolín AA, Deu-Pons J, Perez-Llamas C, et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015;27:382–96.
Schuit E, Panagiotou OA, Munafò MR, Bennett DA, Bergen AW, David SP. Pharmacotherapy for smoking cessation: effects by subgroup defined by genetically informed biomarkers. Cochrane Database Syst Rev. 2017;9:CD011823.
Chen L, Baker TB, Miller JP, Bray M, Smock N, Chen J, et al. Genetic variant in CHRNA5 and response to varenicline and combination nicotine replacement in a randomized placebo‐controlled trial. Clin Pharmacol Ther. 2020;108:1315–25.
Marley RJ, Elmer GI, Goldberg SR. The use of pharmacogenetic techniques in drug abuse research. Pharmacol Ther. 1992;53:217–37.
Shorter D, Nielsen DA, Huang W, Harding MJ, Hamon SC, Kosten TR. Pharmacogenetic randomized trial for cocaine abuse: disulfiram and α1A-adrenoceptor gene variation. Eur Neuropsychopharmacol. 2013;23:1401–7.
Shorter DI, Zhang X, Domingo CB, Nielsen EM, Kosten TR, Nielsen DA. Doxazosin treatment in cocaine use disorder: pharmacogenetic response based on an alpha-1 adrenoreceptor subtype D genetic variant. Am J Drug Alcohol Abus. 2020;46:184–93.
Zhang X, Nielsen DA, Domingo CB, Shorter DI, Nielsen EM, Kosten TR. Pharmacogenetics of Dopamine β-Hydroxylase in cocaine dependence therapy with doxazosin. Addict Biol. 2019;24:531–8.
Schottenfeld RS, Chawarski MC, Cubells JF, George TP, Lappalainen J, Kosten TR. Randomized clinical trial of disulfiram for cocaine dependence or abuse during buprenorphine treatment. Drug Alcohol Depend. 2014;136:36–42.
Kosten TR, Wu G, Huang W, Harding MJ, Hamon SC, Lappalainen J, et al. Pharmacogenetic randomized trial for cocaine abuse: disulfiram and dopamine β-hydroxylase. Biol Psychiatry. 2013;73:219–24.
Kosten TR, Domingo CB, Hamon SC, Nielsen DA. DBH gene as predictor of response in a cocaine vaccine clinical trial. Neurosci Lett. 2013;541:29–33.
Liu S, Green CE, Lane SD, Kosten TR, Moeller FG, Nielsen DA, et al. The influence of dopamine β-hydroxylase gene polymorphism rs1611115 on levodopa/carbidopa treatment for cocaine dependence: a preliminary study. Pharmacogenet Genomics. 2014;24:370–3.
Spellicy CJ, Kosten TR, Hamon SC, Harding MJ, Nielsen D. AANKK1 and DRD2 pharmacogenetics of disulfiram treatment for cocaine abuse. Pharmacogenet Genomics. 2013;23:333–40.
Spellicy CJ, Kosten TR, Hamon SC, Harding MJ, Nielsen DA. The MTHFR C677T variant is associated with responsiveness to disulfiram treatment for cocaine dependency. Front Psychiatry. 2013;3:109.
Nielsen DA, Hamon SC, Kosten TR. The κ-opioid receptor gene as a predictor of response in a cocaine vaccine clinical trial. Psychiatr Genet. 2013;23:225–32.
Nielsen DA, Harding MJ, Hamon SC, Huang W, Kosten TR. Modifying the role of serotonergic 5-HTTLPR and TPH2 variants on disulfiram treatment of cocaine addiction: a preliminary study. Genes Brain Behav. 2012;11:1001–8.
Kampangkaew JP, Spellicy CJ, Nielsen EM, Harding MJ, Ye A, Hamon SC, et al. Pharmacogenetic role of dopamine transporter (SLC6A3) variation on response to disulfiram treatment for cocaine addiction. Am J Addict. 2019;28:311–7.
Bauer IE, Graham DP, Soares JC, Nielsen DA. Serotonergic gene variation in substance use pharmacotherapy: A systematic review. Pharmacogenomics. 2015;16:1307–14.
Nielsen DA, Nielsen EM, Dasari T, Spellicy CJ. Pharmacogenetics of addiction therapy. Methods Mol Biol. 2014;1175:589–624.
Patriquin MA, Bauer IE, Soares JC, Graham DP, Nielsen DA. Addiction pharmacogenetics: a systematic review of the genetic variation of the dopaminergic system. Psychiatr Genet. 2015;25:181–93.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Kowalski MH, Qian H, Hou Z, Rosen JD, Tapia AL, Shan Y, et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15:e1008500.
Lopez-Leon S, González-Giraldo Y, Wegman-Ostrosky T, Forero DA. Molecular genetics of substance use disorders: an umbrella review. Neurosci Biobehav Rev. 2021;124:358–69.
Smeland OB, Frei O, Fan CC, Shadrin A, Dale AM, Andreassen OA. The emerging pattern of shared polygenic architecture of psychiatric disorders, conceptual and methodological challenges. Psychiatr Genet. 2020;29:152–9.
Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–24.
Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–60.
King EA, Wade Davis J, Degner JF. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019;15:e1008489.
Acknowledgements
Major financial support for this research was received by BC from the Spanish ‘Ministerio de Ciencia, Innovación y Universidades’ (RTI2018-100968-B-100), the ‘Ministerio de Sanidad, Servicios Sociales e Igualdad/Plan Nacional Sobre Drogas’ (PNSD-2017I050 and PNSD-2020I042) and ‘Generalitat de Catalunya/AGAUR’ (2017-SGR-738). The research leading to these results has also received funding from the European Union H2020 Program [H2020/2014-2020] under grant agreements no 667302 (CoCA) and Eat2beNICE (728018). JC-D was supported by the H2020 CoCA and Eat2beNICE projects and NF-C by ‘Centro de Investigación Biomédica en Red de Enfermedades Raras’ (CIBERER). RC was recipient of a Ramón y Cajal fellowship (RYC-2017-21636).
Author information
Authors and Affiliations
Contributions
All authors conceived and organized the sections of the review, revised the literature, and contributed to write the “Abstract” and the “Concluding remarks”. BC wrote the sections “Clinical definition”, “Epidemiology” and “Shared genetics of CUD with other psychiatric conditions and traits”. NF-C wrote the sections “Neurobiology”, “Heritability: family and twin studies”, “Transcriptomic studies” and “Convergence of expression and genetic studies”. JC-D wrote the sections “Genetic architecture of cocaine addiction” and “Epigenetics”. RC wrote the section “Treatment and pharmacogenetics”.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernàndez-Castillo, N., Cabana-Domínguez, J., Corominas, R. et al. Molecular genetics of cocaine use disorders in humans. Mol Psychiatry 27, 624–639 (2022). https://doi.org/10.1038/s41380-021-01256-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-021-01256-1
This article is cited by
-
Sex-Specific ADNP/NAP (Davunetide) Regulation of Cocaine-Induced Plasticity
Journal of Molecular Neuroscience (2024)
-
Genotype-by-diagnosis interaction influences self-control in human cocaine addiction
Translational Psychiatry (2023)
-
Schizophrenia in the genetic era: a review from development history, clinical features and genomic research approaches to insights of susceptibility genes
Metabolic Brain Disease (2023)
-
Repeated dosing with cocaine produces strain-dependent effects on responding for conditioned reinforcement in Collaborative Cross mice
Psychopharmacology (2023)
-
Systematic Review: Is There a Medicinal Use of Cocaine in Psychiatry?
Current Treatment Options in Psychiatry (2023)
