
Abstract
Smoking is a major heritable and modifiable risk factor for many diseases, including cancer, common respiratory disorders and cardiovascular diseases. Fourteen genetic loci have previously been associated with smoking behaviour-related traits. We tested up to 235,116 single nucleotide variants (SNVs) on the exome-array for association with smoking initiation, cigarettes per day, pack-years, and smoking cessation in a fixed effects meta-analysis of up to 61 studies (up to 346,813 participants). In a subset of 112,811 participants, a further one million SNVs were also genotyped and tested for association with the four smoking behaviour traits. SNV-trait associations with P < 5 × 10−8 in either analysis were taken forward for replication in up to 275,596 independent participants from UK Biobank. Lastly, a meta-analysis of the discovery and replication studies was performed. Sixteen SNVs were associated with at least one of the smoking behaviour traits (P < 5 × 10−8) in the discovery samples. Ten novel SNVs, including rs12616219 near TMEM182, were followed-up and five of them (rs462779 in REV3L, rs12780116 in CNNM2, rs1190736 in GPR101, rs11539157 in PJA1, and rs12616219 near TMEM182) replicated at a Bonferroni significance threshold (P < 4.5 × 10−3) with consistent direction of effect. A further 35 SNVs were associated with smoking behaviour traits in the discovery plus replication meta-analysis (up to 622,409 participants) including a rare SNV, rs150493199, in CCDC141 and two low-frequency SNVs in CEP350 and HDGFRP2. Functional follow-up implied that decreased expression of REV3L may lower the probability of smoking initiation. The novel loci will facilitate understanding the genetic aetiology of smoking behaviour and may lead to the identification of potential drug targets for smoking prevention and/or cessation.
Similar content being viewed by others
Introduction
Smoking is a major risk factor for many diseases, including common respiratory disorders such as chronic obstructive pulmonary disease (COPD) [1, 2], cancer [3] and cardiovascular diseases [4], and is reported to cause 1 in 10 premature deaths worldwide [5]. A greater understanding of the genetic aetiology of smoking behaviour has the potential to lead to new therapeutic interventions to aid smoking prevention and cessation, and thereby reduce the global burden of such diseases.
Previous genome-wide association studies (GWASs) identified 14 common SNVs [1, 6,7,8,9,10,11,12] (with minor allele frequency, MAF >0.01) robustly associated with smoking behaviour-related traits (P < 5 × 10−8). The 15q25 (CHRNA3/5-CHRNB4) region has the largest effect, explaining ~1% and 4–5% of the phenotypic variance of smoking quantity [13] and cotinine, a biomarker of nicotine intake [14], respectively. Overall, genetic loci identified to date explain ~2% of the estimated genetic heritability of smoking behaviour [6], which is reported to be between 40–60% [15,16,17]. A recent study suggested that an important proportion (~3.3%) of the phenotypic variance of smoking behaviour-related traits was explained by rare nonsynonymous variants (MAF <0.01) [18]. Hence, well-powered studies of rare variants are needed.
To investigate the effect of rare coding variants on smoking behaviour, we studied 346,813 participants (of which 324,851 were of European ancestry) from 61 cohorts (Supp. Tables 1 and 2) at up to 235,116 SNVs from the exome array. As we had access to UK Biobank, we also interrogated SNVs present on the UK Biobank and UK BiLEVE Axiom arrays to identify additional associations across the genome beyond the exome array. To our knowledge, these datasets are an order of magnitude larger than the previous studies [6], and constitute the most powerful exome-array study of smoking behaviour to date.
Materials and methods
Participants
Our study combined study-level summary association data from up to 59 studies of European ancestry and two studies of South Asian ancestry from three consortia (Consortium for Genetics of Smoking Behaviour (CGSB), GWAS & Sequencing Consortium of Alcohol and Nicotine use (GSCAN) and the Coronary Heart Disease (CHD) Exome+ consortium), INTERVAL and UK Biobank. In total, up to 324,851 individuals of European ancestry and 21,962 South Asian individuals were analysed in the discovery stage (Fig. 1). Further information about the participating cohorts and consortia is given in Supp. Table 1 and the Supp. Material. All participants provided written informed consent and studies were approved by local Research Ethics Committees and/or Institutional Review boards.

Study design including the discovery and replication stages. NB: Gene-based studies, conditional analyses, and replication in African American ancestry samples not shown here for clarity. *GFG and NAGOZALC studies contributed additional custom content
Phenotypes
We chose to analyse the following four smoking behaviour-related traits because of their broad availability in existing epidemiological and medical studies, as well as their biological relevance for addiction behaviours:
-
i.
Smoking initiation (binary trait: ever vs never smokers). Ever smokers were defined as individuals who have smoked >99 cigarettes in their lifetime, which is consistent with the definition by the Centre for Disease Control [19];
-
ii.
Cigarettes per day (CPD; quantitative trait: average number of cigarettes smoked per day by ever smokers);
-
iii.
Pack-years (quantitative trait; Packs per day x Years smoked, with a pack defined as 20 cigarettes); years smoked is typically formed from age at smoking commencement to current age for current smokers or age at cessation for former smokers.
-
iv.
Smoking cessation (binary trait: former vs current smokers).
In UK Biobank, phenotypes were defined using phenotype codes 1239, 1249, and 2644 for smoking initiation and smoking cessation, and 1239, 3436, 3456 for CPD and pack-years. CPD was inverse normal transformed in the CHD Exome+, INTERVAL and CGSB studies and categorised (1–10, 11–20, 21–30, and 31+ CPD) by the GSCAN studies and UK Biobank (Supp. Table 2). All studies performed an inverse normal transformation of pack-years. Summary statistics of study level phenotype distributions are provided in Supp. Table 1.
Genotyping and quality control
Fifty-nine cohorts were genotyped using exome arrays (up to 235,116 SNVs) and two (UK Biobank and INTERVAL) were genotyped using Axiom Biobank Arrays (up to 820,000 SNVs; Supp. Table 2). In total, ~1.06M SNVs were analysed including ~64,000 SNVs on both the Axiom and Exome Arrays. Furthermore, two studies (NAGOZALC and GFG) genotyped their participants using arrays with custom content, increasing the total number of variants analysed to 1,207,583 SNVs. Individual studies performed quality control (QC; Supp. Material, Supp. Table 2) and additional QC was conducted centrally (i) to ensure alleles were consistently aligned, (ii) that there were no major sample overlaps between contributing studies, and (iii) variants conformed to Hardy–Weinberg equilibrium and call rate thresholds. We also examined the distribution of the effect sizes and test statistics across cohorts to ensure the test statistics were well-calibrated.
Study level analyses
Each study (including the case-cohort studies [20]) undertook analyses of up to four smoking traits using RAREMETALWORKER [21] or RVTESTS [22] (Supp. Table 2), which generated single variant score statistics and their covariance matrices within sliding windows of 1Mb. CPD and pack-years were analysed using linear models or linear mixed models. Smoking initiation and smoking cessation were analysed using logistic models or linear mixed models. All studies adjusted each trait for age, sex, at least three genetic principal components and any study-specific covariates (Supp. Table 2). Chromosome X variants were analysed using the above-described approach, but coding males as 0/2. This coding scheme ensures that on average females and males have equal dosages and so is optimal for genes that are inactivated (due to X chromosome inactivation) and is valid for genes that do not undergo X chromosome activation. Males and females were analysed together adjusting for sex as a covariate.
Single variant meta-analyses
Fixed effects meta-analyses across the individual contributing studies of single variant associations were undertaken using the Cochran-Mantel-Haenszel method in RAREMETAL. Z-score statistics were used in the meta-analysis to ensure that the association results are robust against potentially different units of measurement in the phenotype definitions across studies [23]. We performed genomic control correction on the meta-analysis results. Variants with P < 1 × 10−6 in tests of heterogeneity were excluded. Variants with P ≤ 5 × 10−8 were taken forward for replication. In addition, rs12616219 was also taken forward for replication as its P-value was very close to this threshold (smoking initiation, P = 5.49 × 10−8). None of the rare SNVs were genome-wide significant, therefore we also took forward the rare variant with the smallest association P-value, rs141611945 (P = 2.95 × 10−7; MAF < 0.0001).
Replication and combined meta-analysis of discovery and replication data
As UK biobank genetic data were released in two phases, we took the opportunity to replicate findings from the discovery stage in a further 275,596 individuals made available in the phase two release of UK Biobank genetic data. To avoid potential relatedness between discovery and replication samples, the replication samples were screened and individuals with relatedness closer than second degree with the discovery sample in the UK Biobank were removed [24]. Phenotypes were defined in the same way as the discovery samples (described above). Since the exome array and the UK Biobank Axiom arrays do not fully overlap, we used both genotyped exome variants (approx. 64,000) as well as the additional ~90,000 well-imputed exome array variants from UK Biobank (imputation quality score >0.3) for replication of single variant and gene-based tests. The rare ATF6 variant was absent from the UK Biobank array and is more prevalent in Africans (MAF = 0.01) than Europeans (MAF = 0.0007). Therefore, replication was sought in 1,437 individuals of African American-ancestry from the HRS and COGA studies. Analysis methods for replication cohorts were the same as for discovery cohorts, including methods to analyse chromosome X (Supp. Table 2). The criteria set for the replication were (i) the same direction of effect as the discovery analysis and (ii) P ≤ 0.0045 in the replication studies (Bonferroni-adjusted for eleven SNVs at α = 0.05).
Finally, in order to fully utilise all available data, we carried out a combined meta-analysis of the discovery and replication samples across the exome array content using the same protocols mentioned above.
Conditional analyses
To identify conditionally independent variants within previously reported and novel loci a sequential forward stepwise selection was performed [25]. A 1 MB region was defined around the reported or novel sentinel variant (500 kb either side) and conditional analyses performed with all variants within the region. If a conditionally independent variant was identified, (P < 5 × 10−6; Bonferroni-adjusted for ~10,000 independent variants in the test region) the analysis was repeated conditioning on both the most significant conditionally independent variant and the sentinel variant. This stepwise approach was repeated (conditioning on the variants identified in current and earlier iterations) until there were no variants remaining in the region that were conditionally independent. The same protocol was followed for the novel SNVs identified in this study.
Gene-based analyses
For discovery gene-based meta-analyses, we utilised three statistical methods as part of the RAREMETAL package: the Weighted Sum Test (WST) [26], the burden test [27] and the Sequence Kernel Association test (SKAT) [28]. EPACTS (v.3.3.0) [29] was used to annotate variants (for use in gene-based meta-analyses), as recommended by RAREMETAL. Two MAF cut-offs were used, one used low-frequency (MAF < 0.05) and rare variants, the second only used rare variants (MAF < 0.01). Nonsynonymous, stop gain, splice site, start gain, start loss, stop loss, and synonymous variants were selected for inclusion. A sensitivity analysis to exclusion of synonymous variants was also performed. Gene-level associations with P < 8 × 10−7 were deemed statistically significant (Bonferroni-adjusted for ~20,000 genes and three tests at α = 0.05). To examine if the gene associations were driven by a single variant, the gene tests were conducted conditional on the SNV with the smallest P-value in the gene, using the shared single variant association statistic and covariance matrices [21, 25].
Mendelian randomization analyses
To evaluate the causal effect of SI and CPD on BMI, schizophrenia and educational attainment (EA), we conducted Mendelian randomization (MR) analyses using three complementary approaches available in MR-Base [30]: inverse variance weighted regression [31], MR-Egger [32, 33], and weighted median [34]. We used both the previously reported smoking-associated SNVs and the SNVs from the current report (as provided in Tables 1–3 and Supp. Table 3) as instrumental variables. The BMI [35], schizophrenia [36] and educational attainment [37] data came from previously published publicly available data. To assess possible reverse causation, we also used outcome associated SNVs as instrumental variables and conducted MR analyses using SI and CPD as outcome. We considered P < 0.05/3 = 0.017 as statistically significant (Bonferroni-adjusted for three traits).
In silico functional follow up of associated SNVs
To identify whether the (replicated) SNVs identified here affected other traits, we queried the GWAS Catalog [38] (version: e91/28/02/2018, downloaded on 01/03/18) for genome-wide significant (P < 5 × 10−8) associations using all proxy SNVs (r2 ≥ 0.8) within 2 Mb of the top variant in our study.
eQTL lookups were carried out in the 13 brain tissues available in GTEx V7 [39], Brain xQTL (dorsolateral prefrontal cortex) [40] and BRAINEAC [41] databases, all of which had undergone QC by the individual studies. We did not perform additional QC on these data. In brief, GTEx used Storey’s q-value method to correct the FDR for testing multiple transcripts based upon the empirical P-values for the most significant SNV for each transcript [43, 42]. BRAINEAC calculated the number of tests per transcript and used Benjamini–Hochberg procedure to calculate FDR per transcript using a FDR < 1% as significant. BRAINxQTL used P < 8 × 10−8 as a cut-off for significance for any given transcript. SNVs that met the study specific significance and FDR thresholds, which were in LD (r2 > 0.8 in 1000 Genomes Europeans) with the top eQTL or the sentinel eQTL for a given tissue/transcript combination were considered significant. The genes implicated by these eQTL databases and/or coding changes (e.g., missense and nonsense SNVs) were put into ConsensusPathDB [44] to identify whether these genes were over-represented in any known biological pathways. Replicated missense SNVs were also put into PolyPhen-2 [45] and FATHMM (unweighted) [46] to obtain variant effect prediction.
Results
Single variant associations
In the discovery meta-analyses, we identified 15 common SNVs that were genome-wide significant (P < 5 × 10-8) for one or more of the smoking behaviour traits, of which 9 were novel (Table 1, Supp. Table 3). Seven novel loci were identified for smoking initiation, one for both CPD and pack-years and one for smoking cessation (Figs. 1, 2, Table 1 and Supp. Figure 1). Results for the significant loci were consistent across participating cohorts and there was at least nominal evidence of association (P < 0.05) at the novel loci within each of the contributing consortia (Supp. Table 4). Full association results for all novel SNVs across the four traits are provided in Supp. Table 5. No rare variants were genome-wide significant; the rare variant with the smallest P-value was a missense variant in ATF6, rs141611945 (MAF < 0.0001, CPD P = 2.95 × 10−7).

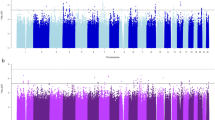
A concentric Circos plot of the association results for smoking initiation (SI; outer ring), cigarettes per day (CPD) and smoking cessation (SC; inner ring) for chromosomes 1–22 (Pack-years results, which can be found in Supp. Figure 1, are omitted for clarity). Each dot represents a SNV, with the X and Y axes corresponding to genomic location in Mb and -log10P-values, respectively. Labels show the nearest gene to the novel sentinel variants identified in the discovery stage and taken forward to replication. The top signals were truncated at 10−10 for clarity. Novel and previously reported signals are highlighted in red and dark blue, respectively. Grey rings on the y-axis increase by increments of 2 (initial ring corresponding to P = 0.001, then 0.00001 etc.); and the outer and inner red rings correspond to the genome-wide significance level (P = 5 × 10−8) and P = 5 × 10−7, respectively. Image was created using Circos (v0.65)
Eleven SNVs (including rs12616219 near TMEM182 with P = 5.49 × 10−8, and the rare variant, rs141611945) were taken forward for replication in independent samples (Table 1). The latest release of European UK Biobank individuals not included in the discovery stage (smoking initiation, n = 275,596; smoking cessation n = 123,851; CPD n = 80,015; pack-years n = 78,897), was used for replication of the common variants (Fig. 1). Five of the common variants replicated (four for smoking initiation and one with CPD and pack-years) at P < 0.0045. Two coding variants (rs11539157, rs1190736) were predicted to be ‘probably damaging’ by PolyPhen-2 and FATHMM. The remaining five SNVs were at least nominally associated (P < 0.01) in the replication samples and had consistent direction of effect across discovery and replication. Replication for the rare variant rs141611945 could not be carried out in UK Biobank as the SNV nor its proxies (r2 > 0.3) were available. Thus we initiated replication in African American samples of the COGA (n = 476) and HRS (n = 961) cohorts (overall MAF≈0.01). The direction of effect was consistent in the two replication cohorts and consistent with the discovery meta-analysis but a meta-analysis of the two replication cohorts yielded a P = 0.28. Further data are required to replicate this association.
We also performed a meta-analysis combining the discovery and replication samples (up to 622,409 individuals). LD score regression showed that the λ (intercept) for all traits was ~1.00, which indicated that confounding factors inflating the results was not an issue [47, 48]. The combined analysis identified 35 additional novel SNV-smoking trait associations, 33 with smoking initiation, one with CPD and one with smoking cessation at P < 5 ×10-8 (Table 2). We note that among our four SNVs that did not replicate, rs216195 (in SMG6) was genome-wide significant in the combined meta-analysis of discovery and replication studies (P = 2.41 × 10−9; Table 2).
We also calculated the phenotypic variance explained for novel and known variants. Results can be found in the ‘Calculation of Phenotypic Variance Explained’ section in the Supplementary Material.
Associations at known smoking behaviour loci
We assessed evidence for associations at the 14 SNVs previously reported for smoking behaviour-related traits. Seven were genotyped on the exome array and proxies (r2 > 0.3; ±2 Mb) were identified for the remaining seven (Supp. Table 3). All showed nominal evidence of association at P < 0.05 and six of these were genome-wide significant in the meta-analysis of the trait for which it was previously reported (Supp. Tables 3 and 5).
Conditional analyses identified five independent associations within three previously reported loci and all five replicated (Table 3). At the 19q13 (RAB4B) locus, there were three variants in or near CYP2A6 associated with CPD independently of the established variant (rs7937) and each other: rs8102683 (conditional P = 4.53 × 10−16), rs28399442 (conditional P = 2.63 × 10−12) and rs3865453 (conditional P = 4.96 × 10−10) and rs28399442 was a low-frequency variant. The same SNVs also showed evidence of independent effects with pack-years, albeit with larger P-values (P < 5 × 10−6; Supp. Table 5). At the TEX41/PABPC1P2 locus, rs11694518 (conditional P = 3.43 × 10−7) was associated with smoking initiation independently of the established variant (rs10427255). At 15q25, rs938682 (P = 7.78 × 10−21) was associated with CPD independently of the established variant (rs1051730) and (in agreement with a previous report [49]) is an eQTL for CHRNA5 in brain putamen basal ganglia tissues in GTEx.
Gene-based association studies
Gene-based collapsing tests using MAF < 0.01 variants, did not identify any associated genes at the pre-specified P < 8 × 10−7 threshold. Of the top four gene associations, three were novel (CHRNA2, MMP17, and CRCP) and one was known (CHRNA5), and had P < 7 × 10−4, with CPD and/or pack-years (Supp. Table 6). Analyses conditional on the variant with the smallest P-value in the gene, revealed the associations at CHRNA2, MMP17 and CRCP were due to more than one rare variant (conditional P < 0.05; Supp. Table 6). In contrast, the CHRNA5 gene association was attributable to a single variant (rs2229961).
Mendelian randomization analyses
We conducted MR analyses to elucidate the potential causal impact of SI and CPD on BMI, schizophrenia and EA using the MR-Egger, median weighted and inverse variance weighted methods. We found a causal association between SI and EA using both the median weighted and inverse variance weighted methods (P < 0.0001; Supp. Table 7) but not with MR-Egger (P = 0.2). There was an association of SI with BMI using MR-Egger only (P = 0.01; Supp. Table 7), but there was evidence of horizontal pleiotropy (P = 0.001) and no support from the other methods. Similarly, increased CPD was only associated with reduced BMI using the weighted median approach (P = 0.009) and not the other methods (P > 0.017). We also tested if schizophrenia, EA or BMI causally influence CPD or SI using SNVs associated with schizophrenia, EA and BMI, respectively, as instrumental variables. No evidence of such reverse causation was found (Supp. Table 7). These results were consistent with previous analyses [50]. There was no evidence of a causal effect of SI on schizophrenia, or CPD on educational attainment (Supp. Table 7).
Functional characterization of novel loci
Using proxies with r2≥0.8 in 1000 Genomes Europeans, we queried the GWAS catalogue [38] (P ≤ 5 × 10−8) for pleiotropic effects of our novel sentinel SNVs. Two, rs11539157 and rs3001723 were previously associated with schizophrenia [36], suggesting shared biological pathways between schizophrenia and smoking behaviours (Table 2). This fits with the known association of smoking with schizophrenia [51]. Two, rs1514175 and rs2947411 have previously been associated with BMI [52], and extreme obesity [53].
eQTL lookups in GTEx V7 (13 Brain tissues with ≥80 samples) [39], Brain xQTL [40] and BRAINEAC [41] databases revealed that the A allele at rs462779, which decreases risk of smoking initiation, also decreased expression of REV3L in cerebellum in GTEx (A allele P = 4.8x10-8; β = −0.40) and was in strong LD with the top eQTL for REV3L in cerebellum (r2 = 0.86 with rs9487668 in 1000 Genomes Europeans). The smoking initiation-associated SNV, rs12780116, was an eQTL for BORCS7 in four brain tissues, and NT5C2 in the cerebellar hemisphere (A allele P = 4.5 × 10−7; β = −0.32) and the cerebellum (P = 5.6 × 10−6; β = −0.415; in strong LD with the top eQTL, r2 = 0.97 with rs11191546). The G allele of a second variant in the region, rs7096169 (intronic to BORCS7 and only in weak LD with rs12780116, r2 = 0.18 in 1000G Europeans) increases smoking initiation and reduces expression of BORCS7 and AS3MT in eight brain tissues (including dorsolateral prefrontal cortex in the Brain xQTL and was the top BORCS7 eSNP in GTEx in the Cerebellar Hemisphere, Cerebellum, and Spinal cord cervical-C1). The same variant also reduced expression of ARL3 in cerebellum in GTEx (Table 2).
Biological pathway enrichment analyses carried out in ConsensusPathDB [44] using the genes implicated by the eQTL databases (Table 2) and/or a coding SNVs (i.e., PJA1, GPR101) showed that the (i) pyrimidine metabolism and (ii) activation of nicotinic acetylcholine receptors pathways are enriched for these smoking behaviour associated genes (false discovery rate <0.01; P < 0.0001).
Discussion
Smoking is the most important preventable lifestyle risk factor for many diseases, including cancers [3, 54], heart disease [4, 55] and many respiratory diseases such as COPD [1, 2]. Not initiating is the best way to prevent smoking-related diseases and genetics can play a considerable part in smoking behaviours including initiation. We have performed the largest exome-wide genetic association study of smoking behaviour-related traits to date involving up to 622,409 individuals, and identified and replicated five associations, including two on the X-chromosome (Table 1). We identified a further 35 novel associations in a meta-analysis of discovery and replication cohorts (Table 2). We validated 14 previously reported SNV-smoking trait associations (Supp. Table 3) and identified secondary independent associations at three loci, including three in the 19q13 region (rs8102683, rs28399442, and rs3865453; Table 3).
Gene-based tests improve power by aggregating effects of rare variants. While no genes reached our Bonferroni-adjusted P-value threshold, we identified three candidate genes with multiple rare variant associations for future replication: calcitonin gene-related peptide-receptor component (CRCP) with CPD and CHRNA2 and MMP17 with pack-years (Supp. Table 6; also see ‘Genes of Interest’ section in Supp. Material). CRCP’s protein product is expressed in brain tissues amongst others and functions as part of a receptor complex for a neuropeptide that increases intracellular cyclic adenosine monophosphate levels [56]. MMP17 encodes a matrix metalloproteinase that is also expressed in the brain and is a member of the peptidase M10 family, and proteins in this family are involved in the breakdown of extracellular matrix in normal physiological processes [57]. Given, we were not able conclusively to identify rare variant associations, even larger studies, are required to identify rare variants associated with smoking behaviours. In addition, phenotypes such as cotinine levels [58] and nicotine metabolism speed [59] could be interrogated using methods such as MTAG [60] to improve power.
As recommended by UK Biobank, we analysed UK Biobank samples by adjusting for genotyping array because a subset of (extreme smokers in) UK Biobank were genotyped on a different array (UK BiLEVE). However, this adjustment could potentially introduce collider bias in analyses of smoking traits. Given that the UK BiLEVE study is relatively small compared to the full study, and the genetic effect sizes for smoking-associated variants are small, we expect the influence of collider bias to be small [61]. Nevertheless, we performed sensitivity analyses to assess the impact of collider bias. Firstly, we performed a meta-analysis excluding the UK BiLEVE samples, and secondly, we re-analysed UK Biobank without adjusting for genotype array. As expected, the estimated genetic effects from these additional analyses were very similar to our reported results suggesting collider bias is not a concern (Suppl. Table 8).
Follow-up of the replicated SNVs in the literature and eQTL databases implicated some potentially interesting genes: NT5C2 is known to hydrolyse purine nucleotides and be involved in maintaining cellular nucleotide balance, and was previously associated with schizophrenia [62]. REV3L, encodes the catalytic subunit of DNA polymerase ζ (zeta) which is involved in translesion DNA synthesis. Previously, polymorphisms in a microRNA target site of REV3L were shown to be associated with lung cancer susceptibility [63]. We showed that decreased expression of REV3L may also lower the probability of smoking initiation. The SNV, rs11776293, intronic in EPHX2, was associated with reduced SI in the combined meta-analysis, and is in LD with rs56372821 (r2 = 0.83), which is associated with reduced cannabis use disorder [64]. rs216195 (in SMG6) was genome-wide significant in the discovery and the combined meta-analysis. SMG6 is a plausible candidate gene as it was previously shown to be less methylated in current smokers compared to never smokers [65]. The combined meta-analysis also identified a rare missense variant in CCDC141, rs150493199 (MAF < 0.01; Table 2). Coding variants in CCDC141 were previously associated with heart rate [66] and blood pressure [67, 68].
Smoking behaviours represent a complex phenotype that are linked to an array of socio-cultural and familial, as well as genetic determinants. Kong et al., recently reported that ‘genetic-nurture’ i.e., effects of non-transmitted parental alleles, affect educational attainment [69]. They also show that there is an effect of educational attainment and genetic nurture on smoking behaviour. Four of our sentinel SNVs (or a strong proxy; r2 > 0.8) were associated with years of educational attainment [37] (rs2292239, rs3001723 (P < 5 × 10−8), rs9320995 (P = 8.90 x 10−7), and rs13022438 (P = 3.79 × 10−6), in agreement with this paradigm and our MR analyses indicated that initiating smoking reduced years in education. Future family studies will be required to disentangle how much of the variance explained in the current analysis is due to direct versus genetic nurturing effects.
Our study primarily focused on European ancestry, but we also included two non-European studies but these non-European studies lacked statistical power on their own to identify ancestry-specific effects. Therefore, we did not perform ancestry-specific meta-analyses. Nevertheless, our results offered cross ancestry replication. One of the associations identified in the conditional analyses, rs8102683 (near CYP2A6), confirmed an association with CPD that was previously identified by Kumasaka et al. in a Japanese population [70] but this is the first time it was associated in Europeans (rs8102683 is also correlated with rs56113850 (r2 = 0.43), a SNV identified previously by Loukola et al. [59] in a genetic association study of nicotine metabolite ratio in Europeans). As more non-European studies become available, it would be of great interest to perform non-European ancestry studies, in order to fine-map causal variants for smoking-related traits.
CPD and pack-years are two correlated measures of smoking. In the ~40,000 individuals from UK Biobank with CPD and pack-years calculated, correlation between CPD and pack-years was 0.640. Interestingly, while pack-years was inversely correlated with smoking cessation (−0.18) i.e., the more years a smoker has been smoking the less likely they were to cease, CPD was positively correlated with smoking cessation (0.13) i.e., heavier smokers were more likely to stop smoking. In contrast, the DBH SNV, rs3025343, (first identified via its association with increased smoking cessation [6]) was associated with increased pack-years (P = 1.29 × 10−14) and increased CPD (P = 2.93 × 10−9) in our study. The association at DBH also represents the first time that a SNV has a smaller P-value for pack-years (n = 131,892) compared to CPD (n = 128,746). These findings may help elucidate the genetic basis of these correlated addiction phenotypes.
We performed the largest exome-wide genetic association study of smoking behaviour-related traits to date and nearly doubled the number of replicated associations to 24 (including conditional analyses) including associations on the X-chromosome for the first time, which merit further study. We also identified a further 35 novel smoking trait associated SNVs in the combined meta-analysis. The novel loci identified in this study will substantially expand our knowledge of the smoking addiction-related traits, facilitate understanding the genetic aetiology of smoking behaviour and may lead to the identification of drug targets of potential relevance to prevent individuals from initiating smoking and/or aid smokers to stop smoking.
References
Wain LV, Shrine N, Miller S, Jackson VE, Ntalla I, Soler Artigas M, et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med. 2015;3:769–81.
Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet. 2017;49:416–25.
McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49:1126–32.
O'Donnell CJ, Nabel EG. Genomics of Cardiovascular Disease. New Engl J Med. 2011;365:2098–109.
Reitsma MB, Fullman N, Ng M, Salama JS, Abajobir A, Abate KH, et al. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990-2015: a systematic analysis from the Global Burden of Disease Study 2015. The Lancet 2017;389:1885–906.
Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42:441–7.
Hancock DB, Reginsson GW, Gaddis NC, Chen X, Saccone NL, Lutz SM, et al. Genome-wide meta-analysis reveals common splice site acceptor variant in CHRNA4 associated with nicotine dependence. Transl Psychiatry. 2015;5:e651.
Siedlinski M, Cho MH, Bakke P, Gulsvik A, Lomas DA, Anderson W, et al. Genome-wide association study of smoking behaviours in patients with COPD. Thorax. 2011;66:894–902.
Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42:448–53.
Timofeeva MN, McKay JD, Smith GD, Johansson M, Byrnes GB, Chabrier A, et al. Genetic polymorphisms in 15q25 and 19q13 loci, cotinine levels, and risk of lung cancer in EPIC. Cancer Epidemiol Biomark Prev. 2011;20:2250–61.
Bloom AJ, Baker TB, Chen L-S, Breslau N, Hatsukami D, Bierut LJ, et al. Variants in two adjacent genes, EGLN2 and CYP2A6, influence smoking behavior related to disease risk via different mechanisms. Hum Mol Genet. 2014;23:555–61.
Thakur GA, Sengupta SM, Grizenko N, Choudhry Z, Joober R. Family-based association study of ADHD and genes increasing the risk for smoking behaviours. Arch Dis Child. 2012;97:1027.
Munafò MR, Flint J. The genetic architecture of psychophysiological phenotypes. Psychophysiology. 2014;51:1331–2.
Keskitalo K, Broms U, Heliovaara M, Ripatti S, Surakka I, Perola M, et al. Association of serum cotinine level with a cluster of three nicotinic acetylcholine receptor genes (CHRNA3/CHRNA5/CHRNB4) on chromosome 15. Hum Mol Genet. 2009;18:4007–12.
Vink JM, Willemsen G, Boomsma DI. Heritability of smoking initiation and nicotine dependence. Behav Genet. 2005;35:397–406.
Carmelli D, Swan GE, Robinette D, Fabsitz R. Genetic Influence on Smoking — A Study of Male Twins. New Engl J Med. 1992;327:829–33.
Kaprio J, Koskenvuo M, Sarna S. Cigarette smoking, use of alcohol, and leisure-time physical activity among same-sexed adult male twins. Prog Clin Biol Res. 1981;69(Pt C):37–46.
Liu DJ, Brazel DM, Turcot V, Zhan X, Gong J, Barnes DR, et al. Exome chip meta-analysis elucidates the genetic architecture of rare coding variants in smoking and drinking behavior. bioRxiv 2017.
Centers for Disease Control and Prevention (CDC). Cigarette smoking among adults--United States, 2007. MMWR Morb Mortal Wkly Rep. 2008;57:1221–6.
Staley JR, Jones E, Kaptoge S, Butterworth AS, Sweeting MJ, Wood AM, et al. A comparison of Cox and logistic regression for use in genome-wide association studies of cohort and case-cohort design. Eur J Human Genet : EJHG. 2017;25:854–62.
Feng S, Liu D, Zhan X, Wing MK, Abecasis GR. RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics. 2014;30:2828–9.
Zhan X, Hu Y, Li B, Abecasis GR, Liu DJ. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics. 2016;32:1423–6.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9
Jiang B, Chen S, Jiang Y, Liu M, Iacono WG, Hewitt JK, et al. Proper Conditional Analysis in the Presence of Missing Data Identified Novel Independently Associated Low Frequency Variants in Nicotine Dependence Genes. bioRxiv 2017.
Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384.
Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34:188–93.
Wu MC. Rare variant association testing for sequencing data using the sequence kernel association test (SKAT). Am J Hum Genet. 2011;89:82–93.
Zhan X, Liu DJ. SEQMINER: An R-Package to Facilitate the Functional Interpretation of Sequence-Based Associations. Genet Epidemiol. 2015;39:619–23.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408.
Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol. 2013;178:1177–84.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25.
Rees JMB, Wood AM, Burgess S. Extending the MR-Egger method for multivariable Mendelian randomization to correct for both measured and unmeasured pleiotropy. Stat Med. 2017;36:4705–18.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421.
Okbay A, Beauchamp JP, Fontana MA, Lee JJ, Pers TH, Rietveld CA, et al. Genome-wide association study identifies 74 loci associated with educational attainment. Nature. 2016;533:539–42.
MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(D1):D896–901.
Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13.
Ng B, White CC, Klein H-U, Sieberts SK, McCabe C, Patrick E, et al. An xQTL map integrates the genetic architecture of the human brain's transcriptome and epigenome. Nat Neurosci 2017; 20:1418-26; advance online publication.
Trabzuni D, Ryten M, Walker R, Smith C, Imran S, Ramasamy A, et al. Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J Neurochem. 2011;119:275–82.
Ongen H, Buil A, Brown AA, Dermitzakis ET, Delaneau O. Fast and efficient QTL mapper for thousands of molecular phenotypes. Bioinformatics. 2016;32:1479–85.
Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–5.
Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB—a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37(suppl_1):D623–8.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Meth. 2010;7:248–9.
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65.
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017;33:272–9.
Wang JC, Cruchaga C, Saccone NL, Bertelsen S, Liu P, Budde JP, et al. Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum Mol Genet. 2009;18:3125–35.
Gage SH, Jones HJ, Taylor AE, Burgess S, Zammit S, Munafo MR. Investigating causality in associations between smoking initiation and schizophrenia using Mendelian randomization. Sci Rep. 2017;7:40653.
Kelly C, McCreadie R. Cigarette smoking and schizophrenia. Adv Psychiatr Treat. 2000;6:327–31.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48.
Wheeler E, Huang N, Bochukova EG, Keogh JM, Lindsay S, Garg S, et al. Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet. 2013;45:513–7.
Hecht SS. Tobacco Smoke Carcinogens and Lung Cancer. JNCI: J Natl Cancer Inst. 1999;91:1194–210.
Ockene IS, Miller NH. Cigarette Smoking, Cardiovascular Disease, and Stroke. A Statement Healthc Prof Am Heart Assoc. 1997;96:3243–7.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419.
O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–45.
Ware JJ, Chen X, Vink J, Loukola A, Minica C, Pool R, et al. Genome-Wide Meta-Analysis of Cotinine Levels in Cigarette Smokers Identifies Locus at 4q13.2. Sci Rep. 2016;6:20092.
Loukola A, Buchwald J, Gupta R, Palviainen T, Hallfors J, Tikkanen E, et al. A Genome-Wide Association Study of a Biomarker of Nicotine Metabolism. PLoS Genet. 2015;11:e1005498.
Turley P, Walters RK, Maghzian O, Okbay A, Lee JJ, Fontana MA, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet. 2018;50:229–37.
Munafo MR, Tilling K, Taylor AE, Evans DM, Davey Smith G. Collider scope: when selection bias can substantially influence observed associations. Int J Epidemiol. 2018;47:226–35.
Aberg KA, Liu Y, Bukszár J, et al. A comprehensive family-based replication study of schizophrenia genes. JAMA Psychiatry. 2013;70:573–81.
Zhang S, Chen H, Zhao X, Cao J, Tong J, Lu J, et al. REV3L 3'UTR 460 T>C polymorphism in microRNA target sites contributes to lung cancer susceptibility. Oncogene. 2013;32:242–50.
Demontis D, Rajagopal VM, Als TD, Grove J, Pallesen J, Hjorthoj C, et al. Genome-wide association study implicates CHRNA2 in cannabis use disorder. bioRxiv 2018.
Steenaard RV, Ligthart S, Stolk L, Peters MJ, van Meurs JB, Uitterlinden AG, et al. Tobacco smoking is associated with methylation of genes related to coronary artery disease. Clin Epigenetics. 2015;7:54.
van den Berg ME, Warren HR, Cabrera CP, Verweij N, Mifsud B, Haessler J, et al. Discovery of novel heart rate-associated loci using the Exome Chip. Hum Mol Genet. 2017;26:2346–63.
Warren HR, Evangelou E, Cabrera CP, Gao H, Ren M, Mifsud B, et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat Genet. 2017;49:403–15.
Hoffmann TJ, Ehret GB, Nandakumar P, Ranatunga D, Schaefer C, Kwok PY, et al. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat Genet. 2017;49:54–64.
Kong A, Thorleifsson G, Frigge ML, Vilhjalmsson BJ, Young AI, Thorgeirsson TE, et al. The nature of nurture: effects of parental genotypes. Science. 2018;359:424–8.
Kumasaka N, Aoki M, Okada Y, Takahashi A, Ozaki K, Mushiroda T, et al. Haplotypes with copy number and single nucleotide polymorphisms in CYP2A6 locus are associated with smoking quantity in a Japanese population. PLoS ONE 2012;7:e44507.
Acknowledgments
Author acknowledgements are included in the Supplementary material.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Conflict of interest
Paul W. Franks has been a paid consultant for Eli Lilly and Sanofi Aventis and has received research support from several pharmaceutical companies as part of European Union Innovative Medicines Initiative (IMI) projects. Neil Poulter has received financial support from several pharmaceutical companies that manufacture either blood pressure lowering or lipid lowering agents or both and consultancy fees. Peter Sever has received research awards from Pfizer. Mark J. Caulfield is Chief Scientist for Genomics England, a UK government company. Adam Butterworth reports grants from Merck and Pfizer and grants outside of this work from AstraZeneca, Biogen, and Novartis, and personal fees from Novartis. John Danesh reports grants, personal fees and non-financial support from Merck Sharp & Dohme (MSD), grants, personal fees and non-financial support from Novartis, grants from British Heart Foundation, grants from European Research Council, grants from NIHR, grants from NHS Blood and Transplant, grants from Pfizer, grants from UK MRC, grants from Wellcome Trust, grants from AstraZeneca, outside the submitted work. Laura J. Bierut is listed as an inventor on Issued U.S. Patent 8,080,371,“Markers for Addiction” covering the use of certain SNPs in determining the diagnosis, prognosis, and treatment of addiction.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Erzurumluoglu, A.M., Liu, M., Jackson, V.E. et al. Meta-analysis of up to 622,409 individuals identifies 40 novel smoking behaviour associated genetic loci. Mol Psychiatry 25, 2392–2409 (2020). https://doi.org/10.1038/s41380-018-0313-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-018-0313-0
This article is cited by
-
Deep sequencing of candidate genes identified 14 variants associated with smoking abstinence in an ethnically diverse sample
Scientific Reports (2024)
-
Polygenic profiles define aspects of clinical heterogeneity in attention deficit hyperactivity disorder
Nature Genetics (2024)
-
Genetic control of DNA methylation is largely shared across European and East Asian populations
Nature Communications (2024)
-
Multi-ancestry meta-analysis of tobacco use disorder identifies 461 potential risk genes and reveals associations with multiple health outcomes
Nature Human Behaviour (2024)
-
Rare coding variants in CHRNB2 reduce the likelihood of smoking
Nature Genetics (2023)
