
Abstract
High-grade serous carcinoma of uterine adnexa (HGSC) is the most frequent histotype of epithelial ovarian cancer and has a poor 5-year survival rate due to late-stage diagnosis and the poor efficacy of standard treatments. Novel biomarkers of cancer outcome are needed to identify new targetable pathways and improve personalized treatments. Cell-surface screening of 26 HGSC cell lines by high-throughput flow cytometry identified junctional adhesion molecule 1 (JAM-A, also known as F11R) as a potential biomarker. Using a multi-labeled immunofluorescent staining coupled with digital image analysis, protein levels of JAM-A were quantified in tissue microarrays from three HGSC patient cohorts: a discovery cohort (n = 101), the Canadian Ovarian Experimental Unified Resource cohort (COEUR, n = 1158), and the Canadian Cancer Trials Group OV16 cohort (n = 267). Low JAM-A level was associated with poorer outcome in the three cohorts by Kaplan–Meier (p = 0.023, p < 0.001, and p = 0.036, respectively) and was an independent marker of shorter survival in the COEUR cohort (HR = 0.517 (0.381–703), p < 0.001). When analyses were restricted to patients treated by taxane–platinum-based chemotherapy, low JAM-A protein expression was associated with poorer responses in the COEUR (p < 0.001) and OV16 cohorts (p = 0.006) by Kaplan–Meier. Decreased JAM-A gene expression was an indicator of poor outcome in gene expression datasets including The Cancer Genome Atlas (n = 606, p = 0.002) and Kaplan–Meier plotter (n = 1816, p = 0.024). Finally, we observed that tumors with decreased JAM-A expression exhibited an enhanced epithelial to mesenchymal transition (EMT) signature. Our results demonstrate that JAM-A expression is a robust prognostic biomarker of HGSC and may be used to discriminate tumors responsive to therapies targeting EMT.
Similar content being viewed by others

Introduction
Epithelial ovarian cancer (EOC) is the fifth leading cause of cancer-related deaths for women in North America and the leading cause of death from gynecological malignancies [1,2,3]. Among the described EOC, high-grade serous carcinoma of uterine adnexa (HGSC), also referred to as tubo-ovarian HGSC, is the most common and lethal histotype [3, 4]. Several studies have described four molecular subtypes within HGSC based on gene expression profiling—differentiated, proliferative, mesenchymal, and immunoreactive—but their association with prognosis is controversial [5,6,7,8]. Identifying novel markers of prognosis is needed to better stratify patients and discern a priori those with aggressive disease. Finding new prognostic biomarkers will help identify pathways and cellular processes involved in cancer progression and may lead to the development of personalized treatments [9].
Analysis of the cancer cell surfaceome can identify cell-surface proteins that are involved in pathways essential for cancer cell progression and, therefore, represent potentially relevant biomarkers [10, 11]. Cell surface proteins can be direct targets for therapeutic antibodies or used to deliver antibody-directed therapies in the form of antibody-drug conjugates. Recent studies have implicated junctional adhesion molecule-A (JAM-A/F11R) in cancer. Junctional adhesion molecules are type-I transmembrane glycoproteins expressed on the surface of endothelial and epithelial cells as well as leukocytes and platelets. JAM-A plays a role in epithelial tight junction (TJ) assembly at the apical part of the lateral membrane in polarized epithelial and endothelial cells and is involved in leukocyte transmigration, platelet activation, angiogenesis, and cell morphology [12,13,14]. Loss of TJ cohesion is required for cancer cells to detach from the epithelium and migrate, leading to invasion and ultimately metastasis [15]. Conflicting roles for JAM-A involvement in tumor progression have been described in the literature [13]. High expression of JAM-A is associated with improved outcome or inhibition of tumor progression in melanoma [16], renal cell carcinoma [17], endometrial carcinoma [18], and pancreatic cancer [19]. However, high JAM-A expression is associated with tumor progression in non-small cell lung cancer [20] and with poor prognosis in nasopharyngeal cancer [21], glioblastoma [22], and multiple myeloma [23]. Divergent roles have been observed for JAM-A in gastric and breast cancer. JAM-A promotes proliferation and inhibits apoptosis of gastric cancer cell lines [24], but low JAM-A expression correlates with poor prognosis in gastric cancer [25]. JAM-A decreases migration of metastatic breast cancer cells [26] yet overexpression is associated with poor prognosis in two cohorts of 270 and 444 breast cancer patients [27, 28]. In a cohort of 44 patients with various EOC histotypes, JAM-A tended to associate with unfavorable prognosis [29]. The role of JAM-A in HGSC prognosis has never been clearly assessed and still requires further elucidation.
In this study, we examined cell-surface proteins across a panel of human ovarian cancer-derived cell lines and identified JAM-A as highly expressed at the surface of EOC cell lines. We evaluated JAM-A protein expression as a prognostic biomarker in three cohorts: (1) a discovery cohort of 101 HGSC cases, (2) the Canadian Ovarian Experimental Unified Resource (COEUR) cohort composed of 1158 HGSC cases, and (3) the OV16 cohort of 267 HGSC cases from the Canadian Cancer Trials Group (CCTG). Immunofluorescence staining and digital image analysis (DIA) were used to quantify JAM-A expression. JAM-A mRNA expression was also identified as a biomarker of prognosis in the The Cancer Genome Atlas (TCGA, n = 606) and Kaplan–Meier plotter (n = 1816) datasets. Finally, we studied the correlation between JAM-A expression and epithelial to mesenchymal transition (EMT).
Material and methods
Patient samples
HGSC tissue microarrays (TMAs) in the form of formalin-fixed paraffin-embedded (FFPE) tissues were obtained from the CHUM, the Terry Fox Research Institute (TFRI)-COEUR and the CCTG. FFPE tumor specimens were collected during debulking surgery of patients. Informed patient consent was obtained prior to sample collection. Inclusion criteria for the study were (1) HGSC histopathology and (2) no neoadjuvant chemotherapy prior to surgery. Clinicopathological features of the three cohorts are summarized in Table 1.
Tumors from 101 patients recruited at the CHUM between 1993 and 2012 were used to build the discovery cohort TMA. All tumor samples were evaluated by a gynecologic-oncologic pathologist (KR) at the CHUM, who assigned histopathological histotype and tumor grade according to the criteria established by the International Federation of Gynecology and Obstetrics (FIGO). The disease stage was determined at time of surgery. Two tissue punches per patient were added to the TMA. Fifteen cases of normal fallopian tube tissues from women who had undergone salpingo-oophorectomy and did not have a gynecological malignancy were added to the TMA as controls and were also evaluated by gynecologic-oncologic pathologist KR.
For the TFRI-COEUR cohort, eight TMA blocks were constructed from 1158 HGSC tumor donors, which were obtained between 1991 and 2017 from ten tumor banks across Canada, including the CHUM [30]. Seventy-seven patient cases were common between the discovery and the COEUR cohorts. Each tumor was represented by duplicate tissue punches in the TMA. Two pathologists (MK and KR) performed a double central review of FFPE TMA blocks. Sixty-five specimens were reviewed as non-HGSC histotype, which were subsequently excluded from the analysis.
For the CCTG OV16 cohort, 819 patients were recruited between 2005 and 2015 for a randomized, phase-III clinical trial of sequential cisplatin-topotecan and carboplatin-paclitaxel (CP) versus CP in first line chemotherapy for advanced EOC [31]. A total of 267 patient samples were used to build eight TMA blocks and to analyze JAM-A expression, including 128 patients in the sequential cisplatin-topotecan and CP arm and 139 patients in the standard CP arm. Tumor tissues were reviewed by pathologists after haematoxylin and eosin staining at the Department of Pathology and Molecular Biology at the Queen’s University. Areas of tumor were marked on FFPE blocks by a pathologist and quadruplicate tissue punches were added in the TMA. Eligible patients had confirmed stage IIB to IV epithelial ovarian, fallopian, or peritoneal cancer with microscopic or macroscopic residual disease.
Datasets for gene expression study
The Cancer Genome Atlas dataset (TCGA)
JAM-A gene expression analysis was performed on the 606 HGSC tumor samples included in the Ovarian Serous Cystadenocarcinoma dataset from TCGA (Firehose Legacy, n = 606). Fragments per kilobase of transcript per million expression of JAM-A was extracted from the Human Protein Atlas database. Proteomic data used in this publication were generated by the Clinical Proteomic Tumor Analysis Consortium (CPTAC, NCI/NIH, https://proteomics.cancer.gov/programs/cptac) from n = 174 cases from this TCGA cohort. CPTAC protein levels measured by mass spectrometry, and clinical data from the Ovarian Serous Cystadenocarcinoma dataset were downloaded on November 2017 from the open-access cBioPortal for Cancer Genomics (http://www.cbioportal.org) [32, 33].
Kaplan–Meier plotter dataset [34]
The Kaplan–Meier plotter has computed 1816 ovarian cancer patient data with a mean follow-up of 40 months (http://kmplot.com). The database was primarily set up using gene expression data and survival information of ovarian cancer patients downloaded from Gene Expression Omnibus (GEO) (n = 1251) and TCGA (n = 565) (Affymetrix HG-U133A, HG-U133A 2.0, and HG-U133 Plus 2.0 microarrays). Analysis was done with the JetSet best probe set “222354_at” against JAM-A/F11R, including auto selection of best cut-off (135 for OS and 137 for post-progression) and was restricted to serous histology, grade 3, and all stages. All debulk types, chemotherapy, TP53 mutation, and available datasets were included (n = 901 patients analyzed for OS and n = 531 patients analyzed for post-progression survival).
Ovarian cancer database of the Cancer Science Institute Singapore (CSIOVDB) [35]
CSIOVDB contains data on 3431 human ovarian carcinomas including carcinoma of the ovary (91.49%), fallopian tube, peritoneum, and metastasis to the ovary from GEO, ArrayExpress, TCGA, ExpO, and private/in-house data (http://csibio.nus.edu.sg). HGSC is the most highly represented carcinoma in CSIOVDB (73.75%). The data were compiled and normalized, and clinical annotation was extracted. The database has 1868 and 1516 samples with overall survival (OS) and progression-free survival (PFS), respectively. Output of a gene query includes expression profiles in histological and molecular subtypes and survival correlations. Molecular subtypes and EMT status for each tumor is provided [7, 36]. CSIOVDB molecular subtypes were correlated to those previously defined by TCGA [6] and Tothill [5]. CSIOVDB categorized 11.75% of tumors as epithelial-A, 29.04% as epithelial-B, 29.01% as mesenchymal, 19.2% as stem-A, and 8.23% as stem-B subtypes. The database provides integration with the copy number, and DNA methylation from TCGA.
Surfaceome analysis
Cell-surface antibody screens were performed as previously described [37]. See reference for source of antibodies. Briefly, we profiled 26 HGSC (Table S1) cell lines by high-throughput flow cytometry using a library of 371 fluorochrome-conjugated cell-surface antibodies. The library comprised all commercial antibodies available at the time of screening, covering broad families of cell-surface molecules including cell adhesion molecules, immune receptors, cytokine receptors, differentiation markers, and receptor protein kinases. Antibodies were aliquoted into round bottom 96-well plates. Cell suspensions of 0.5–1 × 106 cells per ml were aliquoted by multichannel pipette into plates for a final volume of 100 ml per well and a final antibody dilution of 1:50. Plates were incubated for 20 min on ice in the dark, centrifuged for 5 min at 3506 g, washed twice with 200 ml FC buffer (Hank’s balanced salt solution plus 1% bovine serum albumin and 2 mM EDTA), and resuspended in 50–80 ml FC buffer containing 0.1 mg/mL 49,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). In parallel, aliquots were stained in tubes for fluorescence-minus-one controls, which consisted of DAPI only staining if no co-staining was done. Fluorescence-minus-one controls were generated for each antibody used and compensations were set using BD Plus CompBeads and FACSDiva software. Data collection was performed on a Becton-Dickinson LSR II flow cytometer with ultraviolet (20 mW), violet (25 mW), blue (20 mW), and red (17 mW) lasers, with default filter configuration, utilizing the High-Throughput Sampler attachment. At least 10,000 events were collected per well. FCS 3.0 files were exported to FlowJo version 9.3.
TMA construction and immunofluorescence staining
Discovery and COEUR TMAs were constructed at the Centre de Recherche du CHUM (CRCHUM) using two cores of 0.6 mm diameter FFPE tumor specimens for each patient. CCTG OV16 TMA was constructed at the Queen University using four cores of 0.6 mm diameter FFPE tumor specimens for each patient. Immunostaining was applied on 4 µm TMA sections using the Benchmark XT autostainer (Ventana Medical Systems, Tucson, AZ, US). For each candidate marker, antibody staining conditions were based on the manufacturer’s datasheet. For antigen retrieval, slides were incubated in cell conditioning 1 or 2 buffer (Ventana Medical Systems). Slides were then incubated for 1 h with the candidate primary antibody. A mouse monoclonal antibody (H00050848-M01, Abnova) was used at 1:1000 dilution to detect JAM-A. The epithelium was identified by co-staining epithelial cytokeratins using either a cocktail of mouse antibodies against KRT7 (MS-1352-P, Neomarkers), KRT18 (sc-6259, Santa Cruz Biotechnology), and KRT19 (MS-198-P, Thermo Scientific) or a cocktail of rabbit antibodies against KRT8/18 (FLEX, clone EP17/EP30, Dako). After 45 min of incubation with secondary fluorescent antibodies, cell nuclei were stained with DAPI. TMA slides were then immersed in a solution of Sudan black for 15 min to decrease autofluorescence and fluorescence quenching in the tissues. Slides were mounted with coverslips using Fluoromount aqueous mounting medium (Millipore-Sigma, F4680).
Digital image analysis (DIA)
TMA slides were scanned with the VS-110 microscope using a 20 × 0.75 NA objective and a resolution of 0.3225 µm (Olympus Canada Inc.) linked to an OlyVIA® image viewer software (xvViewer.exe). After importing scanned images and identifying each TMA tissue punch into Visiopharm® (VP) software (Visiopharm), fluorescent staining of the different markers were quantified with the VP software for automated DIA. VP detection algorithms enabled the delimitation of the region of interest (ROI) “whole tissue” by recognizing DAPI staining and discriminating for epithelial and stromal structures using epithelial cytokeratin staining (ROIs “epithelium” and “stroma”). When markers were expressed in the cell nuclei, the delimitation of nuclei was performed using DAPI (ROI “nuclei”). We discriminated the ROI “nuclei” from extranuclear components (membrane, cytoplasm, and extracellular matrix) in the epithelium and stroma. For each tissue, marker expression was quantified in each image pixel of the different ROIs; the mean fluorescence intensity (MFI) of all pixels within the selected ROIs was calculated. After analysis, manual review was performed and damaged tissue sections and necrotic or red cell infiltrated zones from the ROIs were excluded. For the COEUR and OV16 TMA datasets, which both comprised eight TMA slides, tumor MFI data of each slide were normalized on the average fluorescence intensity of all TMA slides comprised in one cohort in order to compensate for possible intra-experiment variations of mean fluorescence between slides during scanning process.
Statistics
Statistical analyses were conducted using IBM Statistics SPSS 23 and Graphpad Prism 5. For survival analysis, Kaplan–Meier and univariate and multivariate Cox proportional hazard regression models were used. Multivariate analyses were performed in association with known and available prognostic indicators, including FIGO stages (dichotomous variable), debulking/residual disease at surgery (dichotomous variable), age of patients at diagnosis (continuous variable), age of FFPE blocks of tumor tissues (continuous variable), CA125 levels at surgery (continuous variable), BRCA status (categorized variable), and chemotherapy regimen (categorized variable). ROC curves and maximum log-likelihood approach were used to define cut-off points to dichotomize patients into high and low JAM-A protein expression groups. The same cut-off point was used to perform all the statistical analyses within a cohort. Correlation studies were performed using nonparametric Spearman correlation. The nonparametric Mann–Whitney test was used to compare the mean between two groups. P value < 0.05 was considered statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Surfaceome screening identified new potential biomarkers of HGSC
To identify novel molecular markers of HGSC, we examined cell-surface proteins across a panel of human HGSC-derived cell lines. We screened 26 HGSC cell lines (Table S1) by high-throughput flow cytometry using a library of 371 fluorophore-conjugated antibodies targeting cell adhesion molecules, immune receptors, cytokine receptors, differentiation markers, and receptor protein kinases [10, 37]. This approach allowed for rapid and direct detection of proteins expressed on the cell surface. Markers that occurred in more than 20% of cell lines (6 of 26) and in high abundance (more than 90% of cell population with positive staining) were selected for downstream analysis. In total, 60 cell-surface markers met our filtering criteria for high expression and penetrance (Fig. 1a). Candidates were further prioritized using several criteria: (1) high number of positive cell lines from the flow cytometry screening, (2) high expression levels in HGSC tissues compared with normal fallopian tube tissues from the Human Protein Atlas database, and (3) novelty in ovarian cancer. Notably, five candidates (ERBB2, EGFR, epithelial cell adhesion molecule (EPCAM), FAS, and CD44) were excluded from the study as their roles in ovarian cancer have been extensively studied. Using these criteria, twelve candidates were selected (Table S2) and their expression levels were measured in primary HGSC tissues. To determine protein expression levels, we performed immunofluorescence staining for each candidate using a discovery cohort TMA composed of 101 HGSC cases in duplicate and 15 normal FT epithelial tissues (Fig. 1b and Table 1). Each tissue section was subjected to DIA using Visiopharm® software algorithms to detect relevant ROIs. ROIs include epithelial cell nuclei, epithelial extranuclear component, stromal cell nuclei, and stromal cell extranuclear component. We calculated MFI of each candidate for all ROIs across the TMA and we correlated MFIs with clinical characteristics to identify potential prognostic biomarkers. In particular, JAM-A was found in high abundance in 21 of 26 cell lines (Fig. 1a), met all the selection criteria and gave the most promising results as a prognostic predictor in our discovery cohort.

a Classification of cell-surface markers that present in high abundance (more than 90% of cell population with positive staining) in more than 20% of cell lines (6/26). b TMA slides were stained by immunofluorescence using antibodies directed against each candidate and keratins to discriminate keratin-positive tumor epithelial cells from stroma. Candidate expression was quantified using digital image analysis (DIA) to identify relevant regions of interest (ROIs). DAPI staining was used for whole tissue detection. Nuclei and extranuclear components in epithelium and stroma were discriminated using DAPI staining. Mean fluorescence intensity (MFI) of each candidate was quantified in different ROIs across the TMA. MFI data were correlated with clinical parameters.
Low JAM-A expression is correlated with poor prognosis
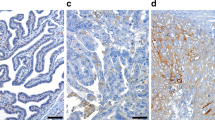
Analysis of JAM-A expression using the DIA quantification method revealed considerable variation in JAM-A expression across HGSC tumors (Fig. 2a). The reproducibility of DIA quantification and the homogeneity of JAM-A staining within a single tumor tissue were confirmed by the high correlation of JAM-A expression observed between duplicate samples (Spearman R = 0.845, p < 0.001) (Fig. 2b). Consistent with the literature, JAM-A was predominantly found at the apical pole of fallopian tube epithelial cells whereas in HGSC epithelial cells, JAM-A expression was disorganized, indicating a loss of cell polarity (Fig. 2c) [14]. Comparison of mean JAM-A expression in the HGSC tissues with normal FT showed no significant difference. Interestingly, we observed high levels of JAM-A in a subset of the HGSC tissues (13.95%) compared with the fallopian tube tissues, suggesting that JAM-A may be a target for antibody-directed drug conjugates in these tumors (Fig. 2d). Correlation of JAM-A expression with clinical parameters revealed an association of low JAM-A expression with both shorter PFS and shorter OS by Kaplan–Meier (Fig. 2e) and univariate Cox regression analyses (Table 2). This association was not significant by Multivariate Cox regression analysis probably due to the small size of the discovery cohort (Table 2). Together, these data suggest that JAM-A expression pattern is deregulated in HGSC and that low JAM-A expression may be a biomarker of poor outcome.

a Representative images of tissue staining corresponding to the first, 25th, 50th, 75th, and 99th percentiles of JAM-A levels in tumor epithelial tissue obtained by DIA. Large images show DAPI (blue) and JAM-A (red) staining. Inset images show DAPI (blue), JAM-A (red), and KRTs (yellow) staining to visualize epithelial structures. b Correlation of JAM-A levels between duplicate tissues for each tumor (n = 101). Spearman Rho and p value are indicated. c JAM-A staining in fallopian tube and HGSC tissues. Large images show DAPI (blue) and JAM-A (red) staining. Inset images show DAPI (blue), JAM-A (red), and KRTs (yellow) staining to visualize epithelial structures. Higher magnifications of the highlighted region are shown (right)—DAPI, JAM-A and KRTs (top), and DAPI and JAM-A (bottom). Arrows indicate JAM-A staining at the apical pole of fallopian tube epithelial cells. d JAM-A protein level in HGSC (n = 101) and fallopian tube (n = 15) tissues from the discovery TMA. Each point represents the mean JAM-A level stained in duplicate. e Kaplan–Meier curves of JAM-A dichotomized into low and high JAM-A expression for progression-free survival (left) and overall survival (right). Log rank p value is indicated. Number of patients and median number of survival months are indicated for each group.
Low JAM-A expression is associated with features of aggressive tumors and poor prognosis in the COEUR cohort
We next evaluated JAM-A expression in the COEUR cohort, consisting of 1158 HGSC cases (Table 1). Two exclusion criteria were applied to the cohort: (1) tumor specimens that were not considered HGSC histotype after secondary review (n = 65) and (2) tumor specimens from patients who received chemotherapy before surgery (n = 65). In addition, damaged or absent tissues and tissues with <5% epithelial content were excluded from analysis (n = 160). In total, 256 cases were excluded, 34 of which met two or more exclusion criteria, and the remaining 902 cases were used for analysis. JAM-A expression was significantly decreased in tumors of patients diagnosed in late stages 3 and 4 compared with early stages 1 and 2 (Fig. 3a). JAM-A expression was also significantly decreased in tumors of patients with marked residual disease after surgery compared with tumors with minimal or no residual disease (Fig. 3b). Kaplan–Meier curves showed that JAM-A expression was weakly correlated with PFS and strongly associated with OS and post-progression survival (Fig. 3c). These results were corroborated by univariate and multivariate Cox regression analyses indicating that JAM-A expression was an independent biomarker of OS and post-progression survival (Table 3). These data show that low JAM-A predicts poor outcome and is particularly associated with decreased survival time in patients with recurrent disease. JAM-A was the best independent predictor of survival after first progression (p < 0.001) among all studied variables including FIGO stage or residual disease as observed by multivariate analysis (Table 3). Collectively, these data suggest that JAM-A may be involved in modulating HGSC tumor aggressiveness and progression after relapse.

Boxplots illustrating JAM-A MFI distribution according to early (1 and 2) and late (3 and 4) FIGO stages (a) and JAM-A MFI distribution according to absence or low rates of residual disease (No RD and RD < 1 cm) and higher rates of residual disease (RD ≥ 1 cm and miliary) (b). The box represents the interquartile (IQ) range and the whiskers represent the highest and lowest values, which are <1.5 times the IQ range. Outliers are 1.5 times (circle) or 2 times (stars) the IQ range. The nonparametric Mann–Whitney test was used to compare the mean between the two groups of each graph. c Kaplan–Meier curves of JAM-A dichotomized into low and high JAM-A expression for progression-free survival (left), overall survival (middle), and post-progression survival (right). Log rank p value is indicated. Number of patients and median number of survival months are indicated for each group of expression.
JAM-A expression is a predictive marker of response to standard treatment
To ascertain whether JAM-A expression was associated with response to treatment, we generated Kaplan–Meier curves restricted to patients who received standard chemotherapy (platinum and taxol) after debulking surgery. JAM-A low expression was significantly associated with a poor outcome (Fig. S1), which was also observed by univariate and multivariate Cox regression analyses (Table S3). These results indicate that patients with low JAM-A expression exhibit a poorer response to standard chemotherapy.
JAM-A is a biomarker of HGSC prognosis at protein and gene expression levels
To determine whether our findings could be generalized to an independent patient cohort, we assessed JAM-A protein expression and clinical correlates in the CCTG OV16 cohort—a randomized phase-III clinical trial of sequential cisplatin-topotecan and carboplatin-paclitaxel versus standard regimen (carboplatin-paclitaxel alone) as first line chemotherapy for advanced EOC (Table 1). Kaplan–Meier analysis confirmed the association between decreased JAM-A protein expression and OS (Fig. 4a). When the analysis was restricted to the standard regimen arm, the association of JAM-A with prognosis was more pronounced (Fig. 4b). This result reinforces the utility of JAM-A protein expression as a marker for response to standard chemotherapy and suggests a possible effect of decreased JAM-A expression in the resistance to platinum and taxol-based chemotherapy.

a, b Kaplan–Meier curves of JAM-A protein expression dichotomized into low and high JAM-A expression for progression-free survival (left) and overall survival times (right) in the CCTG OV16 whole cohort (a) and CCTG OV16 restricted to the carboplatinum + taxol arm (b). Log rank p value is indicated. Number of patients and median number of survival months are indicated for each group of expression. c Correlation of JAM-A protein level z-scores from CPTAC and JAM-A mRNA z-scores (RNA Seq V2 RSEM) from TCGA in overlapping cases (n = 174) retrieved from the TCGA database. Spearman Rho and p value are indicated. d.Kaplan–Meier curves of JAM-A mRNA expression dichotomized into low and high JAM-A expression for progression-free and overall survival times in TCGA (FPKM cut-off = 13.5, 25th percentile). Log rank p value is indicated. Number of patients and median number of survival months are indicated for each group of expression.
We next sought to determine whether JAM-A gene expression was a predictor of patient outcome in HGSC. First, we compared JAM-A protein levels from the CPTAC and JAM-A mRNA levels from the TCGA on 174 common samples and found a significant correlation (Spearman R = 0.46, p < 0.001) (Fig. 4c). Next, we examined JAM-A mRNA levels in 606 HGSC samples from TCGA and observed an association of JAM-A low levels with shorter PFS and OS (Fig. 4d). To confirm the association of JAM-A gene expression with prognosis, we used the Kaplan–Meier plotter dataset (n = 1816 patients). Consistent with our previous findings, low JAM-A expression was associated with shorter OS and PPS (Fig. S2). Altogether, these results show that JAM-A expression is a robust biomarker of HGSC patient prognosis at the protein and gene levels.
Low JAM-A expression is a marker of EMT signature
JAM-A contributes to epithelial TJ formation and epithelial cell cohesion. Inhibition of JAM-A disrupts intercellular TJs [38]. Using the HGSC protein expression data from CPTAC, we similarly observed that low JAM-A expression was associated with a significant decrease in TJ protein expression namely, OCLN, claudin 3 (CLDN3), and zona occludens 1 (Fig. 5a). Next, we hypothesized that inhibition of JAM-A expression was associated with EMT and quantified protein levels of the epithelial marker E-cadherin (E-CADH) and the mesenchymal marker Vimentin (VIM) in our discovery cohort TMA. We observed decreased E-CADH expression and increased VIM expression in tumors with low JAM-A expression (Fig. 5b). Using the protein expression data from CPTAC, Spearman correlation showed that JAM-A protein levels were positively correlated with epithelial markers E-CADH, OCLN, CLDN3, and EPCAM (Fig. 5c). Conversely, JAM-A was negatively associated with mesenchymal proteins such as VIM, alpha smooth muscle actin (α-SMA), fibronectin (FN1), CD44, matrix metalloproteinase 2, 9, 10, and 14 (MMP2, MMP9, MMP10, and MMP14), and transforming growth factor beta 1 (TGF-β1) (Fig. 5c). We then used 3431 human ovarian carcinomas from the CSIOVDB database, which are classified by an EMT score and by molecular subtypes (epithelial-A, epithelial-B, mesenchymal, stem-like-A, and stem-like-B) [7, 36]. CSIOVDB molecular subtypes were correlated to previously defined subtypes [5, 6]: epithelial-A and epithelial-B subtypes are defined by expression of E-CADH, EPCAM, and keratin genes. The mesenchymal subtype is defined by expression of vascular cell adhesion molecule 1, zinc finger E-box binding homeobox 1, twist family bHLH transcription factor 1, TGF-β1, α-SMA, collagen, and FN1. Stem-like-A and Stem-like-B subtypes express leucine rich repeat containing G protein-coupled receptor 5 (LGR5) and prominin 1 (PROM1), with the Stem-A subtype exhibiting higher levels of MYCN proto-oncogene (MYCN), neural cell adhesion molecule 1, N-CADH, and proliferation-related genes [7]. JAM-A gene expression was negatively associated with the EMT score (Fig. 5d) and tumors of the mesenchymal molecular subtype showed the lowest levels of JAM-A mRNA compared with the other molecular subtypes (Fig. 5e). Consistent with these data, the highest levels of JAM-A DNA methylation were found in the mesenchymal tumors (Fig. 5f). Together, these data show that low JAM-A protein and gene expression are associated with EMT and the mesenchymal subtype of HGSC.

a, b Boxplot showing distribution of OCLN, CLDN3, and ZO-1 protein levels in JAM-A low versus high groups (z-score cut-off = −0.67, 25th percentile); protein level z-scores were measured by CPTAC in 174 tumors from TCGA. Boxplot illustrating E-CADH and VIM protein levels distribution in JAM-A low versus high groups (MFI cut-off = 240); protein MFI levels were measured by DIA in the discovery cohort (b). Boxes represents the interquartile (IQ) range and whiskers represent the highest and lowest values, which are <1.5 times the IQ range. Outliers are 1.5 times (circle) or 2 times (star) the IQ range. The nonparametric Mann–Whitney test was used to compare the mean between JAM-A low versus high groups. c Spearman correlations between JAM-A protein levels and protein levels of epithelial markers (no highlight) and mesenchymal markers (gray). Protein levels z-scores were measured by CPTAC in 174 tumors from TCGA. d Spearman correlation between JAM-A gene expression and EMT score in ovarian carcinoma (n = 3431) from CSIOVDB dataset. e JAM-A mRNA expression by CSIOVDB molecular subtypes of ovarian carcinoma (n = 3431). f JAM-A gene methylation from TCGA by CSIOVDB molecular subtypes of ovarian carcinoma (n = 3431). Epi-A, Epi-B, Mes, Stem-A, and Stem-B denote epithelial-A, epithelial-B, mesenchymal, stem-like-A, and stem-like-B subtypes, respectively.
Discussion
JAM-A has been reported to have both tumor-promoting and -suppressing roles depending on cancer type, which may reflect the multiple functions ascribed to JAM-A [13]. In addition to its role in cell–cell adhesion processes, JAM-A also participates in leukocyte transendothelial migration, platelet activation, and angiogenesis [39,40,41,42,43,44], which may account for the tissue-specific effect of JAM-A expression on cancer prognosis. Our study identified low JAM-A expression as a worse prognosis marker in HGSC. Our results were validated in several independent patient cohorts and datasets including the COEUR, the largest cohort of HGSC patients in Canada, and the CCTG OV16 cohort, a phase-III clinical trial. The reproducibility of our results across these large cohorts strongly supports an association between low JAM-A expression and poor prognosis in HGSC. Our study also revealed that high JAM-A expression predicts a better response to standard platinum plus taxol-based chemotherapy. While a potential role of JAM-A in chemoresistance remains unclear and should be further studied, its effects may be occurring via an EMT-dependent mechanism [45,46,47].
Unlike most epithelial solid tumors, which metastasize via hematogenous spread, HGSC cells exfoliate from the primary tumor of fallopian tube (intraepithelial tubal carcinoma), disseminate throughout the peritoneal cavity and seed the mesothelial lining of the peritoneum to form metastatic lesions [48]. In this model of dissemination, the dissolution of TJs and the loss of apico-basal polarity are probably a compulsory step in metastasis of HGSC cells, which may partly explain the effect of low JAM-A expression on prognosis. Disassembly of TJs leads to loss of polarity and loss of contact inhibition, uncontrolled growth, detachment, and invasion [15, 49]. We observed an association between low JAM-A and decreased expression of TJ proteins. In the COEUR cohort, JAM-A protein levels were decreased in tumors of patients diagnosed in advanced stages and in patients with increased residual disease after debulking surgery. Based on these findings, we propose a model whereby JAM-A downregulation promotes TJ dissolution and, therefore, tumor cell detachment and dissemination.
During EMT, E-CADH is replaced by N-cadherin and shedding of the E-CADH extracellular domain disrupts existing cell junctions, stimulates cancer cell detachment, and upregulates matrix metalloproteinases MMP2, MMP9, and MMP14 expression [48, 50]; epithelial cytokeratin intermediate filaments are replaced by VIM [48, 51]. We showed that JAM-A protein expression was positively correlated with E-CADH and negatively correlated with VIM, MMP2, MMP9, and MMP14, suggesting that EMT coincides with a diminution of JAM-A expression in HGSC. JAM-A protein expression was also negatively correlated with CD44, which was shown to predict poor outcome of EOC patients through promoting EMT [52]. We observed that tumors of the mesenchymal molecular subtype showed lower expression of JAM-A mRNA and higher levels of JAM-A DNA methylation. These data implicate low JAM-A as a feature of the mesenchymal subtype, which may be mediated, in part, by increased methylation of its gene. The mesenchymal-like molecular subtype of HGSC was associated with the worst prognosis by several groups [7, 8, 36, 53, 54] that may explain our finding that low JAM-A expression is a marker of poor prognosis.
Despite promising research identifying novel therapeutic targets in HGSC, there have been no significant changes to clinical management of the disease [46, 49, 55]. One approach is to target activated cellular programs, such as EMT, which are involved in tumor progression. Activation of the TGF-β signaling pathway induces EMT, promotes immune suppression within the tumor microenvironment, and contributes to chemotherapy resistance and metastasis [45, 47, 56, 57]; low TGFβ-1 mRNA expression is associated with better prognosis and chemosensitivity [56]; TGFβ-1 downregulates JAM-A transcription and promotes JAM-A protein degradation in breast cancer cells [58]. In Fig. 5c, we highlighted a negative correlation between JAM-A and TGF-β1 protein expression suggesting that TGF-β1 may also downregulate JAM-A expression in HGSC tumors. Several molecules targeting TGF-β1 pathway are in clinical development [59] and further studies are needed to evaluate if low JAM-A could be a relevant marker for identifying HGSC tumors most likely to respond to treatments targeting the TGF-β pathway. Molecular mechanisms and pathways responsible for JAM-A inhibition in HGSC need to be clearly identified. Such findings could reveal possible actionable targets to prevent TJ dissociation, which is a critical step in cell dissemination and invasion in HGSC.
A study was recently published about JAM-A gene expression in a cohort of 44 EOC where JAM-A mRNA was measured by Q-PCR from FFPE tumor samples [29]. In this cohort, the authors found that high expression of JAM-A was associated with worse OS by Kaplan–Meier (p = 0.004) and univariate Cox regression (p = 0.008) but not in multivariate analysis. While this conclusion is the opposite of what we observed, this discrepancy may be related to the relatively small size of the study cohort (n = 44 patients) and to its heterogeneity. Indeed, the study involved 33 serous tumors. While not specifically attributed to any specific histotype, the study reported 26 tumors of grade 2 + 3, which at the most, suggests 26 cases of HGSC. We have not studied JAM-A in other ovarian cancer histotypes, but their results may be biased by different levels of JAM-A in different histotypes with varying outcomes. This observation raises the interest to further study JAM-A expression in other histotypes and to systematically validate the prognosis value of marker expression in independent cohorts with a significantly larger number of patients.
There were two major limitations to our study. JAM-A expression was determined by multi-labeled immunofluorescent staining followed by DIA to localize and quantify JAM-A with the highest levels of definition and objectivity. This strength may limit the transfer of our results into the clinic where immunohistochemistry (IHC) and visual quantifications are still the standard procedure for pathologists to analyze marker expression in patient tumors. However, the antibody we used against JAM-A was validated by IHC in our hands and in the literature [17, 19, 25]; JAM-A expression could be further studied in one of our cohorts using IHC in order to transpose the MFI cut-off point into a visual IHC cut-off usable by pathologists. The second limitation was the lack of certain variables in patient clinical data, which limited our Cox regression analyses of the studied cohorts. Notably, presence and levels of ascites correlate with a poor prognosis in ovarian cancer patients [60], but this information was not available. Since we hypothesize that JAM-A inhibition may increase HGSC cell detachment and dissemination, it would be of interest to correlate JAM-A expression with volumes of ascites and particularly with levels of detached tumor cells in ascites in future studies.
In summary, we propose JAM-A expression as an independent biomarker of poor outcome in HGSC. JAM-A expression may have clinical utility to improve early prognostic stratification of patients and to propose more efficient and personalized therapies. This study highlights a possible role of epithelial TJs loss in HGSC tumor progression and provides a strategy for biomarker validation.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29.
Committee CCSA. Canadian Cancer Statistics 2018. Toronto, ON: Canadian Cancer Society; 2018.
Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. Lancet. 2019;393:1240–53.
Prat J. Ovarian carcinomas: five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. 2012;460:237–49.
Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res. 2008;14:5198–208.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Tan TZ, Miow QH, Huang RY, Wong MK, Ye J, Lau JA, et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol Med. 2013;5:1051–66.
Konecny GE, Wang C, Hamidi H, Winterhoff B, Kalli KR, Dering J, et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J Natl Cancer Inst. 2014;106.
Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: evolution of management in the era of precision medicine. CA Cancer J Clin. 2019.
Medrano M, Communal L, Brown KR, Iwanicki M, Normand J, Paterson J, et al. Interrogation of functional cell-surface markers identifies CD151 dependency in high-grade serous ovarian cancer. Cell Rep. 2017;18:2343–58.
Paterson J, Ailles LE. High throughput flow cytometry for cell surface profiling. Methods Mol Biol. 2018;1678:111–38.
Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–27.
Zhao C, Lu F, Chen H, Zhao X, Sun J, Chen H. Dysregulation of JAM-A plays an important role in human tumor progression. Int J Clin Exp Pathol. 2014;7:7242–8.
Mandell KJ, Babbin BA, Nusrat A, Parkos CA. Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J Biol Chem. 2005;280:11665–74.
Martin TA. The role of tight junctions in cancer metastasis. Semin Cell Dev Biol. 2014;36:224–31.
Ghislin S, Obino D, Middendorp S, Boggetto N, Alcaide-Loridan C, Deshayes F. Junctional adhesion molecules are required for melanoma cell lines transendothelial migration in vitro. Pigment Cell Melanoma Res. 2011;24:504–11.
Gutwein P, Schramme A, Voss B, Abdel-Bakky MS, Doberstein K, Ludwig A, et al. Downregulation of junctional adhesion molecule-A is involved in the progression of clear cell renal cell carcinoma. Biochem Biophys Res Commun. 2009;380:387–91.
Koshiba H, Hosokawa K, Kubo A, Tokumitsu N, Watanabe A, Honjo H. Junctional adhesion molecule A [corrected] expression in human endometrial carcinoma. Int J Gynecol Cancer. 2009;19:208–13.
Fong D, Spizzo G, Mitterer M, Seeber A, Steurer M, Gastl G, et al. Low expression of junctional adhesion molecule A is associated with metastasis and poor survival in pancreatic cancer. Ann Surg Oncol. 2012;19:4330–6.
Zhang M, Luo W, Huang B, Liu Z, Sun L, Zhang Q, et al. Overexpression of JAM-A in non-small cell lung cancer correlates with tumor progression. PLoS One. 2013;8:e79173.
Tian Y, Tian Y, Zhang W, Wei F, Yang J, Luo X, et al. Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis. 2015;36:41–48.
Lathia JD, Li M, Sinyuk M, Alvarado AG, Flavahan WA, Stoltz K, et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 2014;6:117–29.
Solimando AG, Brandl A, Mattenheimer K, Graf C, Ritz M, Ruckdeschel A, et al. JAM-A as a prognostic factor and new therapeutic target in multiple myeloma. Leukemia. 2018;32:736–43.
Ikeo K, Oshima T, Shan J, Matsui H, Tomita T, Fukui H, et al. Junctional adhesion molecule-A promotes proliferation and inhibits apoptosis of gastric cancer. Hepatogastroenterology. 2015;62:540–5.
Huang JY, Xu YY, Sun Z, Wang ZN, Zhu Z, Song YX, et al. Low junctional adhesion molecule A expression correlates with poor prognosis in gastric cancer. J Surg Res. 2014;192:494–502.
Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008;68:2194–203.
McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, et al. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer. 2009;125:1343–51.
Murakami M, Giampietro C, Giannotta M, Corada M, Torselli I, Orsenigo F, et al. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS One. 2011;6:e21242.
Ivana B, Emina M, Marijana MK, Irena J, Zoran B, Radmila J. High expression of junctional adhesion molecule-A is associated with poor survival in patients with epithelial ovarian cancer. Int J Biol Markers. 2019. https://doi.org/10.1177/1724600819850178.
Le Page C, Rahimi K, Kobel M, Tonin PN, Meunier L, Portelance L, et al. Characteristics and outcome of the COEUR Canadian validation cohort for ovarian cancer biomarkers. BMC Cancer. 2018;18:347.
Hoskins P, Vergote I, Cervantes A, Tu D, Stuart G, Zola P, et al. Advanced ovarian cancer: phase III randomized study of sequential cisplatin-topotecan and carboplatin-paclitaxel vs carboplatin-paclitaxel. J Natl Cancer Inst. 2010;102:1547–56.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2012;2:401–4.
Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012;19:197–208.
Tan TZ, Yang H, Ye J, Low J, Choolani M, Tan DS, et al. CSIOVDB: a microarray gene expression database of epithelial ovarian cancer subtype. Oncotarget. 2015;6:43843–52.
Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6:1279–93.
Gedye CA, Hussain A, Paterson J, Smrke A, Saini H, Sirskyj D, et al. Cell surface profiling using high-throughput flow cytometry: a platform for biomarker discovery and analysis of cellular heterogeneity. PLoS One. 2014;9:e105602.
Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci. 2000;113:2363–74.
Bazzoni G. The JAM family of junctional adhesion molecules. Curr Opin Cell Biol. 2003;15:525–30.
Mandell KJ, Parkos CA. The JAM family of proteins. Adv Drug Deliv Rev. 2005;57:857–67.
Xu Z, Jin B. A novel interface consisting of homologous immunoglobulin superfamily members with multiple functions. Cell Mol Immunol. 2010;7:11–19.
Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, et al. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009;122:268–77.
Iden S, Misselwitz S, Peddibhotla SS, Tuncay H, Rehder D, Gerke V, et al. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J Cell Biol. 2012;196:623–39.
Severson EA, Parkos CA. Structural determinants of junctional adhesion molecule A (JAM-A) function and mechanisms of intracellular signaling. Curr Opin Cell Biol. 2009;21:701–7.
Marchini S, Fruscio R, Clivio L, Beltrame L, Porcu L, Fuso Nerini I, et al. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur J Cancer. 2013;49:520–30.
Deng J, Wang L, Chen H, Hao J, Ni J, Chang L, et al. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget. 2016;7:55771–88.
Mitra T, Prasad P, Mukherjee P, Chaudhuri SR, Chatterji U, Roy SS. Stemness and chemoresistance are imparted to the OC cells through TGFbeta1 driven EMT. J Cell Biochem. 2018;119:5775–87.
Klymenko Y, Kim O, Stack MS. Complex determinants of epithelial: mesenchymal phenotypic plasticity in ovarian cancer. Cancers. 2017;9.
Antony J, Thiery JP, Huang RY. Epithelial-to-mesenchymal transition: lessons from development, insights into cancer and the potential of EMT-subtype based therapeutic intervention. Phys Biol. 2019.
Moffitt L, Karimnia N, Stephens A, Bilandzic M. Therapeutic targeting of collective invasion in ovarian cancer. Int J Mol Sci. 2019;20.
Bilyk O, Coatham M, Jewer M, Postovit LM. Epithelial-to-mesenchymal transition in the female reproductive tract: from normal functioning to disease pathology. Front Oncol. 2017;7:145.
Zhou J, Du Y, Lu Y, Luan B, Xu C, Yu Y, et al. CD44 expression predicts prognosis of ovarian cancer patients through promoting epithelial-mesenchymal transition (EMT) by regulating snail, ZEB1, and caveolin-1. Front Oncol. 2019;9:802.
Bonome T, Levine DA, Shih J, Randonovich M, Pise-Masison CA, Bogomolniy F, et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68:5478–86.
Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J Clin Invest. 2013;123:517–25.
Newsted D, Banerjee S, Watt K, Nersesian S, Truesdell P, Blazer LL, et al. Blockade of TGF-beta signaling with novel synthetic antibodies limits immune exclusion and improves chemotherapy response in metastatic ovarian cancer models. Oncoimmunology 2019;8:e1539613.
Komiyama S, Kurahashi T, Ishikawa M, Tanaka K, Komiyama M, Mikami M, et al. Expression of TGFss1 and its receptors is associated with biological features of ovarian cancer and sensitivity to paclitaxel/carboplatin. Oncol Rep. 2011;25:1131–8.
Cao L, Shao M, Schilder J, Guise T, Mohammad KS, Matei D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene. 2012;31:2521–34.
Wang Y, Lui WY. Transforming growth factor-beta1 attenuates junctional adhesion molecule-A and contributes to breast cancer cell invasion. Eur J Cancer. 2012;48:3475–87.
Alsina-Sanchis E, Figueras A, Lahiguera A, Gil-Martin M, Pardo B, Piulats JM, et al. TGFbeta controls ovarian cancer cell proliferation. Int J Mol Sci. 2017;18.
Szender JB, Emmons T, Belliotti S, Dickson D, Khan A, Morrell K, et al. Impact of ascites volume on clinical outcomes in ovarian cancer: a cohort study. Gynecol Oncol. 2017;146:491–7.
Acknowledgements
LC, MM, and FS were supported by the Selective Therapy Program from the TFRI and the Ontario Institute for Cancer Research. LC was supported by a post-doctoral fellowship from the Fond de recherche Québec—Santé (FRQS). MM was supported by Ovarian Cancer Canada’s Teal Heart Scholarship. A-MMM and DP are researchers of CRCHUM/ICM, which receive support from the FRQS and the Réseau de Recherche sur le cancer (RR cancer). We thank Jacqueline Chung for English editing. We thank Anne-Marie Fortier, Senthil Muthuswamy, and Shakeel Virk for helpful discussions and information. We thank the molecular pathology core facility of the CRCHUM for performing the discovery and COEUR TMA construction and tissue scanning. We thank the Department of Pathology and Molecular Biology at the Queen’s University for performing the OV16 TMA construction. Tumor banking for the discovery cohort was supported by the Banque de tissus et de données of the RR cancer of the FRQS affiliated with the Canadian Tumor Repository Network. Tumor banking for the OV16 cohort was performed by the CCTG Tumor Tissue Data Repository. This study uses resources provided by the COEUR biobank funded by the TFRI and managed and supervised by the CHUM. The Consortium acknowledges contributions to its COEUR biobank from Institutions across Canada (for a full list see www.tfri.ca/COEUR/members). We thank the generosity of patients included in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AO is a consultant/advisory board member for Immunogen, AstraZeneca, Tesaro, and Clovis. The other authors declare that they have no conflict of interest.
Ethics approval
Ethical approval for the study was obtained from the Centre hospitalier de l’Université de Montreal (CHUM) institutional ethics committee (Comité d’éthique de la Recherche du CHUM).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Communal, L., Medrano, M., Sircoulomb, F. et al. Low junctional adhesion molecule-A expression is associated with an epithelial to mesenchymal transition and poorer outcomes in high-grade serous carcinoma of uterine adnexa. Mod Pathol 33, 2361–2377 (2020). https://doi.org/10.1038/s41379-020-0586-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0586-0
