
Abstract
The homologous recombination deficiency (HRD) score integrates three DNA-based measures of genomic instability, and has been understudied in prostate cancer. Given the recent FDA approval of two PARP inhibitors for prostate cancer, HRD score analysis could help to refine treatment selection. We assessed HRD score (defined as the sum of loss-of-heterozygosity, telomeric allelic imbalance, and large-scale state transitions) in three cohorts of primary prostate cancer, including a Johns Hopkins University (JHU) cohort with germline mutations in BRCA2, ATM, or CHEK2 (n = 64), the TCGA cohort (n = 391), and the PROGENE cohort (n = 102). In the JHU cohort, tumors with germline BRCA2 mutations had higher HRD scores (median = 27) than those with germline ATM or CHEK2 mutations (median = 16.5 [p = 0.029] and 9 [p < 0.001], respectively). For TCGA tumors without underlying HR pathway mutations, the median HRD score was 11, significantly lower than ovarian carcinoma lacking BRCA1/2 mutations (median = 28). In the absence of HR gene mutations, the median HRD score was unexpectedly higher among prostate cancers with TP53 mutations versus those without (17 vs. 11; p = 0.015); this finding was confirmed in the PROGENE cohort (24 vs. 16; p = 0.001). Finally, among eight BRCA2-altered patients who received olaparib, progression-free survival trended longer in those with HRD scores above versus below the median (14.9 vs. 9.9 months). We conclude that HRD scores are low in primary prostate cancer and higher in cases with germline BRCA2 or somatic TP53 mutations. Germline BRCA2-altered cases have significantly higher HRD scores than germline ATM-altered or CHEK2-altered cases, consistent with the lower efficacy of PARP inhibitors among the latter.
Similar content being viewed by others


Introduction
The goal of precision oncology is to harness knowledge about the underlying molecular biology of a patient’s cancer, and to leverage the inherited genomics of the patient, in order to select the most appropriate therapy for that patient at a given point in time. The recognition that germline or somatic mutations in DNA-repair genes are present in about one-quarter of patients with recurrent or advanced prostate cancer [1] presents an opportunity to engage in the precision medicine revolution for this tumor type in a substantial fraction of patients. To this end, the clearest example of a genomically-informed therapy in this disease is the use of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with metastatic castration-resistant prostate cancer (mCRPC) harboring a mutation in a gene that is directly or indirectly involved in homologous recombination DNA repair [2]. Indeed, a number of PARP inhibitors have demonstrated impressive clinical activity in mCRPC patients with homologous recombination (HR) gene mutations, even in very advanced disease settings [3,4,5,6,7]. These efforts have culminated in the recent FDA approval of rucaparib for mCRPC patients with germline or somatic BRCA1 or BRCA2 mutations, and of olaparib for mCRPC patients with mutations in at least one of 14 HR-related genes (BRCA1/2, ATM, and 11 others).
While it was initially hoped that all HR gene mutations would result in favorable clinical activity with PARP inhibitor treatment, it is now emerging that there are substantial differences in response rates to PARP inhibitors according to the specific HR gene involved. For example, BRCA2-altered prostate cancers broadly appear to derive the greatest clinical benefit, while ATM- and CHEK2-altered cancers generally show little or no benefit from PARP inhibitor treatment [3, 7, 8], with some exceptions. Moreover, even a significant proportion of BRCA2-mutated prostate cancers do not demonstrate clinical responses to PARP inhibition [3, 7]. Conversely, a small number of mCRPC patients without detectable HR gene mutations may experience a clinical benefit from PARP inhibitor treatment [2, 9]. Therefore, having a functional readout of a cancer’s HR status (i.e., proficient versus deficient) might prove useful to better select patients for PARP inhibitor treatment—or to exclude them from this therapy—if validated in clinical trials.
One such readout, the homologous recombination deficiency (HRD) score [10], is derived by measuring genome-wide loss-of-heterozygosity (LOH), telomeric allelic imbalance, and large-scale state transitions using targeted somatic next-generation DNA sequencing to estimate the extent of underlying genomic scarring due to HR deficiency. However, this assay has been understudied in prostate cancer. To explore the HRD score in prostate cancer, we chose to focus on patients treated at Johns Hopkins with known germline mutations in BRCA2, ATM, and CHEK2. These are the three most commonly altered DNA-repair genes in prostate cancer, both at the germline and somatic levels, collectively accounting for roughly three-quarters of all “DNA repairome” mutations in this disease [1, 11]. More specifically, germline mutations in BRCA2, ATM, and CHEK2 are found in 5–6%, 1–2%, and 1–2% of mCRPC patients, respectively [11, 12] (with the prevalence of each being roughly double at the somatic level). These genes are also particularly relevant because they are all included in the molecular eligibility criteria of virtually all PARP inhibitor trials involving mCRPC patients [13].
Given the different response rates to PARP inhibitors among the three groups, we hypothesized that patients with germline BRCA2 mutations would harbor primary tumors with the highest HRD scores, that patients with germline CHEK2-altered prostate cancers would have the lowest HRD scores (similar to wild-type cases), and that germline ATM-altered cancers would have intermediate HRD scores. We also hypothesized that biallelic inactivation would be most common for BRCA2 and least common for CHEK2, and that prostate cancers with biallelic mutations would have higher HRD scores than those with mono-allelic mutations. Finally, we examined HRD scores and their relationship to underlying HR gene mutations in two independent primary prostate tumor cohorts to determine if our findings were generalizable to the broader sporadic prostate cancer population.
Materials and methods
Patients and tissue samples
This study was conducted with Johns Hopkins institutional review board approval, and in accordance with the US common rule under a waiver of consent. Six patient sets were included: (1) The first set included 17 primary prostate tumors from Johns Hopkins University (JHU) with available radical prostatectomy tissue and known pathogenic germline mutations in BRCA2. Of these, nine cases had germline BRCA2 mutations detected during sequencing of benign seminal vesicle or leukocyte DNA performed as a part of previously-described studies [14] or inferred based on variant-allele fraction in tumor sequencing studies [15], while the remaining eight had BRCA2 mutations detected using clinical-grade germline sequencing platforms (Invitae, or Color Genomics) from saliva samples [8]. (2) The second patient set included primary prostate tumors with available radical prostatectomy tissue at JHU with known pathogenic germline (n = 21) or somatic mutations in ATM (n = 11), described previously [16]. (3) The third patient set included 15 JHU radical prostatectomy cases with germline CHEK2 mutations that were either pathogenic or likely pathogenic. Of these, 11 were detected during sequencing of benign seminal vesicle or leukocyte DNA performed as a part of previously-described studies [14] or based on variant-allele fraction in tumor sequencing studies [15], while four were discovered using clinical-grade germline sequencing platforms as above. (4) The fourth patient set was derived from re-analysis of the TCGA PRAD study, which has been previously described [17]. (5) The fifth set included patients collected through the French genetic cohort PROGENE (CeRePP). Samples selected were from patients who subsequently developed metastatic disease after radical prostatectomy [18]. (6) The last set was a previously-published dataset of 167 high-grade ovarian serous carcinomas with available HRD scores [19], which was used as a non-prostate cancer comparator.
DNA isolation
On standard histologic sections, tumor tissue was macro-dissected, guided by hematoxylin-and-eosin staining. 5 × 10 μm sections from formalin-fixed paraffin-embedded (FFPE) tumor samples were used for DNA extraction. Sections from FFPE tissue were first incubated in Proteinase K followed by DNA extraction performed using the Promega Maxwell 16 LEV FFPE Plus kit (AS1290, Promega, Madison, WI) according to the manufacturer’s instructions.
Targeted next-generation sequencing (NGS)
The HRD Plus assay (Myriad Genetics, Salt Lake City, UT) has been previously described in detail for BRCA1/2 sequencing [20]. Briefly, the assay uses a custom method employing IDT’s xGen hybridization-capture technology (Integrated DNA Technologies, Coralville, IA). A custom enrichment panel was developed, which targets 54,091 single-nucleotide polymorphisms (SNP) distributed across the complete human genome. The panel also includes an additional 490 probes targeting the complete coding region of BRCA2, ATM, CHEK2, and TP53. A detailed description of the panel design and development is provided in Timms et al. [21] and the assay process is described in Patel et al. [20]. DNA sequencing was performed on an Illumina HiSeq2500 using a 200 cycle HiSeq Rapid SBS Kit v2 and a HiSeq Rapid PE Cluster Kit v2.
DNA sequence analysis and interpretation
Average coverage alignment to the target regions and removal of non-clonal reads was ATM: 682 (range: 142–1307); BRCA2: 609 (148–1511); CHEK2: 589 (115–1154). Novel variants identified by tumor sequencing using the HRD Plus assay were classified using a process which is consistent with the published standards and guidelines for clinical testing from the American College of Medical Genetics and Genomics [22]. Variants are classified into one of five categories: deleterious, suspected deleterious, variant of uncertain significance, favor polymorphism, and polymorphism. Both deleterious and suspected deleterious variants were considered pathogenic here. In the case of TP53, pathogenic or likely pathogenic inactivating point mutations (missense, nonsense, frameshift, splice-site variants), as well as large-scale rearrangements or homozygous deletions were classified as deleterious. Variant classifications are stored in a classification database and can be retrieved each time they are observed during routine testing.
Calculation of HRD scores (with a range of 0 to 100) from the HRD Plus assay is described in Timms et al. [21]. In the TCGA cohort, HRD scores were calculated from SNP microarray data (Affymetrix GenomeWideSNP6 array) from frozen, rather than FFPE, tissue. Both tumor and corresponding normal tissue was analyzed. Probes on the arrays were used for determination of copy number, and SNP data was used to calculate allele dosage. Data from normal samples was used to generate parameters for analysis. For each probe on the microarray the signal per probe was adjusted on the average signal intensity per sample. Next the adjustment coefficient was calculated so that on average among normal samples the signal from each probe was equal to 2 (expected copy number among normal samples). Analysis of SNPs on the array was similar to the analysis performed for the NGS assay. Signals from both alleles were also adjusted on the average signal intensity per normal sample. Two parameters were calculated: both relative and absolute signal intensity of different alleles. The goal was to obtain an average signal intensity of 2 for all genotypes and average allele dosage of 0.5 for genotype AB in the normal tissue samples. The probes and SNPs which were the least reliable were removed from the analysis. Tumor samples were then analyzed and HRD scores generated using the same analysis process as was used for the NGS assay. Our previous studies comparing HRD scores from paired frozen tissues on microarray vs. FFPE tissues by NGS have shown that highly concordant results are obtained with both assays [21].
Immunohistochemistry
ERG protein immunohistochemistry was performed as previously described on standard histologic slides or tissue microarray spots using a rabbit monoclonal anti-ERG antibody (Clone EPR3864; Ventana Medical Systems) [23], and was scored dichotomously using a previously genetically-validated scoring system [24].
Statistical analysis
Statistical analyses are mainly descriptive, and were performed using GraphPad Prism 8.4 software. HRD scores were compared using the Mann–Whitney U test, or the Kruskal–Wallis test where multiple comparisons were made.
Results
HRD score in prostate cancers with germline mutations in BRCA2, ATM, or CHEK2
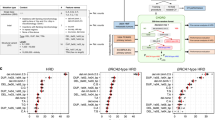
We first examined a JHU cohort of primary prostate cancers from men with known pathogenic germline mutations in BRCA2 (n = 17) found on clinical- or research-grade germline sequencing platforms. In total, 82% (n = 14) had evaluable HRD scores, with a median of 27 (Fig. 1A, Supplementary Table S1). Of these, 71% (10/14) had apparent biallelic alterations in BRCA2, based on presence of LOH or two detectable pathogenic alterations, and these cases had a median HRD score of 29 compared to a median of 23 for cases lacking evidence of biallelic alteration (p = 0.17; Supplementary Fig. S1).

A HRD scores in JHU cohort of patients by BRCA2, ATM, and CHEK2 status. B HRD scores in TCGA cohort of primary prostate tumors by TP53, ATM, and BRCA2 status. C HRD scores in PROGENE cohort by TP53, ATM, and BRCA2 status. D HRD scores in ovarian cancer cohort [19] by BRCA1 and BRCA2 status.
Next, we queried HRD score in a previously-reported cohort of primary prostate cancers from men with known pathogenic germline mutations in ATM (n = 21), of which 16 (76%) had evaluable HRD scores (Supplementary Table S2) [16]. In this group, the median HRD score was 16.5, which was significantly lower than that observed in the germline BRCA2-altered cohort (adjusted p = 0.029; Fig. 1A). Among cases with assessable LOH status, 53% (8/15) had presumed biallelic ATM inactivation based on presence of LOH or two pathogenic alterations. As with BRCA2 cases, cases with biallelic ATM inactivation had numerically higher HRD scores compared to those without evidence of biallelic inactivation (median HRD scores 20.5 vs. 12, p = 0.058; Supplementary Fig. S2A). We previously validated and reported ATM immunohistochemistry results in a partially overlapping cohort of patients [16], and 75% of the cases here with evaluable HRD scores had ATM protein loss. Consistent with the previously-reported enrichment of cases with biallelic ATM loss among the group with ATM protein loss [16], we found that cases with ATM protein loss similarly had numerically higher HRD scores compared to those without protein loss (median HRD score of 20 vs. 10.5, p = 0.057; Supplementary Fig. S2B).
In the aforementioned ATM immunohistochemistry study [16], we also reported 11 cases with somatic ATM mutation (detected by loss of ATM protein on immunohistochemistry and confirmed on tumor DNA sequencing, Supplementary Table S3). Of 10 evaluable for HRD score, the median score was 19.5 (Fig. 1A), similar to that seen with germline ATM mutations (adjusted p > 0.99), and 75% of these with evaluable LOH status (6/8) were presumed biallelic alterations.
Next, we examined 15 cases with germline CHEK2 alterations. Given the low frequency of these alterations, we included both deleterious and suspected deleterious CHEK2 mutations in our study (Supplementary Table S4). Notably, all cases were presumed to be mono-allelic, based on lack of LOH and absence of additional somatic CHEK2 mutations. Overall, 87% (13/15) of these cases had evaluable HRD scores, with a median score of 9, which was significantly lower than BRCA2-mutated cases (adjusted p < 0.0001) but not significantly different from ATM germline (adjusted p = 0.46) or ATM somatic mutation cases (adjusted p = 0.40) (Fig. 1A).
HRD score in primary prostate cancers without HR pathway gene mutations
Next, we sought to explore the baseline HRD score for primary prostate cancers without germline HR pathway gene mutations by examining the TCGA primary prostate tumor cohort [17]. There were 385 TCGA prostate cancer cases without BRCA2, ATM, or CHEK2 mutations, and these demonstrated a median HRD score of 11 (Fig. 1B). Cases with pathogenic BRCA2 (n = 1) and ATM mutations (n = 5) had numerically higher median HRD scores compared to those without HR gene mutations; however, the very small numbers of BRCA2/ATM-altered cases precluded meaningful comparisons in this cohort. TCGA cases without BRCA2, ATM, or CHEK2 mutations had significantly lower HRD scores compared to JHU cases with BRCA2 mutations (adjusted p < 0.0001) or somatic ATM mutations (adjusted p = 0.039), but were not significantly different from JHU cases with germline ATM mutations (adjusted p = 0.136) or germline CHEK2 mutations (adjusted p > 0.99) Unexpectedly, when exploring the association of other common somatic genomic alterations with HRD score, we observed that among cases lacking BRCA2/ATM/CHEK2 mutations, those with pathogenic TP53 mutations (n = 12) had significantly higher HRD scores compared to cases without TP53 mutation (n = 373) (median HRD score of 17 vs. 11, p = 0.015).
We then validated these findings in the TCGA cohort using an independent PROGENE cohort of 102 primary prostate cancers from patients who subsequently developed metastases [18] (Fig. 1C). The median HRD score for cases lacking BRCA2, ATM, or CHEK2 mutations was 18 in this cohort, which was significantly higher than that seen in the TCGA cohort (adjusted p = 0.0002), potentially due to the selection for subsequently metastatic tumors in the PROGENE cohort. As observed in the TCGA cohort, tumors with BRCA2 (n = 6) or ATM mutations (n = 3) had higher median HRD scores than those without such HR gene mutations, though the low numbers precluded meaningful statistical comparisons. Notably, and consistent with our prior observation, among cases lacking BRCA2/ATM/CHEK2 mutations, the median HRD score was significantly higher for those with TP53 mutations (n = 27; median HRD = 24) compared to those without (n = 66; median HRD = 16) (p = 0.0013).
In order to explore differences in HRD scores among BRCA-associated prostate and non-prostate cancers, we compared HRD scores observed in primary prostate cancer with those from a previously-published cohort of high-grade serous ovarian carcinoma [19], another tumor type with a high prevalence of HR gene mutations. Including only cases with TP53 mutations (94% of the cohort) [25], the median HRD score for cases lacking BRCA2, ATM, or CHEK2 mutations (n = 133) was 28, compared to 55 for case with pathogenic BRCA2 mutations (n = 15; p = 0.0006) and 65 for those with pathogenic BRCA1 mutations (n = 19; p < 0.0001) (Fig. 1D).
Associations between HRD scores and Gleason grade, ERG fusion status, androgen receptor activity, and percent genome altered
We also sought to examine potential correlations between HRD scores and a number of other histologic and molecular parameters. To do so, we used data from the TCGA cohort. First, we explored the relationship between Gleason grade and HRD scores. We found that there was a significant association between HRD and Gleason Grade Group, both in the overall TCGA cohort and in the subset lacking HR gene and TP53 mutations (Supplementary Fig. S3A, B). This is consistent with previous reports by our group and others suggesting that prostate cancers with HR deficiency are more likely to demonstrate higher Gleason grades [16, 26].
Next, we assessed the percentage of the genome altered (PGA), defined as the fraction of the genome (from whole-exome sequencing analysis) that is affected by copy number gains or losses, as previously described [27]. To this end, we observed a significant correlation between HRD scores and PGA in both the overall TCGA cohort and the subset lacking HR gene and TP53 mutations (Supplementary Fig. S3C, D). However, although these two measures of genomic instability are broadly correlated, they are not identical; in particular, there were a number of cases with high PGA and low HRD (scores of <20).
We then assessed the relationship between ERG gene fusions and HRD scores, due to prior preclinical reports suggesting that presence of ERG fusions may sensitize to PARP inhibition in prostate cancer [28], although this has not been borne out in prospective clinical trials [29]. Using the TCGA cohort, we found (paradoxically) that ERG-fusion positive tumors had significantly lower HRD scores than ERG-negative tumors, both in the whole TCGA cohort and in the subset of cases without HR gene or TP53 mutations (Supplementary Fig. S3E, F). However, in the JHU cohorts, there were no significant associations between ERG-fusion status as determined by immunohistochemistry and HRD scores in prostate tumors from patients with germline ATM or BRCA2 mutations (Supplementary Fig. S4).
Finally, we aimed to understand the relationship between AR signaling and HRD scores in primary prostate cancers. While we did not detect any activating AR gene mutations in any of the three cohorts (as expected in this hormone-naïve population), we were able to evaluate androgen receptor activity (AR-A) transcription signatures using the weighted gene expression of nine AR-responsive genes, as previously described [30]. Thus, using the HRD scores from the TCGA cohort generated here and the matching AR-A scores from the same cohort generated previously [30], we observed a significant negative correlation between AR-A scores and HRD scores, both in the overall TCGA cohort and in the subset lacking HR gene and TP53 mutations (Supplementary Fig. S3G, H).
HRD score and efficacy of PARP inhibitors among prostate cancers with germline BRCA2 mutations
Finally, we explored (in a very preliminary fashion) the potential association between HRD scores and clinical outcomes to PARP inhibitors in a group of patients with pathogenic BRCA2 mutations who received olaparib therapy. To this end, 8 of the BRCA2-altered patients included in the present study (Supplementary Table S1) with evaluable HRD scores had received olaparib treatment as part of routine clinical practice, and their clinical characteristics are summarized in Supplementary Table S5. In these patients, we analyzed best PSA responses (proportional PSA decrease compared to baseline) and clinical/radiographic progression-free survival (PFS; time to clinical or radiographic progression or death, whichever occurred first) after stratifying the group above or below the median HRD score (which was 25 among these eight patients). Accordingly, in the four patients with HRD scores above this median, the mean PSA reduction was 93% (range, 84–100% reduction) and the median PFS was 14.9 months (range, 10.1–19.8 months). Conversely, among the four patients with HRD scores below the median, the mean PSA reduction was 91% (range, 89–93% reduction) and the median PFS was only 9.9 months (range, 6.8–11.0 months). This preliminary exploratory analysis suggests that higher HRD scores may potentially be associated with improved clinical outcomes to PARP inhibition in prostate cancer, but must be further substantiated.
Discussion
The recent FDA approval of rucaparib for mCRPC patients with germline or somatic BRCA1/2 mutations, and of olaparib for mCRPC patients with mutations in one of 14 HR-related genes, heralds a new era for the precision treatment of advanced prostate cancer. Similar FDA approvals are soon expected for other PARP inhibitors, including niraparib and talazoparib. While these agents represent a welcome addition to the prostate cancer therapeutic arsenal, the current method of selection of patients for these drugs—based on the presence or absence of a germline/somatic mutation in a particular HR gene—is likely imperfect. Pan-cancer analyses have demonstrated that only biallelic inactivation of HR genes leads to mutational signatures and genomic scarring consistent with underlying DNA-repair deficiency [31, 32]. Importantly, evidence of biallelic alteration is present in only 70–80% of prostate cancers with germline BRCA2 mutations [31], consistent with estimates that as many as 20% of prostate cancers in germline BRCA2-mutation carriers are sporadic and unrelated to HR deficiency [33]. Thus, selection of prostate cancer patients for PARP inhibitor therapy using only germline mutation data is likely to include a significant proportion of patients who may not benefit from this approach; and even with somatic sequencing, it can be difficult to assess LOH status in a significant proportion of cases as seen in the current study. In addition, not all HR gene mutations result in identical consequences for DNA repair; some may not be associated with PARP inhibitor sensitivity at all [34]. Moreover, it is clear that some prostate cancer patients without apparent HR gene mutations may have underlying genomic scarring consistent with deficient DNA-repair processes, potentially signifying PARP inhibitor sensitivity [35]. In this context, additional methods to quantify functional HR status [36, 37] could help to further refine optimal selection of mCRPC patients for treatment with PARP inhibitors, if validated in prospective clinical trials. Of note, since HRD scores are not a functional readout of HR deficiency/proficiency, they might erroneously predict PARP inhibitor sensitivity in some contexts (e.g., in BRCA2-associated cancers in which reversion mutations have occurred that restore the open reading frame of BRCA2) where a functional HR assay could prove to be more useful. Finally, the use of HRD assays may also have utility in other contexts, such as the use of platinum-based chemotherapies for mCRPC [38] or other synthetic-lethal approaches.
Here, we show that HRD scores vary significantly in prostate cancers from patients harboring germline mutations in BRCA2, ATM, and CHEK2. Assessed using a targeted NGS panel in a CLIA-accredited lab, HRD scores were highest in tumors from germline BRCA2-altered patients, intermediate in germline ATM-altered patients, and lowest in germline CHEK2-altered patients (akin to wild-type prostate cancers). Further, while HRD scores were generally higher in tumors with biallelic (compared to mono-allelic) inactivation of BRCA2 or ATM, the prevalence of biallelic mutations did not significantly differ in these two patient cohorts. In other words, the higher HRD scores in BRCA2-mutated prostate cancers (compared to ATM-mutated cancers) could not be explained simply by a higher proportion of biallelic events in the former versus the latter, although this may contribute in some contexts. Remarkably, none of the germline CHEK2-altered cases showed biallelic mutations in tumor tissue, and these had lower HRD scores. These findings were generally replicated in two independent sporadic prostate cancer cohorts, where BRCA2 mutations were associated with numerically higher HRD scores than ATM mutations, and both groups had higher HRD scores than wild-type (i.e., BRCA2/ATM/CHEK2 negative) prostate cancers.
An unexpected finding of our study was that prostate cancers harboring somatic TP53 mutations demonstrated higher HRD scores than wild-type cases in both of the independent cohorts examined. TP53 mutations in prostate cancer are associated with other hallmarks of genomic instability, such as chromothripsis [39], perhaps partially explaining this phenomenon. Unfortunately, it was not possible for us to evaluate the effect of TP53 mutations on HRD scores in the ovarian cancer cohort, since virtually all cases of that disease are characterized by TP53 inactivation [25]. Although the co-existence of BRCA1/2 and TP53 mutations in the vast majority of ovarian cancers might contribute to the higher median HRD scores in that tumor type when compared to BRCA2-altered prostate cancers, ovarian tumors also harbor many other non-BRCA1/2 HR pathway gene mutations that almost certainly increase the HRD scores among BRCA1/2 wild-type patients [40]. In prostate cancer, it is tempting to speculate that the co-occurrence of BRCA2 and TP53 mutations might portend a more favorable prognosis in the context of PARP inhibitor treatment, although preliminary clinical data do not suggest that mCRPC patients with TP53 mutations are more sensitive to PARP inhibition [29, 41]. Finally, in contrast to BRCA1/2, TP53 and ATM mutations are mutually exclusive in many tumor types [42,43,44,45], which may also contribute to the relatively lower HRD scores in ATM-mutated tumors.
Our data on HRD scores are concordant with the observed clinical activity of PARP inhibitors in prostate cancer. While BRCA2-altered mCRPC patients derive a clear and consistent benefit from both olaparib and rucaparib [2, 3, 5,6,7], the efficacy of these and other PARP inhibitors in men with ATM mutations is more modest [3, 4, 8], and in those with CHEK2 mutations is low with some rare exceptions [3, 4]. The “synthetic lethality” hypothesis [46] predicts greatest efficacy of PARP inhibitors in cancers with biallelic HR gene deficiency, especially biallelic inactivation of genes directly involved in mediating HR DNA repair. The fact that BRCA2-mutated prostate cancers have higher HRD scores than ATM-mutated cancers which, in turn, have higher HRD scores than CHEK2-mutated cancers might be one plausible (yet simplistic) explanation for the clinical effects seen. Furthermore, the notion that even among BRCA2-altered prostate cancers, those with the highest HRD scores may respond the best to PARP inhibitor therapy (as suggested by our preliminary clinical findings) implies that knowledge about HR functional status may serve as a treatment-selection marker. This hypothesis remains to be confirmed, and should be explored in the context of one or more of the completed or ongoing clinical trials.
Our study has a number of limitations. First, HR gene mutations were assessed using a panel-based or whole-exome sequencing approach, which may miss complex somatic genomic rearrangements or alternative mechanisms of HR gene inactivation, such as methylation. Thus, it is possible that some cases classified as lacking HR gene mutations in the TCGA and PROGENE cohorts may have actually harbored underlying HR gene inactivation and we may have misclassified some cases with biallelic alterations among the JHU cohort. We were also unable to study HRD scores in prostate cancer samples with other important HR gene mutations such as BRCA1, PALB2 or RAD51, due to the relative rarity of these alterations in prostate cancer. Second, we acknowledge that our analysis of HRD scores and clinical outcomes to PARP inhibition is based on very small numbers of patients (n = 8) using primary tumor samples rather than metastatic samples for determination of HRD scores. Accordingly, we lacked statistical power to derive meaningful conclusions, and were we unable to define optimal HRD cut-points for clinical use (we simply stratified the cohort by the median HRD score in order to maximize the number of patients in each group). Therefore, these clinical data should only be considered hypothesis-generating, and require confirmation in prospective clinical trials.
In conclusion, optimal treatment selection towards or against PARP inhibitor use (and potentially also platinum-based chemotherapies) in prostate cancer requires further refinement [47]. While the selection of patients based on the presence or absence of a particular HR gene mutation (e.g., BRCA2) is a good starting point—and has led to FDA approval of two PARP inhibitors—this only represents the beginning of the precision oncology era in prostate cancer. We propose that HRD assays may augment clinical utility, and deserve further exploration and prospective validation. We envision that such HRD assays would be used in conjunction with, and not instead of, germline and somatic genetic analysis of the key HR genes. The ability to further tailor our treatment recommendations based on additional validated genomic or transcriptomic functional assays remains our challenge for the future.
References
Chung JH, Dewal N, Sokol E, Mathew P, Whitehead R, Millis SZ, et al. Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. JCO Precis Oncol. 2019;3. https://doi.org/10.1200/PO.18.00283.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–708.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091–102.
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res. 2020;26:2487–96.
Smith MR, Sandhu SK, Kelly WK, Scher HI, Efstathiou E, Lara P, et al. Phase II study of niraparib in patients with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD): preliminary results of GALAHAD. J Clin Oncol. 2019;37. https://doi.org/10.1200/JCO.2019.37.7_suppl.202.
Bono JSD, Mehra N, Higano CS, Saad F, Buttigliero C, Mata M, et al. TALAPRO-1: a phase II study of talazoparib (TALA) in men with DNA damage repair mutations (DDRmut) and metastatic castration-resistant prostate cancer (mCRPC)—First interim analysis (IA). J Clin Oncol. 2020;38. https://doi.org/10.1200/JCO.2020.38.6_suppl.119.
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. 2020;38:3763–72.
Marshall CH, Sokolova AO, McNatty AL, Cheng HH, Eisenberger MA, Bryce AH, et al. Differential response to olaparib treatment among men with metastatic castration-resistant prostate cancer harboring BRCA1 or BRCA2 versus ATM mutations. Eur Urol. 2019;76:452–8.
Antonarakis ES, Wang H, Teply BA, Kelly WK, Willms J, Sullivan R, et al. Interim results from a phase 2 study of olaparib (without ADT) in men with biochemically-recurrent prostate cancer after prostatectomy, with integrated biomarker analysis. J Clin Oncol. 2019;37. https://doi.org/10.1200/JCO.2019.37.15_suppl.5045.
Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous recombination deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22:3764–73.
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–53.
Nicolosi P, Ledet E, Yang S, Michalski S, Freschi B, O’Leary E, et al. Prevalence of germline variants in prostate cancer and implications for current genetic testing guidelines. JAMA Oncol. 2019;5:523–8.
Marshall CH, Antonarakis ES. Therapeutic targeting of the DNA damage response in prostate cancer. Curr Opin Oncol. 2020;32:216–22.
Na R, Zheng SL, Han M, Yu H, Jiang D, Shah S, et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71:740–7.
Velho PI, Lim D, Wang H, Park JC, Kaur HB, Almutairi F, et al. Molecular characterization and clinical outcomes of primary gleason pattern 5 prostate cancer after radical prostatectomy. JCO Precis Oncol. 2019;3. https://doi.org/10.1200/PO.19.00081.
Kaur H, Salles DC, Murali S, Hicks JL, Nguyen M, Pritchard CC, et al. Genomic and clinical-pathologic characterization of ATM-deficient prostate cancer. Clin Cancer Res. 2020;26:4869–81.
Cancer Genome Atlas Research N. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–25.
Leon P, Cancel-Tassin G, Drouin S, Audouin M, Varinot J, Comperat E, et al. Comparison of cell cycle progression score with two immunohistochemical markers (PTEN and Ki-67) for predicting outcome in prostate cancer after radical prostatectomy. World J Urol. 2018;36:1495–500.
Stronach EA, Paul J, Timms KM, Hughes E, Brown K, Neff C, et al. Biomarker assessment of HR deficiency, tumor BRCA1/2 mutations, and CCNE1 copy number in ovarian cancer: associations with clinical outcome following platinum monotherapy. Mol Cancer Res. 2018;16:1103–11.
Patel JN, Braicu I, Timms KM, Solimeno C, Tshiaba P, Reid J, et al. Characterisation of homologous recombination deficiency in paired primary and recurrent high-grade serous ovarian cancer. Br J Cancer. 2018;119:1060–6.
Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16:475–83.
Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23.
Morais CL, Herawi M, Toubaji A, Albadine R, Hicks J, Netto GJ, et al. PTEN loss and ERG protein expression are infrequent in prostatic ductal adenocarcinomas and concurrent acinar carcinomas. Prostate. 2015;75:1610–9.
Chaux A, Albadine R, Toubaji A, Hicks J, Meeker A, Platz EA, et al. Immunohistochemistry for ERG expression as a surrogate for TMPRSS2-ERG fusion detection in prostatic adenocarcinomas. Am J Surg Pathol. 2011;35:1014–20.
Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156.
Guedes LB, Antonarakis ES, Schweizer MT, Mirkheshti N, Almutairi F, Park JC, et al. MSH2 loss in primary prostate cancer. Clin Cancer Res. 2017;23:6863–74.
Lalonde E, Ishkanian AS, Sykes J, Fraser M, Ross-Adams H, Erho N, et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 2014;15:1521–32.
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19:664–78.
Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol. 2018;36:991–9.
Spratt DE, Alshalalfa M, Fishbane N, Weiner AB, Mehra R, Mahal BA, et al. Transcriptomic heterogeneity of androgen receptor activity defines a de novo low AR-active subclass in treatment naive primary prostate cancer. Clin Cancer Res. 2019;25:6721–30.
Jonsson P, Bandlamudi C, Cheng ML, Srinivasan P, Chavan SS, Friedman ND, et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature. 2019;571:576–9.
Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet. 2017;49:1476–86.
Hughley R, Karlic R, Joshi H, Turnbull C, Foulkes WD, Polak P. Etiologic index: a case-only measure of BRCA1/2-associated cancer risk. N Engl J Med. 2020;383:286–8.
Poti A, Gyergyak H, Nemeth E, Rusz O, Toth S, Kovacshazi C, et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019;20:240–52.
Sztupinszki Z, Diossy M, Krzystanek M, Borcsok J, Pomerantz MM, Tisza V, et al. Detection of molecular signatures of homologous recombination deficiency in prostate cancer with or without BRCA1/2 mutations. Clin Cancer Res. 2020;26:2673–80.
Galbiati A, Beausejour C, d’Adda, di Fagagna F. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell. 2017;16:422–7.
Guidugli L, Carreira A, Caputo SM, Ehlen A, Galli A, Monteiro AN, et al. Functional assays for analysis of variants of uncertain significance in BRCA2. Hum Mutat. 2014;35:151–64.
Pomerantz MM, Spisak S, Jia L, Cronin AM, Csabai I, Ledet E, et al. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer. 2017;123:3532–9.
Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, et al. Genomic hallmarks and structural variation in metastatic prostate. Cancer Cell. 2018;174:758–69.
Lheureux S, Lai Z, Dougherty BA, Runswick S, Hodgson DR, Timms KM, et al. Long-term responders on olaparib maintenance in high-grade serous ovarian cancer: clinical and molecular characterization. Clin Cancer Res. 2017;23:4086–94.
Markowski MC, Antonarakis ES. BRCA1 versus BRCA2 and PARP inhibitor sensitivity in prostate cancer: more different than alike? J Clin Oncol. 2020;38:3735–9.
Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J Natl Cancer Inst. 2018;110:1030–4.
Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–8.
Greiner TC, Dasgupta C, Ho VV, Weisenburger DD, Smith LM, Lynch JC, et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc Natl Acad Sci USA. 2006;103:2352–7.
Pettitt AR, Sherrington PD, Stewart G, Cawley JC, Taylor AM, Stankovic T. p53 dysfunction in B-cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood. 2001;98:814–22.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Markowski MC, Antonarakis ES. PARP inhibitors in prostate cancer: time to narrow patient selection? Expert Rev Anticancer Ther. 2020;20:523–6.
Acknowledgements
This work was supported by the Patrick C. Walsh Prostate Cancer Research Fund (ESA, TLL), the Prostate Cancer Foundation (ESA), NIH/NCI Prostate SPORE P50 CA58236; and the NCI Cancer Center Support Grant 5P30 CA006973-52. ALR is supported by a grant from the Breast Cancer Research Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
TLL has received research support from Roche/Ventana Medical Systems and DeepBio for other studies. ESA has served as a paid consultant/advisor for Janssen, Pfizer, Sanofi, Dendreon, Merck, Bristol-Myers Squibb, AstraZeneca, Clovis, Eli Lilly and Amgen; has received research funding to his institution from Janssen, Johnson & Johnson, Sanofi, Dendreon, Genentech, Novartis, Merck, Bristol-Myers Squibb, AstraZeneca, and Constellation; and is a co-inventor of an AR-V7 biomarker technology that has been licensed to Qiagen. JSL and KMT are employees and shareholders in Myriad Genetics. RB has received research support from Myriad Genetics and AstraZeneca.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lotan, T.L., Kaur, H.B., Salles, D.C. et al. Homologous recombination deficiency (HRD) score in germline BRCA2- versus ATM-altered prostate cancer. Mod Pathol 34, 1185–1193 (2021). https://doi.org/10.1038/s41379-020-00731-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00731-4
This article is cited by
-
A novel targeted NGS panel identifies numerous homologous recombination deficiency (HRD)-associated gene mutations in addition to known BRCA mutations
Diagnostic Pathology (2024)
-
Combination of genomic instability score and TP53 status for prognosis prediction in lung adenocarcinoma
npj Precision Oncology (2023)
-
Integrated analyses reveal the prognostic, immunological features and mechanisms of cuproptosis critical mediator gene FDX1 in KIRC
Genes & Immunity (2023)
-
HRD effects on first-line adjuvant chemotherapy and PARPi maintenance therapy in Chinese ovarian cancer patients
npj Precision Oncology (2023)
-
Innovative molecular subtypes of multiple signaling pathways in colon cancer and validation of FMOD as a prognostic-related marker
Journal of Cancer Research and Clinical Oncology (2023)
