
Abstract
Alveolar Rhabdomyosarcoma (ARMS) is an aggressive pediatric cancer with about 80% of cases characterized by either a t(1;13)(p36;q14) or t(2;13)(q35;q14), which results in the formation of the fusion oncogenes PAX7-FOXO1 and PAX3-FOXO1, respectively. Since patients with fusion-positive ARMS (FP-RMS) have a poor prognosis and are treated with an aggressive therapeutic regimen, correct classification is of clinical importance. Detection of the translocation by different molecular methods is used for diagnostics, including fluorescence in situ hybridization and RT-PCR or NGS based approaches. Since these methods are complex and time consuming, we developed specific monoclonal antibodies (mAbs) directed to the junction region on the PAX3-FOXO1 fusion protein. Two mAbs, PFM.1 and PFM.2, were developed and able to immunoprecipitate in vitro-translated PAX3-FOXO1 and cellular PAX3-FOXO1 from FP-RMS cells. Furthermore, the mAbs recognized a 105 kDa band in PAX3-FOXO1-transfected cells and in FP-RMS cell lines. The mAbs did not recognize proteins in fusion-negative embryonal rhabdomyosarcoma cell lines, nor did they recognize PAX3 or FOXO1 alone when compared to anti-PAX3 and anti-FOXO1 antibodies. We next evaluated the ability of mAb PFM.2 to detect the fusion protein by immunohistochemistry. Both PAX3-FOXO1 and PAX7-FOXO1 were detected in HEK293 cells transfected with the corresponding cDNAs. Subsequently, we stained 26 primary tumor sections and a rhabdomyosarcoma tissue array and detected both fusion proteins with a positive predictive value of 100%, negative predictive value of 98%, specificity of 100% and a sensitivity of 91%. While tumors are stained homogenously in PAX3-FOXO1 cases, the staining pattern is heterogenous with scattered positive cells only in tumors expressing PAX7-FOXO1. No staining was observed in stromal cells, embryonal rhabdomyosarcoma, and fusion-negative rhabdomyosarcoma. These results demonstrate that mAbs specific for the chimeric oncoproteins PAX3-FOXO1 and PAX7-FOXO1 can be used efficiently for simple and fast subclassification of rhabdomyosarcoma in routine diagnostics via immunohistochemical detection.
Similar content being viewed by others

Introduction
Chromosomal translocations in carcinogenesis can result in alterations of gene structure including truncations and the formation of novel chimeric genes. Many chimeric genes have been described as important contributors to the tumorigenesis of leukemias and sarcomas, and show striking associations with distinct prognostic subgroups.
Rhabdomyosarcoma (RMS) is the most common pediatric soft tissue sarcoma accounting for about 5–8% of all childhood malignancies [1]. Based on the latest WHO-defined histological criteria, RMS tumors are further subdivided into four different subgroups, including embryonal RMS (ERMS), spindle cell/sclerosing RMS, alveolar RMS (ARMS) and pleomorphic RMS, with the latter one only found in the adult population [2]. Most ARMS are characterized by the presence of translocations t(2;13)(q35;q14) or t(1;13)(p36;q14) [3]. These translocations result in fusion genes that generate chimeric proteins containing the DNA-binding domains of PAX3 or PAX7 and the strong transcriptional transactivation domain from FOXO1 [4]. Recently, RMS have been molecularly classified as either fusion-positive (FP-RMS) or negative (FN-RMS) characterized by the presence or absence of PAX3 or PAX7 fusions, respectively [3, 5]. Recent genomic and epigenetic studies have demonstrated that FP-RMS have a low mutational burden and that the PAX3/7-fusion gene is the main oncogenic driver of the disease [3, 5]. It acts as strong transactivator to induce a myogenic program [6] through epigenetic reprograming of the cells locking them in a myoblastic state [7]. The fusion gene is the initiating oncogenic event, and underscores the importance of this chimeric oncogene in FP-RMS.
Molecular subclassification is important in risk stratification of RMS as patients with FP-RMS often present with metastatic disease and have a worse outcome than FN-RMS [8]. Determination of the fusion status is therefore of great importance for RMS therapy. Different methods for direct detection of the translocations are used in the clinics. These include RT-PCR detection of the fusion transcript, FISH detection of FOXO1 rearrangements and cytogenetic detection of the chromosomal translocations. However, these methods are all relatively complex and time consuming when compared to standard immunohistochemistry used in routine tumor diagnostics.
In the present study, we describe the development and characterization of novel monoclonal antibodies (mAbs) specific for the PAX3/7-FOXO1 oncoprotein and evaluate the suitability of immunohistochemical detection of the PAX-FOXO1 fusion protein using this breakpoint specific antibody. These mAbs also serve as valuable tools to study the function of PAX3/7-FOXO1 in the oncogenesis of ARMS.
Materials and methods
Cells
Five FP-RMS cell lines RH-4, RH-5, RH-28, RH-30, RMS-13 (derived from the same patient as RH-30) and two FN-RMS lines, RD and CTR were previously described [9]. The human cervical carcinoma cell line HeLa, the murine fibroblast cell line NIH-3T3, the murine myeloma cell line P3x63Ag8.653, and the human embryonic kidney cell line HEK293T were obtained from ATCC (Manassas, VA). The cell lines were maintained in RPMI-1640 or DME media supplemented with 10% FBS, 1mM l-glutamine and penicillin/streptomycin. Hybridomas and the myeloma cell line P3x63Ag8.653 were maintained in HY media (DMEM supplemented with 10% FBS, 10% NCTC-109, 10 mM HEPES, 0.2 U/ml insulin, 0.45 mM pyruvate, 1 mM oxaloacetate, and 2 mM Glutamax). Cells were grown at 37 °C in a humidified 5% CO2 incubator.
Antibodies
The murine mAbs PFM.1 and PFM.2 were developed using the method described below. The anti-AIB1 mAb AC3 has been previously described [10]. The following primary antibodies were used: Rabbit anti-tubulin IgG (ICN, Costa Mesa, CA), rabbit anti-PAX3 (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-FOXO1 (GeneTex, Irvine, CA). Control mIgG1 protein MOPC21 was purchased from Sigma (St. Louis, MO). Horseradish peroxidase (HRP)-conjugated goat anti-mouse Fc and HRP-goat anti-rabbit H&L were purchased from Jackson Immunoresearch (West Grove, PA).
Production of PAX3-FOXO1 fusion mAbs
mAbs were prepared as previously described [10, 11]. Briefly, female Balb/c mice (6–8 weeks old) were injected i.p. with 100 µg of KLH conjugated with peptide PF corresponding to the PAX3-FOXO1 translocation region aa 100-117 (TIGNGLSPQNSIRHNLSL) in Freund’s adjuvant (Sigma) followed by additional i.p. injections of 100 µg of purified KLH peptide in Freund’s adjuvant at two weeks intervals. Two weeks after the third injection, one mouse received i.p. and i.v. boosts of purified KLH peptide in PBS for 3 consecutive days. A day after the final boost, the spleen of the mouse was removed and fused with the myeloma cell line P3x63Ag8.653 as previously described using 50% PEG with 5% DMSO. Fused cells were resuspended in HY media supplemented with 20% FBS, HAT, and 1× Nutridoma-CS (Roche) and seeded into 96-well plates. Hybridoma colonies were screened by ELISA for secretion of mAbs that bound to ovalbumin coupled peptide. Hybridomas secreting mAbs of interest were subcloned twice by limiting dilution and final hybridoma clones were isotyped using a murine antibody isotyping kit (Roche).
Western blot analysis
Detergent lysates of cell lines were prepared by lysis with RIPA buffer (50 mM Tris-HCl pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS) with 1× Complete protease inhibitor cocktail (Roche). Lysate containing 60 µg of total protein was diluted in 2× non-reducing sample buffer and proteins were separated by SDS-PAGE. Proteins were transferred to Immobilon-P nylon membrane (Millipore, Bedford, MA) and blocked with 5% non-fat milk in PBS for 18 h at 4 °C. Membranes were washed 3× in PBS-T (PBS containing 0.1% Tween-20) and incubated for 1 h at RT with primary antibodies in 1% casein. After washing 3× with PBS-T, membranes were incubated with a 1:20,000 dilution of HRP-goat anti-mouse or HRP-goat anti-rabbit antibody in 5% milk for 1 h at RT. The membranes were washed 3× with PBS-T prior to protein detection using ECL chemiluminescent substrate (GE Healthcare Life Sciences) and exposed to autoradiography film (Hyperfilm-ECL, GE Healthcare Life Sciences). Membranes were stripped of bound IgG by incubating in 62.5 mM Tris-HCl pH 7.5, 2% SDS, 100 mM 2-mercaptoethanol for 30 min at 65 °C. Stripped membranes were washed in PBS and blocked with 5% non-fat dried milk prior to being probed with a subsequent antibody.
Immunoprecipitations
Full-length PAX3-FOXO1 protein was labeled with 35S-methionine using rabbit reticulocyte lysate (Promega, Madison WI) to in vitro-transcribe/translate from a pBluescript plasmid containing the coding regions. A dilution of the in vitro-translated product was incubated with the mAbs and immunoprecipitated with the addition of rabbit anti-mouse IgG and Protein G-Agarose (Invitrogen). Pellets were washed 5× with TEN (50 mM Tris-HCl pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% NP-40) lysis buffer, separated by SDS-PAGE, dried and exposed to autoradiography film. For immunoprecipitation of native PAX3-FOXO1, a detergent lysate of the RMS-13 rhabdomyosarcoma cell line was precleared with Protein A from Staphylococcus aureus Cowan strain (SaC) (Sigma). Protein G-Agarose was incubated with capturing antibody, blocked with 1% BSA, washed and incubated with the precleared lysate. The beads were washed three times with lysis buffer and immune complexes were removed by boiling in sample buffer and separated by SDS-PAGE. Western blot analysis of immune complexes was done using an anti-FOXO1 rabbit antisera and protein detection using ECL chemiluminescent substrate (GE Healthcare Life Sciences).
Patient samples
A total of 20 patients diagnosed with ARMS were retrieved from the files of the Department of Pathology and Molecular Pathology of the University Hospital Zurich, Switzerland from 1991–2012. Tumors were diagnosed according to the WHO Classification of Tumors of Soft Tissue and Bone by two pathologists with expertise in soft tissue pathology (PKB and BB). The series included tumors of 6 female and 14 male patients with an age range from 9 month to 45 years (median age 17 years). The most common site of the primary tumor was the head and neck area, followed by trunk, extremities, and scrotum. Two cases were metastases (bone and lymph node) where the site of the primary tumor was unknown. Five poorly differentiated ERMS with round cell morphology and one pleomorphic RMS were selected as negative controls. The clinical data are summarized in Table 1.
A tissue microarray with 254 cores of 750 µm diameter from 127 additional RMS tumors (14 ARMS with known FOXO1 gene rearrangements, 6 FN-ARMS and 107 ERMS) was constructed. Tumors used were collected at the University Hospital Zurich and at the Kiel Paediatric Tumor Registry. The project has been approved by the local ethics committee (KEK-ZH-Nr. 2013-0430).
Evaluation of specificity of the PAX-FOXO1 antibody in situ
293T cells grown in chamber slides and transfected with plasmids encoding for either PAX3, FOXO1, PAX3-FOXO1, or PAX7-FOXO1 were used for immunofluorescence staining with the PFM.2 antibody. For that, cells were first washed once with PBS and then fixed with 4% formalin for 15 min. After washing once with PBS and quenching formalin with 0.1 M glycine in PBS for 5 min, cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min. Cells were then blocked with 4% horse serum in permeabilization buffer for 15 min followed by incubation with 7.5 μg/ml PFM.2 antibody in the same buffer over night at 4 °C in a humid chamber. After three washing steps with PBS for 5 min, cells were incubated for 1 h with Alexa-488 labeled donkey anti-mouse secondary antibodies (Invitrogen, Basel, Switzerland) diluted 1:250 in PBS. Cells were then washed again three times with PBS as described above. After dipping the slides once into water, cells were embedded with Vectashield with DAPI (Vector Laboratories, Servon, Switzerland) and covered with a cover slip. Staining was evaluated by fluorescence microscopy.
Immunohistochemical staining
Three-micron-thick sections of tissue microarray blocks of formalin-fixed, paraffin-embedded tissues were mounted on glass slides (SuperFrost Plus; Menzel, Braunschweig, Germany), deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E) using standard histological techniques. For the detection of the PAX-FOXO fusion protein, the PFM.2 antibody was used. The staining was performed on a Bond-Max staining system. The dilution of the antibody (1:150) was adjusted after performing titrations. For epitope retrieval, tissue sections were boiled for 45 min in “Bond Epitope Retrieval Solution 2” (Leica, Heerbrugg, Switzerland). For development, the “Bond Polymer Refine Detection” system (Leica, Heerbrugg, Switzerland) was used with rabbit anti-mouse and anti-rabbit polymers following the instructions of the manufacturer. HRP-DAB was used as chromogen. Immunoreactivity for PAX-FOXO1 was nuclear.
Fluorescence in situ hybridization
FISH studies were carried out on interphase nuclei present on FFPE tissue sections of 4 µm thickness from large sections according standard protocols [12]. Detection of FOXO1 was performed with FOXO1 (13q14) dual color, break-apart rearrangement probes (Abbott Molecular/Vysis, IL, USA). The cut-off for the detection of FOXO1 rearrangement was 7% cells with split signals.
RT-PCR
RNA extraction was done as previously described [13]. Briefly, fifteen 5-μm-thick cuts from paraffin-embedded tissue were suspended in 300 μl extraction buffer, composed of 20 mM Tris (pH 8), 20 mM EDTA and 1% SDS (pH 8) and incubated at 95 °C for 10 min. Samples were centrifuged at decreasing temperature down to 4 °C, resulting in a paraffin lid, which was subsequently removed with a separate sterile pipette tip for each sample. The tissue was then digested with 0.3 μg/μl of proteinase K (Roche Applied Science, Switzerland) at 55 °C for 48–72 h using a thermomixer. Samples were centrifuged for 2 min at 4 °C. Two hundred fifty microliters of each supernatant was transferred to a fresh 1.5 ml tube and 750 µl of Trizol LS Reagent (ThermoFisher, Switzerland) was added. Samples were homogenized by centrifugation through a QIA-shredder column (Qiagen, Switzerland) for 2 min followed by standard chloroform/glycogen/isopropanol extraction. RT-PCR was performed using an OneStep RT-PCR Kit (Qiagen, Switzerland). The reactions were done in duplicates (50 ng and 200 ng of sample RNA) in a final volume of 25 μl containing 0.4 mM dNTP, 1× OneStep RT-PCR Buffer, 1 μl enzyme mix and 8 units of the inhibitor RNAse OUTTM (ThermoFisher, Switzerland). The primers used for the fusion transcript-specific amplification were as follows: 5′-TACAGACAGCTTTGTGCCTCC-3′ (PAX3_Ex7up)/5′-TCCTTCATTCTGCACACGA-3′ (FOXO1_Ex2low) and 5′-CCCGCCACAGCTTCTCCA-3′ (PAX7_Ex7up)/5′-CAGTTCCTTCATTCTGCACACG-3′ (FOXO1_Ex2low1). To estimate the RNA quality of samples, the following primers were used for the internal controls (β-actin and importin), both duplex RT-PCR set-ups: 5′-AGCCTCGCCTTTGCCGA-3′ (β-actin-up)/5′-CTGGTGCCTGGGGCG-3′ (β-actin-low1)/5′-GAGGCGTACAGGGATAGCAC-3′ (β-actin-low2) and 5′-GTCAAAACTAGTGAGCTTCGTACTA-3′ (Imp8-up)/5′-CCTATTATACACATCTTCCGGTCA-3′ (Imp8-low1)/5′-AATACTTCTTCATCCCAGTCATC-3′ (Imp8-low2), respectively. The reverse transcription was done for 30 min at 50 °C, followed by a PCR activation step for 15 min at 95 °C and 40 cycles consisting of 1 min at 95 °C, 1 min at 61 °C, and 1 min at 72 °C. The RT-PCR products were analyzed by 2% MetaPhor agarose gel electrophoresis. Bands were excised and purified with MinEluteTM Gel Extraction Kit (Qiagen, Hombrechtikon, Switzerland). Sanger sequencing of the PCR products was done using an ABI 3130XL Sequencer (Applied Biosystems, Zug, Switzerland) using the Big Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Zug, Switzerland) and the same fusion transcript-specific primers as indicated above.
Calculation of sensitivity and specificity
Positive predictive value was calculated using the formula true positive/(true positive + false positive), negative predictive value was calculated as true negative/(true negative + false negative). Sensitivity was calculated using the formula sensitivity = true positives⁄(true positives + false negatives) and specificity was calculated using the formula specificity = true negatives⁄(true negatives + false positives). For the calculations, results from staining of single sections and TMA were pooled. Only ARMS tumors with the proven presence of PAX-FOXO1 fusions were included in the calculations.
Results
Development of anti-PAX3-FOXO1 mAbs
In order to obtain a specific reagent recognizing the chimeric gene product from the t(2;13)(q35;q14) in ARMS, mAbs were developed to a peptide matching amino acids 100–117 of the PAX3-FOXO1 chimeric protein and spanning the junction region (Fig. 1A). Several hundred hybridomas were tested for secretion of mAbs that bound to the peptide using ELISA. A second screening of the hybridomas was conducted for secretion of mAbs that recognized a single band from PAX3-FOXO1-expressing RMS cell lysate by western blot resulting in the identification of two clones designated PFM.1 and PFM.2. Both mAbs were isotyped and found to be mIgG1. To further evaluate the binding specificity of the mAbs, we tested their ability to immunoprecipitate PAX3-FOXO1. Both PFM.1 and PFM.2, but not anti-NCOA3 antibody AC3 nor the IgG1 control MOPC21, were able to immunoprecipitate PAX3-FOXO1 from RMS-13 cellular lysate (Fig. 1B) or the in vitro-transcribed PAX3-FOXO1 fusion protein (Supplementary Fig. 1). These results demonstrate that mAbs PFM.1 and PFM.2 both recognize and bind cellular PAX3-FOXO1.

A The transcription factor PAX3 contains a paired boxed domain (PB), homeodomain-DNA-binding domain (HD) and a PAX3 transactivation domain. FOXO1 contains a forkhead DNA-binding domain (FD) and a transactivation domain. The sequence of peptide PF1 spanning the fusion junction site is shown with the PAX3 amino acids in gray and the FOXO1 amino acids in black. The corresponding PAX7-FOXO1 sequence is shown with identical amino acids underlined. Adapted from [24, 25]. B Recognition of PAX3-FOXO1 by anti-PAX3-FOXO1 peptide mAbs. PAX3-FOXO1 was immunoprecipitated specifically from cellular lysate from the FP-RMS cell line RH-30 by anti-PAX3-FOXO1 mAbs PFM.1 (lane 2) and PFM.2 (lane 3), but not by IgG1 control mAb MOPC21 (lane 1) or by anti-NCOA3 peptide mAb AC3 (lane4). Immunoprecipitated proteins were separated by SDS-PAGE, transferred to nylon membranes and detected using anti-FOXO1 antibodies.
Specificity of anti-PAX3-FOXO1 mAbs
To further characterize and assess the specificity of the novel anti-PAX3-FOXO1 mAbs, we used western blot and immunofluorescence analyses of PAX3-FOXO1 expressing ARMS cell lines. Antibody reactivity of mAb PFM.2 was compared to the reactivity of anti-PAX3 and anti-FOXO1 polyclonal antisera on cells expressing PAX3-FOXO1, PAX3, or FOXO1. We used previously generated NIH-3T3 cells stably expressing either PAX3 or PAX3-FOXO1 (clone PF.1) and control mock-transfected NIH-3T3 cells (clone NIL.C) [14]. Lysates from these cells were analyzed by western blot analysis using mAb PFM.2, anti-PAX3 polyclonal antibodies, and anti-FOXO1 polyclonal antibodies (Fig. 2A). All three antibodies recognized PAX3-FOXO1 from the PAX3-FOXO1 expressing NIH-3T3 cell line PF.1, while only anti-PAX3 antibodies recognized PAX3 from the PAX3 expressing cell line. A similar result was seen using a panel of tumor cell lines whereby only cells expressing the fusion protein displayed a reactive band (Supplementary Fig. 2). These results indicate that mAb PFM.2 does not cross react with either PAX3 or FOXO1 and is specific for the PAX3-FOXO1 fusion protein in FP-RMS cells. Furthermore, both mAbs PFM.1 and PFM.2 recognize PAX3-FOXO1 as a 105 kDa band in FP-RMS cell lines including RH-4, RH-28, RH-30, and RMS-13 but not in the ERMS cell lines CTR or RD (Fig. 2B). To test for recognition of cellular PAX3-FOXO1, FP-RMS cell lines RMS-13 and RH-28 and the FN-RMS cell line RD were stained with the anti-fusion mAbs (Fig. 3). The antibodies demonstrated nuclear staining in only the FP-RMS cell line and not the FN-RMS cell line. These results validate expression of PAX3-FOXO1 in the nucleus of ARMS cells and establishes the mAbs PFM.1 and PFM.2 as specific antibodies capable of detecting the PAX3-FOXO1 oncoprotein. A summary of the reactivity of mAbs PFM.1 and PFM.2 with PAX3-FOXO1 is depicted in Table 2.

A Antibody reactivity of mAb PFM.2 was compared to anti-PAX3 and anti-FOXO1 Abs by western blot analysis of NIH-3T3 cells stably-transfected with vector alone (NIL.C), PAX3 (PAX3.1), or PAX3-FOXO1 (PF.1). MAb PFM.2 recognizes only PAX3-FOXO1 and not PAX3 in NIH-3T3. Immunoblots were re-probed with an anti-tubulin antibody as a loading control. B Expression of PAX3-FOXO1 in rhabdomyosarcoma cell lines. MAbs PFM.1 and PFM.2 recognize PAX3-FOXO1 on FP-RMS cell lines RH-4, RH-28, RH-30, and RMS-13 but not on FN-RMS cell lines RD and CTR. Immunoblots were re-probed with an anti-tubulin antibody as a loading control.

FP-RMS cell lines RMS-13 and RH-28, and the FN-RMS cell line RD were incubated with mAbs PFM.1 or PFM.2. Antibody binding was visualized using a Cy3-goat anti-mouse antibody and DAPI was used to visualize the nucleus.
Immunohistochemical analysis using anti-PAX3-FOXO1 mAbs
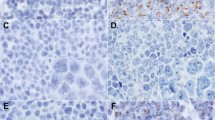
We further evaluated the specificity of PFM.2 to detect PAX-FOXO1 by immunohistochemical analysis. Since PAX3-FOXO1 and PAX7-FOXO1 breakpoint regions are highly similar, we tested reactivity toward both fusion proteins. To this end, 293T cells were transfected with expression plasmids encoding PAX3, PAX3-FOXO1, PAX7-FOXO1, or FOXO1. Transfected cells were then used for immunofluorescence staining with either the PFM.2 antibody, or antibodies directed against wild-type PAX3 and FOXO1 proteins. These analyses demonstrated that the PFM.2 antibody detects both PAX3-FOXO1 and PAX7-FOXO1 fusion proteins with similar sensitivity (Fig. 4). In contrast, no reactivity was detected with wild-type FOXO1 protein and reactivity with wild-type PAX3 protein was detected only after a ten times longer exposure (data not shown). Therefore, the PFM.2 antibody is specific for both fusion proteins in immunofluorescence staining.

293T cells transfected with indicated wild-type or fusion proteins were stained with mAb PFM.2 or antibodies directed against PAX3 or FOXO1. Scale bar, 100 μm.
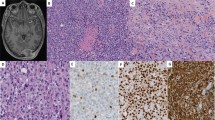
We next tested the PFM.2 antibody in immunohistochemical staining of paraffin sections of RMS tumors. Staining was specific to the tumor cells and not the neighboring stromal cells (Fig. 5A–D). Staining was nuclear in the tumor cells (Fig. 5B), which also stained positive with desmin (Fig. 5C) and myogenin (Fig. 5D). We then selected 20 ARMS cases, which showed a distinct and nuclear expression of AP2β in more than 50% of the tumor cells (Table 1), a fusion protein target gene which can serve as surrogate marker for the presence of any PAX-translocation in ARMS [15]. Indeed, we detected the fusion protein in 18 of 20 of these tumors by RT-PCR and/or FISH (Table 1). Eighteen of 18 FP-RMS tumors were positive in immunohistochemical staining with the PFM.2 antibody, exhibiting a moderate to strong expression exclusively in the nucleus as expected for the fusion protein. In contrast, the two FISH and RT-PCR negative cases were also negative in the IHC staining (Table 1 and Fig. 6, lower right case) and these two cases were therefore diagnosed as fusion-negative ARMS. Overall, the results from the PFM.2 IHC staining were in complete concordance with the results from FOXO1-FISH analysis and RT-PCR.

PAX3-FOXO1 was detected in a tumor sample from a patient with alveolar rhabdomyosarcoma containing the t(2;13) using mAb PFM.2. Sections were stained with A Hematoxylin and Eosin, B PFM.2, C desmin, and D myogenin.

Tissue sections from the four depicted tumors were stained with Hematoxylin and Eosin (upper left picture), with the mAb PFM.2 (upper right picture), an antibody directed against AP2β (lower right picture) and by FISH using a FOXO1 break-apart probe (bap) (lower left picture). The depicted tumors are positive for PAX3-FOXO1 (upper left case), PAX7-FOXO1 (upper right case), or negative for known fusion proteins in poorly differentiated ERMS (lower left case) and translocation negative ARMS (lower right case).
Interestingly, while in PAX3-FOXO1 positive cases the staining pattern with the PFM.2 antibody was homogenous with most of the cells positive for the fusion protein (Fig. 6, upper left case), in four out of five PAX7-FOXO1 cases the staining pattern was very heterogeneous with only scattered cells positive for the fusion (Fig. 6, upper right case). The small fraction of positive cells was intensely stained, while all other cells showed no staining. Importantly, however, FISH staining detected the rearrangement and amplification of the FOXO1 locus also in the unstained cells, demonstrating that these are fusion-positive cancer cells as well (Fig. 6). Taken together, these data suggest that some PAX7-FOXO1 positive ARMS are composed of a mixture of cells with high and low (or absent) expression of the fusion protein, despite carrying the underlying chromosomal abnormality whereas expression of the PAX3-FOXO1 protein appears more uniform. The functional consequences of this observation are currently unclear. In all five ERMS cases and in the one pleomorphic RMS case analyzed, no PFM.2 IHC staining was detected (Fig. 6, lower left case). All these cases were negative in RT-PCR and FISH for PAX-FOXO1 translocations.
To further determine specificity and sensitivity of the PFM.2 antibody, we used a tissue microarray with 254 cores representing 127 RMS tumors (14 ARMS with known FOXO1 gene rearrangements, 6 FN-ARMS and 107 ERMS). On this tissue array, 11 out of 14 fusion-positive ARMS were reactive with the PFM.2 antibody, while none of the ERMS and the FN-ARMS was stained. Hence, taking staining of both single sections and of the tissue array into account, a positive predictive value of 100%, negative predictive value of 98%, specificity of 100%, and a sensitivity of 91% was calculated (Table 3).
Discussion
This report describes the development of novel immunological reagents directed specifically to the PAX-FOXO1 chimeric protein. Detection of the PAX3 or PAX7 translocations is the current gold standard for the diagnosis of ARMS and fusion-positivity has therapeutic consequences. The most common methods used for this purpose in the clinics include detection of the fusion transcript by RT-PCR or NGS-based approaches and FISH detection of FOXO1 rearrangements, the most frequent fusion partner of the PAX proteins in ARMS. These methods are relatively complex and time-consuming when compared to the immunohistochemical methods used in the standard diagnostic process. We therefore embarked on developing mAbs specific for the chimeric protein. Using a conjugated peptide spanning the junction region of PAX3-FOXO1 as the sole immunogen, we generated two unique hybridoma clones secreting mAbs specific for the fusion protein. The mAbs designated PFM.1 and PFM.2 did not cross react with either PAX3 or FOXO1 and were shown to recognize PAX3-FOXO1 by its reactivity in immunoprecipitation and western blot analyses. In addition, the antibody was also successfully applied in chromatin immunoprecipitations (ChIPseq) experiments recently [7, 11].
These results indicate that the anti-PAX3-FOXO1 mAbs will be a valuable reagent in studying the protein product PAX3-FOXO1 of the t(2;13)(q35;q14) in ARMS. Interestingly, PFM.2 can also recognize the protein product PAX7-FOXO1 of the t(1;13)(p36;q14) by immunofluorescence, which differs from PAX3-FOXO1 in its junction region spanned by peptide PF by three amino acids (Fig. 1). Therefore, these three amino acids might not be included in the actual epitope recognized by the PFM.2 antibody.
Several groups have developed both polyclonal and mAbs with specificity to the chimeric proteins BCR-ABL [16, 17], E2A/PBX1 [18], AML-ETO [19], and SS18-SSX [20]. Sang et al. [18], developed mAbs to the E2A (TCF3)-PBX1 fusion protein, which results from the t(1;19)(q23;p13.3) in childhood acute lymphoblastic leukemia (ALL). However, initial attempts to raise mAbs using conjugated peptide as the sole immunogen were unsuccessful. Instead, they substituted their last two immunizations using a recombinant GST-fusion protein containing a 20 aa fragment of TCF3 and a 144 aa fragment of PBX1 to obtain hybridomas secreting mAbs to the junction region. These mAbs were shown to be immunoreactive in several assays including western blot analysis and immunohistochemistry [18]. Recently, rabbit mAbs were developed to the SS18-SSX fusion protein expressed from the t(X;18)(p11;q11) translocation that is found in synovial sarcoma patients [20]. These mAbs were found to be selective and specific for the fusion protein in immunoblotting, immunoprecipitation, chromatin immunoprecipitation and immunohistochemistry. Furthermore, the activity of these mAbs in IHC analysis supports the utilization of fusion-specific mAbs to replace or complement molecular genetic or cytogenetic diagnosis.
Here, we evaluated the immunohistochemical detection of PAX3/7-FOXO1 fusion proteins as simple and rapid alternative method for diagnostics of ARMS using the breakpoint specific antibody PFM.2. By testing the specificity of the PFM.2 antibody under native conditions using cells transfected with PAX3-FOXO1, PAX7-FOXO1 or wild-type PAX3, and FOXO1 for immunofluorescence staining, we found that breakpoint specific antibody preferentially reacts with the fusion proteins and only very weakly with PAX3. PAX3-FOXO1 and PAX7-FOXO1 fusion proteins were detected with similar sensitivity. By staining both single sections from a cohort of RMS tumors as well as a tissue array of RMS, we found that the PFM.2 antibody specifically stains PAX3/7-FOXO1 fusion-positive tumors but not ERMS or fusion-negative tumors, resulting in a maximal positive predictive value and specificity of 100%. We and others have recently evaluated different PAX3/7-FOXO1 target genes as markers for ARMS by immunohistochemistry. The most specific one among these markers is AP2β, with a specificity of more than 90%. Hence, specificity of the PFM.2 antibody determined here is at least as high or might be even higher. In contrast, the sensitivity found here was somewhat lower when compared to AP2β, reaching 91%. However, the number of cores per tumor on the tissue array used for calculation of the sensitivity was only two, and even though the core size of 750 µm should contain sufficient tumor, some positive tumors might have been missed. Indeed, some tumors containing PAX7-FOXO1 translocations were found to be heterogenous and contain only few scattered positive cells, which might not be represented on two small tissue punches. Therefore, the calculated sensitivity might be rather an underestimate. Further testing of the antibody using a larger number of tumor samples would help determine the utility of using IHC as a diagnostic for fusion-positive RMS.
The underlying biology causing the different staining patterns between PAX3-FOXO1 and PAX7-FOXO1 positive cases is not entirely clear. Interestingly, this pattern is not recapitulated in the available PAX7-FOXO1 positive cell lines, which grow as a homogenous population of positive cells in vitro (data not shown). It remains to be investigated whether culture conditions have selected a subset of cells or whether staining conditions in vivo restrict access of the antibody. However, additional differences between PAX3- and PAX7-FOXO1 ARMS tumors have been described. First, the PAX7-FOXO1 fusion gene is amplified in most cases, while this is only the case in 9% of the tumors with a PAX3-FOXO1 gene. Furthermore, markers for active cell cycle and apoptosis differed significantly between PAX3-FOXO1 and PAX7-FOXO1 FP-RMS tumors [21]. Finally, clinical outcome of patients with PAX7-FOXO1 has been reported to be significantly better when compared to PAX3-FOXO1 cases [22] although pooled hazard ratios did not reach statistical significance [23]. Therefore, the difference in expression pattern of the fusion proteins detected here might be an additional reflection of a much broader biological difference between these two ARMS subtypes.
Taken together, our data suggest that the PFM.2 antibody is a suitable tool for rapid and technically simple detection of PAX-FOXO1 fusions by immunohistochemistry and might be used in routine diagnostics of RMS. Since PAX3/7-FOXO1 fusion proteins are drivers of RMS and the PFM.2 antibody is specific for PAX3/7-FOXO1 fusion detection, we would expect its use to be prioritized to cases where RMS is suspected. We envision the utility of PFM.2 as an additional antibody used along with anti-desmin and anti-myogenin to help diagnosis FP-RMS samples by IHC, particularly in laboratories that routinely perform IHC but are not equipped with tools for molecular analysis. Positive PFM.2 staining would indicate FP-RMS, which could be confirmed with RT-PCR or FISH to identify the fusion partner. If a sample showed negative staining with PFM.2, and had a round cell morphology compatible with the diagnosis of ARMS, then we would recommend follow-up molecular diagnostics to confirm or exclude the presence of a fusion. Nonetheless, the use of the PAX3/7-FOXO1 antibody PFM.2 in IHC of suspected ARMS tumors would greatly contribute to the diagnosis of fusion-positive RMS.
References
Pappo AS. Rhabdomyosarcoma and other soft tissue sarcomas in children. Curr Opin Oncol. 1996;8:311–6.
WHO Classification of Tumours Editorial Board. Soft tissue and bone tumours: WHO classification of tumours. 5th ed. Lyon: IARC Press; 2020.
Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–31.
Barr FG, Galili N, Holick J, Biegel JA, Rovera G, Emanuel BS. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;3:113–7.
Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013;24:710–24.
Khan J, Simon R, Bittner M, Chen Y, Leighton SB, Pohida T, et al. Gene expression profiling of alveolar rhabdomyosarcoma with cDNA microarrays. Cancer Res. 1998;58:5009–13.
Gryder BE, Yohe ME, Chou HC, Zhang X, Marques J, Wachtel M, et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017;7:884–99.
Rudzinski ER, Anderson JR, Lyden ER, Bridge JA, Barr FG, Gastier-Foster JM, et al. Myogenin, AP2beta, NOS-1, and HMGA2 are surrogate markers of fusion status in rhabdomyosarcoma: a report from the soft tissue sarcoma committee of the children’s oncology group. Am J Surg Pathol. 2014;38:654–9.
Hinson AR, Jones R, Crose LE, Belyea BC, Barr FG, Linardic CM. Human rhabdomyosarcoma cell lines for rhabdomyosarcoma research: utility and pitfalls. Front Oncol. 2013;3:183.
Azorsa DO, Meltzer PS. Production and characterization of monoclonal antibodies to the steroid receptor coactivator AIB1. Hybridoma. 1999;18:281–7.
Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, et al. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010;70:6497–508.
Nishio J, Althof PA, Bailey JM, Zhou M, Neff JR, Barr FG, et al. Use of a novel FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of alveolar rhabdomyosarcoma. Lab Investig. 2006;86:547–56.
Bode B, Frigerio S, Behnke S, Senn B, Odermatt B, Zimmermann DR, et al. Mutations in the tyrosine kinase domain of the EGFR gene are rare in synovial sarcoma. Mod Pathol. 2006;19:541–7.
Khan J, Bittner ML, Saal LH, Teichmann U, Azorsa DO, Gooden GC, et al. cDNA microarrays detect activation of a myogenic transcription program by the PAX3-FKHR fusion oncogene. Proc Natl Acad Sci USA. 1999;96:13264–9.
Wachtel M, Runge T, Leuschner I, Stegmaier S, Koscielniak E, Treuner J, et al. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. J Clin Oncol. 2006;24:816–22.
van Denderen J, ten Hacken P, Berendes P, Zegers N, Boersma W, Grosveld G, et al. Recognition of the ALL-specific BCR-ABL junction in P190bcr-abl by monoclonal antibody ER-FP1. Leukemia. 1994;8:1503–9.
van Denderen J, Hermans A, Meeuwsen T, Troelstra C, Zegers N, Boersma W, et al. Antibody recognition of the tumor-specific bcr-abl joining region in chronic myeloid leukemia. J Exp Med. 1989;169:87–98.
Sang BC, Shi L, Dias P, Liu L, Wei J, Wang ZX, et al. Monoclonal antibodies specific to the acute lymphoblastic leukemia t(1;19)-associated E2A/pbx1 chimeric protein: characterization and diagnostic utility. Blood. 1997;89:2909–14.
Erickson PF, Dessev G, Lasher RS, Philips G, Robinson M, Drabkin HA. ETO and AML1 phosphoproteins are expressed in CD34+ hematopoietic progenitors: implications for t(8;21) leukemogenesis and monitoring residual disease. Blood. 1996;88:1813–23.
Baranov E, McBride MJ, Bellizzi AM, Ligon AH, Fletcher CDM, Kadoch C, et al. A novel SS18-SSX fusion-specific antibody for the diagnosis of synovial sarcoma. Am J Surg Pathol. 2020;44:922–33.
Collins MH, Zhao H, Womer RB, Barr FG. Proliferative and apoptotic differences between alveolar rhabdomyosarcoma subtypes: a comparative study of tumors containing PAX3-FKHR or PAX7-FKHR gene fusions. Med Pediatr Oncol. 2001;37:83–9.
Sorensen PH, Lynch JC, Qualman SJ, Tirabosco R, Lim JF, Maurer HM, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2002;20:2672–9.
Kubo T, Shimose S, Fujimori J, Furuta T, Ochi M. Prognostic value of PAX3/7-FOXO1 fusion status in alveolar rhabdomyosarcoma: systematic review and meta-analysis. Crit Rev Oncol Hematol. 2015;96:46–53.
Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, et al. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995;15:1522–35.
Sublett JE, Jeon IS, Shapiro DN. The alveolar rhabdomyosarcoma PAX3/FKHR fusion protein is a transcriptional activator. Oncogene. 1995;11:545–52.
Acknowledgements
The authors thank M. Storz, S. Behnke, and A. Wethmar (Pathology Zurich) for excellent technical assistance. We also thank F. Barr (National Cancer Institute) for providing the PAX3-FOXO1 and PAX7-FOXO1 plasmids. This work was funded by the Intramural Research Program of the National Institutes of Health. PKB was supported by a grant from the Cancer League Kt. Zürich to BWS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Azorsa, D.O., Bode, P.K., Wachtel, M. et al. Immunohistochemical detection of PAX-FOXO1 fusion proteins in alveolar rhabdomyosarcoma using breakpoint specific monoclonal antibodies. Mod Pathol 34, 748–757 (2021). https://doi.org/10.1038/s41379-020-00719-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00719-0
This article is cited by
-
KDM3B inhibitors disrupt the oncogenic activity of PAX3-FOXO1 in fusion-positive rhabdomyosarcoma
Nature Communications (2024)
-
PAX3-FOXO1 uses its activation domain to recruit CBP/P300 and shape RNA Pol2 cluster distribution
Nature Communications (2023)
-
Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Soft Tissue Tumors
Head and Neck Pathology (2022)
