
Abstract
Although relatively uncommon, pathologists may encounter minimal inflammatory foci in the absence of typical structural heart disease; however, the clinicopathological significance of minimal inflammatory foci, including correlation with sudden unexpected death, is unexplored. From 1072 serial autopsy subjects, cases with unexplained minimal inflammatory foci, the extent of which was under 1% of the whole examined ventricle, were extracted to exclude cases with borderline/focal myocarditis resulting from local, systemic infection, or autoimmune mechanisms. Immunohistochemistry and genetic analysis targeting viral genomes and heart disease-related genes using next generation sequencing were performed. We detected 10 cases with unexplained minimal inflammatory foci (five males, five females, aged 15–68 years). The cause and/or manner of death were sudden unexpected death (6 cases, 60%), sudden unexpected death with epilepsy (1 case, 10%), drowning in a hot bath (1 case, 10%), and suicide (2 cases, 20%). In none of these cases was pathogen-derived DNA or RNA detected. In 8 of the 10 cases (80%), 17 possible pathogenic genetic variants causative for arrhythmogenic right ventricular cardiomyopathy or dilated cardiomyopathy; DSP was the most frequently involved gene (three cases with two different variants), followed by LAMA4 and MYBPC3 (two cases, two variants for each gene), LDB3 (two cases, one variant), and the remaining 10 variants occurred in seven cases (DSC2, RYR2, SOS1, SCN5A, SGCD, LPL, PKP2, MYH11, GATA6, and DSG2). All mutations were missense mutations. DSP_Lys1581Glu and DSC2_p.Thr275Met were classified according to American College of Medical Genetics and Genomics consensus statement guidelines as pathogenic or likely pathogenic for arrhythmogenic cardiomyopathy in three patients (30%). The remaining 15 variants were classified as potentially pathogenic variants. Unexplained minimal inflammatory foci may be an early sign of inherited cardiomyopathy, and such cases might already have arrhythmogenic potential that can lead to sudden unexpected death. Detection of minimal inflammatory foci by careful pathological examination may indicate the value of conducting comprehensive genetic analysis, even if significant structural abnormalities are not evident.
Similar content being viewed by others

Introduction
Sudden cardiac death, a major cause of sudden unexpected death, occurs most commonly with structural heart disease in both younger and older subjects [1, 2]. Methods for detailed postmortem investigation of sudden unexpected death have been proposed [3]; however, in some sudden unexpected death cases the evaluation of pathogenicity is complicated by the lack of any observable gross and/or microscopic abnormality that fulfills the diagnostic criteria. Such sudden unexpected death cases older than 1 year are classified as sudden unexpected death syndrome or sudden arrhythmogenic death syndrome, which is defined as death due to a possible arrhythmogenic event but without clear evidence of pathological or toxicological abnormality [4, 5].
Postmortem genetic analysis using next generation sequencing provides useful information relating to the genetics of inherited heart diseases, such as channelopathies and cardiomyopathy [6], and such investigation of sudden unexpected death cases has been termed, molecular autopsy [7, 8]. We recently presented the benefit of combined pathological and genetic evaluation for sudden unexpected death and sudden unexpected death with epilepsy cases [9, 10]. We suggest that minute inflammatory foci of unknown etiology might predict the association of sudden unexpected death and sudden unexpected death with epilepsy with possible pathogenic cardiomyopathy-related genetic variants, including genes associated with dilated cardiomyopathy and arrhythmogenic cardiomyopathy, even if the clinical symptoms appeared just before death. Mutations in a number of genes that encode proteins that function in the cardiac sarcomere, desmosome, cytoskeleton, nuclear lamina, mitochondria and in ion flux-handling, can cause cardiomyopathy, which may result in contractile dysfunction and/or an arrhythmogenic event [11, 12].
In this study, we attempted to explore the cause and clinicopathological significance of unexplained minimal lesions by comprehensive genetic analysis using next generation sequencing for a series of autopsy cases with minimal inflammatory foci lack significant structural heart disease.
Methods
Subjects
All autopsies performed in our department from 2010 to 2016 were evaluated (n = 1345). Of these cases, heart specimens from 1072 subjects (0–101 years old, male; 667, female; 405, mean age 62.6 ± 21.0) without severe injury or postmortem degradation were examined. A natural cause of death was recorded for 262 cases (male: 192, female: 70), and 435 cases (male: 292, female: 143) suffered accidental traumatic death, for example from a fall, traffic accident, burning, drowning, or hypothermia. Suicide or homicide accounted for 342 cases (male: 166, female: 176), and there were 33 cases (male: 19, female: 14) with undetermined causes of death. The clinical histories of patients were obtained from their families and from police records. Toxicological screening was applied to all cases, and quantitative assessment was also performed as appropriate, in parallel with pathological and serological examination, including quantitative estimation for C-reactive protein. A natural cause of death was recorded for 262 cases (male; 192, female; 70), and 440 cases (male; 295, female; 145) suffered accidental traumatic death, for example from a fall, traffic accident, burning, drowning, or hypothermia.
This study was performed in accordance with the ethical standards established in the 1964 Declaration of Helsinki and approved by the Ethics Committee of Toyama University.
Examination of cardiac pathology
Pathological examination was routinely conducted by reference to recently proposed criteria and methods used by cardiovascular pathologists [3, 13]. The hearts were excised and dissected free from the great vessels. Heart weight, including epicardial coronary arteries and epicardial fat, was measured to the nearest gram. The right and left ventricles were cut at 1 cm intervals parallel to the levels of the papillary muscle from the apex. The hearts were then cut open along the direction of blood flow and any abnormality in the myocardium, endocardium, or valves was noted. Sections at the level of the papillary muscle and the level just above the apex were subjected to a thorough histological examination after dividing into 5–8 blocks each 3 × 3 cm. Both atria were also cut into sections. The major epicardial coronary arteries and the left main, left anterior descending, left circumflex, and right coronary artery were cut transversely at 5 mm intervals. In all cases, the right ventricular out flow tract of 2–4 block and the conduction system, including the sinoatrial node divided into 2–4 blocks, the atrioventricular conduction system divided into 4–6 blocks was histologically examined (Supplemental Fig. 1) [14].
Cases with structural heart disease (e.g., congenital heart disease, myocardial infarction, overt myocarditis, hypertrophic cardiomyopathy, arrhythmogenic cardiomyopathy, coronary artery disease, including atherosclerosis of the coronary artery with luminal narrowing beyond 70% or coronary artery anomalies, obesity cardiomyopathy, idiopathic left ventricular hypertrophy, etc.) were excluded (Supplemental Table 1) [3, 13]. Among the included cases, we detected cases with minimal inflammatory foci in the ventricular myocardium. In present study, we defined minimal inflammatory foci as inflammatory foci in the ventricle that constituted <1% of the whole examined area to exclude cases with borderline/focal myocarditis resulting from local, systemic infection, or autoimmune mechanisms. The total area of a pathological lesion was measured using a BX51 microscope (Olympus) equipped with a digital camera (DP73, Olympus) and analyzed using Olympus cellSens imaging software (version 1.7, Olympus, Tokyo, Japan).
Immunohistochemical examination
Infiltrating cell populations were assessed by immunohistochemistry. The T-lymphocyte marker, CD3 (F7.2.38, Dako, Glostrup, Denmark), the B-lymphocyte marker, CD20 (L26, Dako), the neutrophil marker, myeloperoxidase (Dako), and the macrophage marker, CD68 (KPI, Dako) were examined. Sections 6-µm thick were deparaffinized, and antigen retrieval was performed in a microwave oven for 20 min at 98 °C (in 10 mM citrate buffer, pH 6.0). Immunostaining was performed using the Leica Bond-IV automation and Leica Refine detection kits (Leica Biosystems, Bannockburn, IL). Subsequently, all sections were counterstained with hematoxylin.
Library preparation and sequencing for next generation sequencing analysis
Viral genomes
Extraction of total nucleic acids from serum samples was performed with a QIAamp MinElute Virus Spin Kit (Qiagen, Germantown, MD, USA) in accordance with the manufacturer’s instructions. Sequencing libraries were prepared using the ScriptSeq v2 RNA-Seq Library Preparation kit (Illumina, San Diego, CA USA) and sequenced on a MiSeq sequencer with the MiSeq reagent kit v3 to generate 2 × 75 bp pair-end protocol (Illumina). For identification of virus-derived sequences from the paired end next generation sequencing reads, the VirusTAP pipeline (https://gph.niid.go.jp/cgi-bin/virustap/index.cgi) was used [15].
Inherited genetic defects
Genomic DNA was extracted from whole blood samples from subjects with minimal inflammatory foci using a QIAamp DNA Mini Kit (Qiagen). Next generation sequencing of 404 cardiovascular disorder-related genes (Life Technologies, Carlsbad, CA, USA), which are listed in Supplemental Table 2, was performed using an Ion PGM system (Life Technologies). Minimal coverage of 20 reads was defined as the cut-off value. All known and potential variants were classified as candidate pathogenic variants and were validated by Sanger sequencing. Detailed methods used for sequence analysis are previously reported [9, 10].
Variant classification of inherited genetic defects
The allelic frequency in the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org) was determined for all detected variants and those with a minor allele frequency of >0.1% in the general population were removed. Variants were then verified using combined annotation-dependent depletion (CADD), which is a tool for scoring variants and variants with a score <15 were filtered out [16]. We then classified the remaining variants as one of the following: pathogenic, likely pathogenic, or variant of uncertain significance, according to the American College of Medical Genetics and Genomics consensus statement guidelines [17]. To evaluate the pathogenic potential of variant of uncertain significance, we used the in silico predictive algorithm: GAVIN (https://molgenis20.gcc.rug.nl) and the disease mutation database: Clinvar disease mutation database (http://www.ncbi.nlm.nih.gov/clinvar), which accurately classify variants for clinical diagnostic purposes.
Statistical analysis
Differences in the frequency of rare variants versus control variants archived in the human genetic variation database (HGVD; http://www.hgvd.genome.med.kyoto-u.ac.jp) [18] and the integrative Japanese genome variation database (iJGVD; https://ijgvd.megabank.tohoku.ac.jp) [19] were assessed using Fisher’s exact test with p < 0.05 being statistically significant.
Results
Subjects
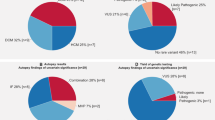
From 1072 serial autopsy cases, the cases of coronary artery disease (191 cases), deformity of aortic valve with calcification (20 cases), hypertrophic cardiomyopathy (7 cases), dystrophic myopathy (3 cases), overt myocarditis (3 cases), sarcoidosis (4 cases), amyloidosis (25 cases), irradiation pericarditis (1 case), infectious endocarditis (1 case), Rosai-Dorfman syndrome (1 case), idiopathic left ventricular hypertrophy (57 cases), obesity cardiomyopathy (14 cases), and atthythmogenic cardiomyopathy (1 case) were excluded. In remaining 744 cases, 10 cases fulfilled the criteria of minimal inflammatory foci of the present study (five males and five females, aged 15–68 years). A summary of all cases is described in Table 1. Eight of ten cases did not have family of sudden unexpected death within first degree relative, and we cannot obtain the data of other two cases. Some autopsy data for Cases 6 and 9 have already been reported [9]. With respect to cause of death, six cases (60%) were judged as sudden unexpected death syndrome, and the other causes of death were judged as sudden unexpected death with epilepsy (one case), drowning in a bathtub because of a sudden intrinsic loss of consciousness (one case) suicidal drowning (one case), and drug intoxication with a high blood concentration of olanzapine (one case). Four of the ten cases showed abnormal electrocardiogram in the clinical record, and Cases 5 (ventricular premature contraction) and 6 (Saddleback-type ST elevation in V2) had received routine follow up but had not received treatment. Two cases showed mild elevation of C-reactive protein and one had localized bronchitis and the other liver fibrosis, possibly because of excessive alcohol consumption. Next generation sequencing that mapped to a specific viral genome were not detected in all serum samples. Sequencing result is shown in Supplemental Table 3.
Gross and microscopic features
Mild to moderate fatty infiltration of right ventricle without significant interstitial fibrosis that did not fulfill the criteria of arrhythmogenic cardiomyopathy was identified in six cases. Four cases did not show significant pathological change (Fig. 1a).

Gross and microscopic appearance of hearts. a Gross appearance of Case 4 with fatty infiltration of the right ventricle. b, c Fatty infiltration with mild interstitial fibrosis in the right ventricular free wall of Case 4 (b) and outflow tract of Case 1 (c). d High power view of the left ventricle with mild interstitial fibrosis of Case 6 (f–n) Minimal inflammatory foci in all extracted cases. Shows Case 1–Case 10, respectively. Minimal necrosis can be seen in Cases 3 and 5–9. Scale bars: 500 µm (b), 200 µm (c, d), 50 µm (e–n)
None of the cases had obvious replacement fibrosis. In six cases with fatty infiltration of right ventricle, four cases (Cases 1–4) additionally showed mild to moderate interstitial fibrosis in the right ventricle and three cases (Cases 4–6) showed minor fibrosis in the left ventricle. Only Case 4 showed fibrosis in the both ventricles. (Fig. 1b–d), and three cases (Cases 8–10) did not show significant interstitial fibrosis in the heart. The minimal number of inflammatory cells in one focus was 20. Necrosis was visible in six of ten cases (Cases 3, 5–9). The distribution of minimal inflammatory foci was varied; minimal inflammatory foci were detected in the free wall of the left ventricle in seven cases, in the interventricular septum in no cases, in the right ventricular free wall in one case (Case 2) and in the right ventricular outfloe tract in five cases (Cases 1–4, 10) (Fig. 1e–n). Myeloperoxidase positive neutrophils were observed in six cases that had necrosis. Both T and B cells were also frequently found. CD68-positive macrophages were found in all cases (Fig. 2). No cases with eosinophil-predominant inflammation were evident, and minimal inflammatory foci were not found in the cardiac conduction system.

Pathological appearance including immunohistochemistry. (a–c) Case 2; (a) hematoxylin–eosin, (b) myeloperoxidase, (c) CD3. (d–f) Case 9; (d) hematoxylin–eosin, (e) myeloperoxidase, (f) CD68. Scale bars: 50 μm
Cardiovascular disorder-related genes variants
In two (20%) of the ten cases with unexplained minimal inflammatory foci, no potential pathogenic mutation was identified. In eight cases (80%), a total of 17 rare variants in 14 different genes were identified, which are causative genes variants for arrhythmogenic cardiomyopathy or dilated cardiomyopathy; DSP was the most frequently involved gene (three cases with two different variants), followed by LAMA4 and MYBPC3 (two cases, two variants for each gene), LDB3 (two cases, one variant) and the remaining 10 variants occurred in seven cases (DSC2, RYR2, SOS1, SCN5A, SGCD, LPL, PKP2, MYH11, GATA6, and DSG2) (Fig. 3). All mutations were missense mutations. Two of the 17 rare variants were classified according to American College of Medical Genetics and Genomics consensus statement guidelines as pathogenic or likely pathogenic for arrhythmogenic cardiomyopathy in three patients (30%). These were DSP_Lys1581Glu and DSC2_p.Thr275Met. The remaining 15 variants were classified as potentially pathogenic variants. The prevalence of gene variants detected in the present study and in Japanese controls was compared. One variant (RYR2_p.Tyr4939Phe) were not found in gnomAD, HGVD, or iJGVD, and the prevalence of one variant (LDB3_p.Asp673Asn) was significantly higher than in control cases (Table 2, Supplement Table 4). Two or more rare heterozygous variants were found in six of the 10 cases (60%), and three cases (30%) carried three or more variants (Fig. 4).

Detected variant classification strategy. MAF minor allele frequency, VUS variant of uncertain significance

The number of variants in each gene subset
Discussion
Many pathologists have experience of minimal inflammatory foci-like lesions in autopsy cases, however, the pathological significance and the etiology of such restricted lesion remained to be determined. We assume that cases with restricted minimal inflammatory foci of uncertain etiology are likely to be evaluated as “myocarditis of uncertain significance” [20] or as sudden arrhythmogenic death syndrome because the lesion is overlooked or disregarded. In the present study, we selected the minimal inflammatory foci cases using relatively strict conditions to avoid selecting cases with infection or systemic inflammatory disease. Our postmortem investigation indicated that of 10 cases with unexplained minimal inflammatory foci, eight suffered sudden unexpected death after the onset of some sort of arrhythmogenic event and two cases were suicide victims. Therefore, the etiology of undetermined minimal inflammatory foci may correlate with the occurrence of sudden unexpected death even if clinical signs and gross abnormalities are not evident. Under conditions of coronary vessel disease, where infection and the other primary inflammatory disease is carefully ruled out, applied genetic analysis shows that restricted minimal inflammatory foci of uncertain etiology may be a pathological sign of early inherited arrythmogenic cardiomyopathy or dilated cardiomyopathy, which can result in sudden unexpected death, including drowning in a bathtub or sudden unexpected death with epilepsy.
Interpretation of pathogenicity of variants detected using next generation sequencing remains difficult in some cases because the number of rare variants detected was higher than we expected. Determination of pathogenicity of the variants mainly relies on the prevalence of the variants in large control cohorts, such as gnomAD, segregation in family members, in silico predictive programs, and experimental investigations [21, 22]. Segregation studies or experimental investigations are useful for evaluating the pathogenicity of detected variants in some cases; however, in many autopsy departments or laboratories it may be difficult to apply appropriate segregation analysis or experimental techniques. Other investigators have also commented that future approaches to validate the pathogenicity of variants will rely more strongly on in silico predictive tools and population frequency [23, 24]. Although we should always note the considerable “background noise” of innocent variants in the genetic analysis of heart disease, including sudden cardiac death [25,26,27], the present study shows that detailed pathological examination for the heart of sudden unexpected death cases might be useful for evaluating the pathogenicity of detected variants, and may be especially useful to avoid false positive results. In contrast, additional pathological examination targeting minimal inflammatory foci might be essential when possible pathogenic variants associated with cardiomyopathy are detected, even if initial routine pathological examination concludes that the heart is within normal limits. In addition, an unpredictable cardiomyopathy-related genetic variant might be present in some patients diagnosed as mild/borderline myocarditis on myocardial biopsy.
Arrhythmogenic cardiomyopathy is characterized by a high incidence of ventricular arrhythmia and sudden cardiac death. Pathogenesis of arrhythmogenic cardiomyopathy involves disruption of cell–cell adhesion and/or altered intracellular signaling resulting from mutations in various desmosome-related genes [28]. A series of ultrastructural alterations, which decease mechanical coupling between cells, resulting in detachment of cardiomyocytes with subsequent cell death [27, 29], may contribute to the typical gross and microscopic pathology of advanced arrhythmogenic cardiomyopathy, which includes islands of surviving myocytes, inflammatory infiltration, and fibro-fatty tissue [30].
Te Riele et al. examined the relatives of arrhythmogenic cardiomyopathy patients and revealed that almost one-third already showed electrical instability preceding detectable structural change. Such electrical instability may cause sudden cardiac death at any time during the course of arrhythmogenic cardiomyopathy arrhythmogenic cardiomyopathy [31]. In none of the six cases with a desmosome-related genetic variant was advanced fibrofatty infiltration evident; however, distribution of minimal inflammatory foci may be consistent with the pericardial side of the right ventricle in advanced arrhythmogenic cardiomyopathy [32]. In addition, examination of the right ventricular outflow tract, which, routinely, is less frequently examined but is a common site of arrhythmogenic cardiomyopathy, might be significant for detecting minimal inflammatory foci in sudden unexpected death cases with arrhythmogenic cardiomyopathy-related variants. Involvement of the left ventricle, as found in two of the six cases with desmosome-related variants, is also found in a large number of arrhythmogenic cardiomyopathy cases [28, 32, 33]. In addition, the present study showed that fatty infiltration of right ventricle, which mimics early arrhythmogenic cardiomyopathy, may not be relevant to arrhythmogenic cardiomyopathy because fatty infiltration of right ventricle was also found in the case without desmosome gene-related variants. Both detailed pathological examination targeting minimal inflammatory foci and genetic analysis may be essential for detecting early cases of arrhythmogenic cardiomyopathy.
We also note that desmosome molecules including plakophilin 2 (PKP2) synergistically interact with gap junctions and with the voltage-gated sodium channels, and that failure of these interactions associated with desmosome-related genetic variants can create a substrate for arrhythmias by decreasing the amplitude and kinetics of the Na+ current, even in the early phase of the disease [28, 34]. These observations may also explain the clinical and electrophysiological similarities between arrhythmogenic cardiomyopathy and Brugada syndrome cases, which sometimes have desmosome-related variants but do not show significant structural abnormality [35]. In this regard, differentiation between early arrhythmogenic cardiomyopathy and Brugada syndrome may be complicated when typical arrhythmogenic cardiomyopathy pathology is not evident, as in the present cases. “Possible Brugada syndrome with minimal inflammatory foci related to desmosome gene variation” might be a more appropriate autopsy diagnosis rather than “early or preclinical arrhythmogenic cardiomyopathy” when typical arrhythmogenic cardiomyopathy pathology is absent. In Case 6 with a history of saddleback type ST elevation, the rare variant of PKP2 may be associated with possible Brugada syndrome-related arrhythmia, as shown in a previous report [36]. It is also interesting that RYR2 has been shown to be a possible Brugada syndrome-causing gene [37]. An overlap of such genes might contribute to the occurrence of this sudden unexpected death, possibly through Brugada syndrome.
The etiology of dilated cardiomyopathy is complex, involving structural heart disease, hypertension, inflammatory diseases, and toxins [38]; therefore, careful investigation to rule out “secondary dilated cardiomyopathy-associated minimal inflammatory foci” should be performed ahead of genetic investigation. However, recent studies have revealed mutations in various genes may regulate the pathogenesis of dilated cardiomyopathy [38]. Variants of sarcomere-related genes, involved with the cytoskeleton, extracellular matrix, sarcoplasmic reticulum, nuclear envelope, and nucleus, were found in the present study, and are also considered to be dilated cardiomyopathy-causing genes [22,23,24], even if the number of the reported cases has been small. Although the specific pathology of early dilated cardiomyopathy has not been explored, mutations in these genes may generate vulnerability in myocytes to mechanical stress, and may lead to necrosis of myocytes [39], which could subsequently trigger an inflammatory response and fibrosis [40].
Dilated cardiomyopathy is associated with an increased risk of arrhythmogenic events and sudden cardiac death [41]; however, prevalence of early dilated cardiomyopathy in sudden unexpected death autopsy cases has not been explored because of a lack of pathological diagnostic criteria of early dilated cardiomyopathy. Spezzacatene et al. identified a subset of dilated cardiomyopathy patients with a prominent arrhythmic phenotype (arrhythmogenic dilated cardiomyopathy) in the early stages of the disease, characterized by frequent ventricular arrhythmias and unexplained syncope in the absence of overt heart failure [42]. A few studies have aimed to detect early dilated cardiomyopathy. Mahon et al. showed most asymptomatic relatives of dilated cardiomyopathy patients with mild left ventricular enlargement already showed infiltration of inflammatory cells, at levels that were similar to those of patients with established disease [43]. Additionally, previous results indicated that MYBPC3 is considered one of the major hypertrophic cardiomyopathy-causing genes [44]. Lynch et al. have recently demonstrated that a proinflammatory response occurs in a form of genetic dilated cardiomyopathy caused by MYBPC3 mutation and this is likely a response to cardiac myocyte damage resulting from contractile dysfunction and mechanical stress [45]. The present study shows that minimal inflammatory foci might be a sign of early dilated cardiomyopathy in cases with various disease-causing variants, including in MYBPC3, and that some such cases may already have arrhythmogenic potential that can lead to sudden unexpected death. We should also note that the SCN5A variant p.Leu1988Arg, seen in Case 5, is associated with Long QT syndrome [46], and that other SCN5A variants may be causative of Brugada syndrome or Long QT syndrome depending on kind of variants. No abnormal QT interval was found before death in Case 5. Therefore, rather than evaluating SCN5A p.Leu1988Arg, we assessed two other dialted cardiomyopathy-causing variants that may contribute to ventricular premature contraction as a complication of minimal inflammatory foci associated with cardiomyopathy.
The present study has some limitations. We did not carry out a genetic investigation of “control cases” who died from known causes with minimal inflammatory foci in the heart or who died suddenly without minimal inflammatory foci. Full investigations of such age-matched, sex-matched, and comorbidity-matched control cases might establish the importance of our present findings. Arrhythmogenic events and related sudden unexpected death usually require both an abnormal myocardial substrate and an instigating trigger, such as exercise, being asleep, or emotional stress [47, 48]. However, many of the cases with minimal inflammatory foci in the present study were young adults and/or cases of unwitnessed death; therefore, we could not fully discuss the clinical appearance or precursor state of sudden unexpected death with respect to minimal inflammatory foci because of insufficient clinical and radiological data, and information immediately before death. Also we cannot explore the correlation between the kind of genetic variants and microscopic appearance including the location of the minimal inflammatory foci because of both paucity of the examined cases and the restricted lesion in examined cases. On the other hand, we found multiple variants in six cases. A significant proportion of patients with hypertrophic cardiomyopathy harbor multiple mutations [49]; however, the significance of multiple variants in arrhythmogenic cardiomyopathy and dilated cardiomyopathy remains to be determined. In addition, we could not find any pathogenic variant in one case with minimal inflammatory foci. Future investigation using advanced genetic analysis methods with appropriate control cases may more clearly reveal the pathogenesis of unexplained minimal inflammatory foci.
In a retrospective analysis of a series of autopsy cases, the cause of death in many cases with unexplained minimal inflammatory foci was evaluated as a possible arrhythmogenic event. Also, a genetic investigation using next generation sequencing showed that eight of 10 cases with unexplained minimal inflammatory foci had potential pathogenic variants of arrhythmnogenic cardiomyopathy or dilated cardiomyopathy-related genes. These findings show that minimal inflammatory foci are a tentative pathological finding in early stages of inherited cardiomyopathy and that some cases might have a risk of sudden cardiac death. Detection of minimal inflammatory foci by careful histopathological evaluation may promote comprehensive genetic investigation targeting cardiomyopathy-related genetic variants. Such an approach might provide a reliable diagnosis for sudden unexpected death autopsy cases, even if significant structural heart disease is not evident.
References
Vaartjes I, Hendrix A, Hertogh EM, et al. Sudden death in persons younger than 40 years of age: incidence and causes. Eur J Cardiovasc Prev Rehabil. 2009;16:592–6.
Virmani RBA, Farb A. Sudden cardiac death. Cardiovasc Pathol. 2001;10:211–8.
Basso C, Aguilera B, Banner J, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017;471:691–705.
Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–63.
Behr ER, Dalageorgou C, Christiansen M, et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 2008;29:1670–80.
Lubitz SA, Ellinor PT. Next-generation sequencing for the diagnosis of cardiac arrhythmia syndromes. Heart Rhythm. 2015;12:1062–70.
Basso C, Carturan E, Pilichou K, et al. Sudden cardiac death with normal heart: molecular autopsy. Cardiovasc Pathol. 2010;19:321–5.
Lahrouchi N, Raju H, Lodder EM, et al. Utility of post-mortem genetic testing in cases of sudden arrhythmic death syndrome. J Am Coll Cardiol. 2017;69:2134–45.
Hata Y, Kinoshita K, Mizumaki K, et al. Postmortem genetic analysis of sudden unexplained death syndrome under 50 years of age: a next-generation sequencing study. Heart Rhythm. 2016;13:1544–51.
Hata Y, Yoshida K, Kinoshita K, et al. Epilepsy-related sudden unexpected death: targeted molecular analysis of inherited heart disease genes using next-generation DNA sequencing. Brain Pathol. 2017;27:292–304.
Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genom Hum Genet. 2005;6:185–216.
Jacoby D, McKenna WJ. Genetics of inherited cardiomyopathy. Eur Heart J. 2012;33:296–304.
de Noronha SV, Behr ER, Papadakis M, et al. The importance of specialist cardiac histopathological examination in the investigation of young sudden cardiac deaths. Europace. 2014;16:899–907.
Nishida N, Chiba T, Ohtani M, et al. Relationship between cardiopulmonary resuscitation and injuries of the cardiac conduction system: pathological features and pathogenesis of such injuries. Crit Care Med. 2006;34:363–7.
Yamashita A, Sekizuka T, Kuroda M. VirusTAP: Viral genome-Targeted Assembly Pipeline. Front Microbiol. 2016;7:32.
Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547–53.
Nagasaki M, Yasuda J, Katsuoka F, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun. 2015;6:8018.
Papadakis M, Raju H, Behr ER, et al. Sudden cardiac death with autopsy findings of uncertain significance: potential for erroneous interpretation. Circ Arrhythm Electro. 2013;6:588–96.
Pugh TJ, Kelly MA, Gowrisankar S, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–8.
McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121:731–48.
Norton N, Robertson PD, Rieder MJ, et al. Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ Cardiovasc Genet. 2012;5:167–74.
McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26.
Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46.
Kapplinger JD, Landstrom AP, Salisbury BA, et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol. 2011;57:2317–27.
Wijeyeratne YD, Behr ER. Sudden death and cardiac arrest without phenotype: the utility of genetic testing. Trends Cardiovasc Med. 2017;27:207–13.
Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376:61–72.
Garcia-Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–21.
Burke AP, Farb A, Tashko G, et al. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different diseases? Circulation. 1998;97:1571–80.
te Riele AS, James CA, Rastegar N, et al. Yield of serial evaluation in at-risk family members of patients with ARVD/C. J Am Coll Cardiol. 2014;64:293–301.
Basso C, Thiene G, Corrado D, et al. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–91.
Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–87.
Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–14.
Corrado D, Zorzi A, Cerrone M, et al. Relationship between arrhythmogenic right ventricular cardiomyopathy and Brugada syndrome: new Insights from molecular biology and clinical implications. Circ Arrhythm Electro. 2016;9:e003631.
Cerrone M, Lin X, Zhang M, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. 2014;129:1092–103.
Seidelmann SB, Smith E, Subrahmanyan L, et al. Application of whole exome sequencing in the clinical diagnosis and management of inherited cardiovascular diseases in adults. Circ Cardiovasc Genet. 2017;10:e001573.
Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390:400–14.
Gulati A, Jabbour A, Ismail TF, et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. 2013;309:896–908.
Mewton N, Liu CY, Croisille P, et al. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57:891–903.
Halliday BP, Cleland JGF, Goldberger JJ, et al. Personalizing risk stratification for sudden death in dilated cardiomyopathy: the past, present, and future. Circulation. 2017;136:215–31.
Spezzacatene A, Sinagra G, Merlo M, et al. Arrhythmogenic phenotype in dilated cardiomyopathy: natural history and predictors of life-threatening arrhythmias. J Am Heart Assoc. 2015;4:e002149.
Mahon NG, Madden BP, Caforio AL, et al. Immunohistologic evidence of myocardial disease in apparently healthy relatives of patients with dilated cardiomyopathy. J Am Coll Cardiol. 2002;39:455–62.
Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–15.
Lynch TLt, Ismahil MA, Jegga AG, et al. Cardiac inflammation in genetic dilated cardiomyopathy caused by MYBPC3 mutation. J Mol Cell Cardiol. 2017;102:83–93.
Yoshinaga M, Kucho Y, Sarantuya J, et al. Genetic characteristics of children and adolescents with long-QT syndrome diagnosed by school-based electrocardiographic screening programs. Circ Arrhythm Electro. 2014;7:107–12.
Peacock J, Whang W. Psychological distress and arrhythmia: risk prediction and potential modifiers. Prog Cardiovasc Dis. 2013;55:582–9.
Hata Y, Kinoshita K, Kudo K, et al. Anomalous origin of the right coronary artery from the left coronary sinus with an intramural course: comparison between sudden-death and non-sudden-death cases. Cardiovasc Pathol. 2015;24:154–9.
Kelly M, Semsarian C. Multiple mutations in genetic cardiovascular disease: a marker of disease severity? Circ Cardiovasc Genet. 2009;2:182–90.
Acknowledgements
The authors thank Ms. Tamae Sasakura, Mr. Noboru Onozuka, Ms. Syuko Matsumori, and Mr. Osamu Yamamoto for their technical assistance. We thank Jeremy Allen, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This work was supported in part by JSPS KAKENHI Grant numbers JP18k10119 to YH and JP17k09263 to NN.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hata, Y., Hirono, K., Yamaguchi, Y. et al. Minimal inflammatory foci of unknown etiology may be a tentative sign of early stage inherited cardiomyopathy. Mod Pathol 32, 1281–1290 (2019). https://doi.org/10.1038/s41379-019-0274-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-019-0274-0
This article is cited by
-
Comprehensive pathological and genetic investigation of three young adult myotonic dystrophy type 1 patients with sudden unexpected death
Journal of Neurology (2023)
-
Myocarditis and inflammatory cardiomyopathy: current evidence and future directions
Nature Reviews Cardiology (2021)
-
A standardized postmortem protocol to assess the real burden of sudden infant death syndrome
Virchows Archiv (2020)
