
Abstract
Undifferentiated solid tumors with small blue round cell histology and expression of CD99 mostly resemble Ewing sarcoma. However, they also may include other tumors such as mesenchymal chondrosarcoma, synovial sarcoma, or small cell osteosarcoma. Definitive classification usually requires detection of entity-specific mutations. While this approach identifies the majority of Ewing sarcomas, a subset of lesions remains unclassified and, therefore, has been termed “Ewing-like sarcomas” or small blue round cell tumors not otherwise specified. We developed an approach for further characterization of small blue round cell tumors not otherwise specified using an array-based DNA-methylation profiling approach. Data were analyzed by unsupervised clustering and t-distributed stochastic neighbor embedding analysis and compared with a reference methylation data set of 460 well-characterized prototypical sarcomas encompassing 18 subtypes. Verification was performed by additional FISH analyses, RNA sequencing from formalin-fixed paraffin-embedded material or immunohistochemical marker analyses. In a cohort of more than 1,000 tumors assumed to represent Ewing sarcomas, 30 failed to exhibit the typical EWS translocation. These tumors were subjected to methylation profiling and could be assigned to Ewing sarcoma in 14 (47%), to small blue round cell tumors with CIC alteration in 6 (20%), to small blue round cell tumors with BCOR alteration in 4 (13%), to synovial sarcoma and to malignant rhabdoid tumor in 2 cases each. One single case each was allotted to mesenchymal chondrosarcoma and adamantinoma. 12/14 tumors classified as Ewing sarcoma could be verified by demonstrating either a canonical EWS translocation evading initial testing, by identifying rare breakpoints or fusion partners. The methylation-based assignment of the remaining small blue round cell tumors not otherwise specified also could be verified by entity-specific molecular alterations in 13/16 cases. In conclusion, array-based DNA-methylation analysis of undifferentiated tumors with small blue round cell histology is a powerful tool for precisely classifying this diagnostically challenging tumor group.
Similar content being viewed by others


Introduction
Ewing sarcoma is a highly malignant tumor that accounts for ~8 % of all bone tumors. Although Ewing sarcoma is a rare disease, it represents the second most common malignant bone tumor in children and adolescents with a peak incidence in the second decade of life [1]. Ewing sarcoma is a prototypical example for an undifferentiated sarcoma with small blue round cell phenotype, which, however, is shared with several other sarcoma entities. Unfortunately, expression of CD99, typical for Ewing sarcoma, is also encountered in several morphological mimics. Therefore, it is not sufficient diagnosing Ewing sarcomas based on histologic criteria and immunohistochemical marker expression alone [2].
A breakthrough in the diagnostics of Ewing sarcoma was the discovery of two highly specific translocations, which both result in a gene fusion between a member of the TET and ETS gene families. The two most common translocations in Ewing sarcoma either lead to a chimeric gene fusion between EWSR1–FLI1 or EWSR1–ERG seen in ~85–90 % and 5–10 % of all cases, respectively [3, 4]. Gene fusions with other TET and ETS family members and rearrangements of EWSR1 with non-ETS family genes have also been described. However, such fusions are exceptionally rare occurring in <1 % of Ewing sarcomas [5, 6].
Molecular analysis of Ewing sarcomas and mimics sharing histological and clinical features by extended FISH analysis and next generation sequencing has separated Ewing sarcomas with canonical translocations from so-called “Ewing-like” sarcomas. Subgroups of “Ewing-like” sarcomas carry a CIC–DUX4 or a BCOR–CCNB3 gene fusion or other specific molecular alterations [7,8,9,10]. However, frequently “Ewing-like” sarcomas remain molecularly undefined due to the rarity and/or diversity of the discriminating molecular features and the lack of established routine techniques to detect them. These tumors usually are placed with a basket category termed small blue round cell tumors not otherwise specified [11].
The methylation status of gene promoters is a strong indicator of the differentiation status along cell lineages [12, 13]. Interestingly, this approach has also been shown very useful in distinguishing the tumor cell origin in a lineage dependent manner [14]. Moreover, this approach allows molecular classification within seemingly morphological homogenous entities [15,16,17,18,19,20,21,22,23]. The considerable stability of epigenetic signatures during tumor disease has been demonstrated for many tumors [18, 24, 25]. Thus, determination of DNA-methylation signatures may also be useful for addressing “Ewing-like” sarcomas or small blue round cell tumors not otherwise specified.
In the present study we aimed at characterization of 30 sarcomas believed to represent Ewing sarcomas but not exhibiting the canonical EWS translocations at initial testing by reference pathology.
Material and methods
Sample selection
The 30 samples, all from different patients, were identified from a series of more than 1,000 patients referenced in the cooperative Ewing’s sarcoma study (CESS) centre in Germany [26,27,28]. Clinical characteristics are given in Supplementary Table S1. The CESS reference pathology in Muenster tested these samples negative for a translocation between EWSR1 with either FLI1 or ERG by applying RT-PCR that covered the most common fusion breakpoints. The diagnostic repertoire has been expanded to EWSR1break-apart and FUS break-apart FISH since 2014. The reference set for building a framework of methylation groups comprised a total of 460 prototypical sarcoma cases from primary manifestations and metastases encompassing classic adamantinoma of the tibia with prominent epithelial component (n = 6), alveolar soft part sarcoma (n = 22), mesenchymal chondrosarcoma (n = 9), clear cell sarcoma of the kidney (n = 12), conventional osteosarcoma (n = 82), dermatofibrosarcoma protuberans (n = 39), desmoplastic small round cell tumor (n = 28), epithelioid sarcoma (n = 17), Ewing sarcoma carrying a gene fusion either between EWSR1–FLI1 (n = 41), EWSR1–ERG (n = 3) or FUS–ERG (n = 1), or showing an EWSR1 break-apart signal in the FISH test (n = 12), infantile fibrosarcoma (n = 13), malignant peripheral nerve sheath tumor (n = 22), malignant rhabdoid tumor (n = 18), embryonal rhabdomyosarcoma (n = 31), alveolar rhabdomyosarcoma (n = 33), small blue round cell tumor with BCOR–CCNB3 fusion (n = 8), small blue round cell tumor with an rearrangement of CIC (n = 10), solitary fibrous tumor (n = 22), and synovial sarcoma (n = 31). Reference cases of genetically defined sarcoma subtypes were molecularly confirmed. The reference set also includes a control group composed of non-neoplastic reactive soft tissue (n = 10). The study was done in concordance with the guidelines set forth by the local ethics committee of the University of Heidelberg and Muenster.
Genomic DNA and total RNA extraction
Representative formalin-fixed paraffin-embedded tumor tissue with highest available tumor content was chosen for extraction of RNA and DNA. Total cellular RNA was obtained using the Maxwell® 16FFPE Plus LEV RNA Kit and genomic DNA using the Maxwell® 16FFPE Plus LEV DNA Kit employing the automated Maxwell device (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The quality and concentration of RNA was determined on an Agilent 2100 Bioanalyzer® (Agilent Technologies, Santa Clara, CA, USA). DNA was quantified using the QuantiFast SYBR Green PCR Kit (Qiagen, Duesseldorf, NW, Germany).
Methylation array data generation and pre-procession
The DNA-methylation status was obtained using the Illumina Infinium HumanMethylation450 (450 k) array or the EPIC array (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions at the Genomics and Proteomics Core Facility of the DKFZ. DNA input quantity from formalin-fixed paraffin-embedded tumor material was 250 ng (recommended by manufacturer). Equal data quality was obtained for DNA input down to 100 ng [24]. The turnaround time for the entire workflow starting with sample preparation, array processing, scanning and data analysis was approximately 5 working days. DNA-methylation data were normalized by performing background correction and dye bias correction shifting of negative control probe mean intensity to zero and scaling of normalization control probe mean intensity to 10,000, respectively. Probes targeting sex chromosomes and probes containing single nucleotide polymorphism that not uniquely matched were removed. In total, 438,370 probes contained on both, the 450 k array and the EPIC array, were used for analysis.
Unsupervised clustering, copy number profiling, and identification of differentially methylated regions
For unsupervised hierarchical clustering, we selected 10,000 probes that showed the highest standard deviation across the beta values. Samples were hierarchically clustered using the Pearson correlation coefficient as distance measure and average linkage. The CpGs were reordered using the Euclidian distance and complete linkage. For unsupervised 2D representation of pairwise sample correlations dimensionality reduction by t-distributed stochastic neighbor embedding was performed using the 20,000 most variable probes, a perplexity of 15 and 3,000 iterations. Copy number profiles were generated using the ‘conumee’ R package (http://www.bioconductor.org) and assessed manually.
Targeted RT-RNA and total RNA sequencing
The TruSeq RNA Access Library Prep Kit for formalin-fixed paraffin-embedded material (Illumina) was applied for total RNA sequencing according to the manufacturer’s instructions. RNA libraries were sequenced on a NextSeq sequencer system (Illumina). Gene fusion transcripts were called from the RNA sequencing data using both deFuse and TopHat-Fusion algorithms [29, 30]. If automated detection of gene fusions was negative, reads of candidate genes and their 3′ and 5′ intergenic neighborhood were manually investigated using the Integrative Genomics Viewer (IGV) [31]. For targeted RNA sequencing the RNA was reverse transcribed and then subjected to PCR amplification using primer pairs covering the breakpoints of the common gene fusions of Ewing sarcoma (EWSR1–FLI1, EWSR1–ERG). BCOR–CCNB3 rearrangements were detected by RT-PCR as well. Primer sequences are listed in Supplementary Table S2.
Fluorescent in situ hybridization
For interphase FISH, the slides were subjected to hybridization with the ZytoLight ® SPEC EWSR1 dual color break-apart probe, the ZytoLight ® SPEC FUS dual color break-apart probe or the ZytoLight ® SPEC SS18 dual color break-apart probe (all ZytoVision, Bremerhafen, HB, Germany) according to the manufacturer’s instructions. A CIC break apart assay was designed and performed using BAC clones RP11-374A11 and RP11-979P13 (Life Technologies, Carlsbad, CA, USA) essentially following protocols as described before [32]. Hybridization signals were visualized with a DM5500 fluorescence microscope (Leica, Wetzlar, HE, Germany), and images were captured on a CCD camera.
Gene panel next generation sequencing
A customized SureSelect XT technology (Agilent) panel covering the coding regions of 130 genes, including BCOR and CIC, was applied to fusion-negative cases where DNA-methylation profiling indicated a BCOR or CIC alteration. Library preparation, quality control, sequencing on a NextSeq sequencer (Illumina) and data processing were exactly performed as previously described [33]. Reads were aligned against the reference genome hg19. Reads covering the BCOR locus (NM_001123383) were visualized in IGV and assessed manually for alterations.
Immunohistochemistry
A representative block was chosen for immunohistochemistry. Four micron paraffin sections were dried at 80 °C for 15 min and stained on a Ventana BenchMark XT immunostainer (Ventana Medical Systems, Tucson, AZ, USA) using standard techniques (Supplementary Table S3).
Results
Clinical features of the study cohort
A total of 30 CESS trial patients with the diagnosis of small blue round cell tumor not otherwise specified after histological and molecular evaluation by an expert panel of pathologists were subjected to DNA-methylation profiling. In total, 26 cases were from the primary tumor, 3 from a metastatic tumor manifestation and 1 case from a recurrence. In total, 18 patients were male, 12 were female. The median age at diagnosis was 17 (range 0–55) years. The primary manifestation site was skeletal in all but two cases. The majority of cases originated at the lower extremity (12/30), thoracic wall (6/30), pelvis (6/30), the upper extremity (4/30), and head and neck (2/30). Clinical data are compiled in Supplementary Table 1.
Epigenetic profiling assigned small blue round cell tumors not otherwise specified to distinct sarcoma subtypes
Genome-wide DNA-methylation profiles were generated of these 30 small blue round cell tumors not otherwise specified. Unsupervised clustering and t-distributed stochastic neighbor embedding analysis (Fig. 1) together with 460 sarcomas from the reference series allotted the 30 small blue round cell tumors not otherwise specified to different methylation classes corresponding to sarcomas with a defined histology and characteristic molecular hallmarks: 14 (47%) assigned to Ewing sarcoma, 6 (20%) to small blue round cell tumors with CIC alteration, 4 (13%) to small blue round cell tumors with BCOR alteration, which is a methylation group composed of small blue round cell tumors with BCOR–CCNB3 fusion and clear cell sarcoma of the kidney with BCOR internal tandem duplication, two (7%) to synovial sarcomas, two (7%) to malignant rhabdoid tumors, and one (3%) to mesenchymal chondrosarcomas. Interestingly, one small blue round cell tumor not otherwise specified (3%) clustered together with classic adamantinomas. Similar results were obtained when varying the number of CpGs used for the analysis (data not shown).

Assignment of 30 small blue round cell tumors not otherwise specified to methylation groups of reference sarcoma sets. a Unsupervised hierarchical clustering analysis, and b t-distributed stochastic neighbor embedding using the 10,000 most variable DNA-methylation probes of array-generated DNA-methylation profiles from the Illumina Infinium HumanMethylation450 or EPIC BeadChip (Illumina, San Diego, USA). Black bars/circles indicate the positions of the 30 small blue round cell tumors not otherwise specified. Abbreviations: Ada adamantinoma (* suspect), ASPS alveolar soft part sarcoma, mCS mesenchymal chondrosarcoma, CCSK clear cell sarcoma of the kidney, CT control tissue of non-neoplastic inflammatory origin, OS conventional osteosarcoma, DFSP dermatofibrosarcoma protuberans, DSRCT desmoplastic small round cell tumor, ES epithelioid sarcoma, EwS Ewing sarcoma, IFS infantile fibrosarcoma, MPNST malignant peripheral nerve sheath tumor, MRT malignant rhabdoid tumor, eRMS embryonal rhabdomyosarcoma, aRMS alveolar rhabdomyosarcoma, SBRCT (BCOR) small blue round cell tumor with BCOR alteration (BCOR–CCNB3 fusion = yellow; internal tandem duplication = yellow with greyish contour), SBRCT (CIC) small blue round cell tumor with CIC alteration, SFT solitary fibrous tumor, SySa synovial sarcoma
Genetic analyses validate the predicted sarcoma subtypes in most cases
To further validate the 30 small blue round cell tumors not otherwise specified, which by the DNA-methylation profiles were assigned to defined sarcoma subtypes, we analyzed these samples for molecular hallmark alterations. The results are compiled in Table 1. Cases falling into the Ewing sarcoma methylation class were tested by FISH, RT-PCR and total RNA sequencing. By applying this procedure, 6 of 14 tumors harbored a gene fusion between EWSR1 and FLI1. The EWSR1–FEV fusion was detected once and so was the EWSR1–ETV1 fusion. Four cases presented with a break-apart signal in the FISH analysis, three cases in EWSR1 and one case in FUS. The gene fusion partner could not be further determined due to lack of sufficient tissue. In two cases, where material was only sufficient for FISH analysis, one case was negative for a break-apart signal in EWSR1 or FUS and the other case was non-determinable.
Cases falling into the small blue round cell tumors with CIC alteration methylation class were tested by FISH and total RNA sequencing. Four of six tumors showed a CIC break-apart signal in the FISH analysis, indicating a rearrangement of the CIC locus. A CIC–DUX4 fusion was revealed by RNA sequencing in two of them. Furthermore, RNA sequencing indicated a CIC–DUX4 fusion in one of the two cases without a CIC break-apart signal.
Cases assigning to the methylation class small blue round cell tumors with BCOR alteration were tested by RT-PCR, total RNA sequencing and panel sequencing of the entire coding region of BCOR if RNA-based methods were negative. Three of four tumors carried the BCOR–CCNB3 fusion. In the fusion-negative case, a BCOR internal tandem duplication spanning 66 bases in exon 15 was detected (Fig. 2).

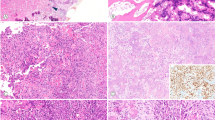
BCOR internal tandem duplication in a skull tumor of an infant. The genomic footprint of BCOR is depicted (a). The duplicated 66 bp sequence in Exon 15, which encodes for the PCGF Ub-like fold discriminator (PUFD) domain at the C-terminus of BCOR, is recognizable by a sharply demarcated doubling of the coverage rate. Rosette formations are prominent in some tumor areas (b). The tumor cells focally present with a vacuolated cytoplasm (c).
Two tumors clustered with the methylation class for synovial sarcomas and were rearranged in the SS18 locus. Histological re-evaluation lead to classification as poorly differentiated synovial sarcoma (Fig. 3a,b).

Histologic phenotypes of two representative Ewing-like sarcomas. Case 83172 (a–c) represents a poorly differentiated synovial sarcoma exhibiting a small blue round cell phenotype (a). FISH analysis revealed a SS18 break-apart signal indicated by two separated green and red signals (b). Interestingly, some tumor cells express CD99 (c). Case 94172 (d, e) exhibits an epithelioid to rhabdoid phenotype (d). Nuclear INI-1 expression is lost (d; inlet). Copy number analysis demonstrated a loss on chromosome arm 22q involving the SMARCB1 locus (e)
Two tumors clustered with the methylation class for malignant rhabdoid tumor and could be demonstrated to be SMARCB1 deficient by copy number analysis and by INI-1 immunohistochemistry (Fig. 3c,d).
One tumor clustered with mesenchymal chondrosarcomas. Molecular testing by RNA sequencing did not reveal the HEY1–NCOA2 fusion being expected in these malignancies. Cartilaginous areas or staghorn-shaped blood vessels, both frequent findings in mesenchymal chondrosarcomas, were absent in this case. Furthermore, the tumor was almost negative for S100 and CD99 expression (Fig. 4).

Histological phenotype of an unusual mesenchymal chondrosarcoma. Case 95322 shows an organoid growth pattern at low-power view (a). The small round tumor cells are arranged in sheets (b). In less cellular parts the tumor shows a vague reticulated growth pattern (c). The tumor is almost negative for S100 (d) and CD99 (e). A copy number analysis demonstrates several whole-chromosome gains (f)
One round cell tumor, which clustered together with adamantinomas, carried an EWSR1–NFATC2 gene fusion. CD99 expression was faint, but yet recognizable positive. The copy number profile, which was calculated from the methylation array data, indicated a complex alteration on chromosome 22q (Fig. 5). A very similar chromosome 22q alteration was found in one of the six classic adamantinomas. The histology of this particular case was unique compared with the other adamantinomas (Supplementary Figure S1). Unfortunately, the EWSR1 break-apart FISH analysis failed in this case.

Histologic phenotype of case with an EWSR1–NFATC2 gene fusion. Case 97480 is a highly cellular round cell tumor with an indistinctive growth pattern (a). The tumor cells faintly express CD99 (b). A copy number analysis demonstrates complex chromosomal alterations, e.g., on chromosome 22, on an otherwise relatively balanced background (c)
Discussion
The designations “Ewing-like” sarcoma or small blue round cell tumor not otherwise specified are highly unsatisfactory because they are employed in diagnostic settings having failed to produce unequivocal evidence for genetically defined sarcoma entities. Here, we demonstrated that methylation analysis is a powerful method, which assigns tumors currently named “Ewing-like” sarcoma and small blue round cell tumor not otherwise specified to well-defined diagnostic categories.
Validation analysis clearly supported the methylation-based diagnostic assignment in 25 of 30 cases. In these, the detection of characteristic alterations was supportive for the prediction of the DNA-methylation-based analysis. It is noteworthy that methylation-based assignment also correctly recognized sarcoma subtypes carrying exceedingly rare mutations, e.g., in Ewing sarcomas.
In only four cases, no characteristic alteration could be detected, probably due to the limitations of the applied methods. These four cases allotted to the methylation class Ewing sarcoma (n = 2), to small blue round cell tumors with CIC alteration (n = 1) and to mesenchymal chondrosarcoma (n = 1). In the latter case, the diagnosis could not be molecularly validated since it lacked the typical HEY1–NCOA2 fusion described as a recurrent event in most, albeit not in all mesenchymal chondrosarcomas [34]. Given the distinctive DNA-methylation signature in mesenchymal chondrosarcoma, we re-classified this case as mesenchymal chondrosarcoma with predominant small round cell component. In the two cases that assigned to the methylation class Ewing sarcoma, molecular validation was restricted to FISH analysis. The case matching with the methylation class small blue round cell tumors with CIC alteration had segmental copy number gains on chromosome arm 19q involving the CIC locus, which may have had an adverse effect on the FISH analysis (data not shown). Therefore, we consider it likely that DNA-methylation-based prediction is also correct for those cases lacking the expected molecular alterations.
However, one outlier, which clustered in close proximity with classic adamantinomas of the tibia, surprisingly contained an EWSR1–NFATC2 gene fusion. This uncommon fusion, first described in 2009, has yet been detected only in single bone tumors presenting with a phenotype similar to Ewing sarcoma [5, 11]. Interestingly, the EWSR1–NFATC2 gene fusion has also been described in one case with a hemangioma of the bone, although the breakpoint in EWSR1 was more distal compared to the Ewing sarcoma-like cases [35]. In the present case, the EWSR1–NFATC2 breakpoint was matching with the one observed in Ewing sarcoma-like cases. The copy number profile of this tumor showed an unusual copy number alteration on chromosome 22q, which was also detected in one reference case classified as classic adamantinoma of the tibia. The histologic pattern of this reference case was well compatible with the histological features described in small blue round cell tumors carrying the EWSR1–NFATC2 fusion [5, 11, 36]. All of this makes not only the initial diagnosis of this particular reference case questionable, but also raise the question of the close epigenetic relation between EWSR1–NFATC2-fused small blue round cell tumors and classic adamantinomas. The unusual “dot-like” cytokeratin expression pattern in EWSR1–NFATC2-fused small blue round cell tumors might at least be suggestive of an epithelial differentiation similar to classic adamantinoma, which is characterized by a prominent epithelial cell component [36]. However, we also cannot exclude a clustering artifact with absolute certainty. Thus, this case remained unsolved.
Methylation-based diagnostic assignment predicted four small blue round cell tumors with BCOR alteration. This methylation class is composed of different tumor subtypes sharing the molecular background of BCOR alteration either by rearrangement, internal tandem duplication or point mutation. Small blue round cell tumors with BCOR–CCNB3, a sarcoma subtype probably being independent from the group of Ewing sarcoma, belongs to this methylation class [37]. Accordingly, the BCOR–CCNB3 fusion was detected in three cases [8, 10]. The fourth case, an infant patient with a small blue round cell tumor in the skull, carried a BCOR internal tandem duplication. This case was re-classified as undifferentiated round cell sarcoma of infancy, in which the BCOR internal tandem duplication has been described as a highly recurrent event [38].
BCOR alterations have been recognized in a growing number of tumor subtypes affecting the soft tissue, the kidney or the central nervous system [22, 38, 39]. Recent studies comparing the molecular and histologic phenotype in these BCOR altered tumors found overlapping features, especially between soft tissue and kidney tumors [38, 40, 41]. At the level of DNA-methylation, we observed a great overlap in tumors from soft tissue and the kidney carrying BCOR alteration. Furthermore, using an extended set of tumor subtypes for DNA-methylation profiling (data not shown), also central nervous system high-grade neuroepithelial tumors with BCOR alteration fall into this methylation class [22]. This is in line with the previously suggested concept that these tumors might constitute a family of tumors sharing BCOR alterations.
Overall, DNA-methylation profiling might potentially have clinical implications in the molecular diagnostics of small blue round cell tumors, as already seen in central nervous system tumors [22, 25]. Four recently published studies observed misleading results of molecular tests widely used in the diagnostics of small blue round cell tumors. A deletion of the SMARCB1 locus on chromosome arm 22q, which located juxtaposed to the EWSR1 locus, can result in a false-positive EWSR1 break-apart FISH signal, although the gene integrity is retained [42]. Decision-making might further be impeded by false negative results in the molecular testing of small blue round cell tumors with CIC alteration [9, 43,44,45]. Two recent articles focused on the performance of molecular tests in small blue round cell tumors with CIC rearrangement observed an overall low performance of break-apart FISH and total RNA sequencing in these tumors [46, 47]. Their results reflect our experience with molecular testing in small blue round cell tumors with CIC alteration. We could demonstrate a CIC rearrangement in four of six cases by break-apart FISH analysis. Copy number profiling of the two break-apart negative cases revealed complex alterations involving the CIC locus on chromosome 19q, which may have had an adverse effect on the FISH analysis (data not shown). Furthermore, two different automated algorithms for fusion discovery from RNA data did not detect the underlying gene fusion in any of these six cases. This may come down to the highly repetitive DNA sequences juxtaposing the breakpoint in CIC. In three cases, the underlying CIC–DUX4 fusion could only be detected by manually reviewing the reads of these genes in IGV.
A more practicable method might be using a surrogate marker. Gene expression profiling revealed an upregulation of ETS transcription factors (ETV1 on chromosome 7p, ETV4 on chromosome 17q, ETV5 on chromosome 3q) in small blue round cell tumors with CIC–DUX4 fusion [47,48,49,50]. Likewise, BCOR overexpression has been proposed as a potential biomarker for diagnosing small blue round cell tumors with BCOR alteration, although diagnostic pitfalls have already been recognized [37, 51].
In conclusion, array-based DNA-methylation molecular profiling is a robust method that proved extraordinary powerful for clarifying the diagnoses of a cohort of tumors initially deemed small blue round cell tumor not otherwise specified. Although it remains to be seen whether DNA-methylation may delineate sarcoma subtypes not investigated in this study, this approach already now can be highly useful in the diagnosis of Ewing sarcoma and mimics.
References
Alava ED, Lessnick SL, Sorensen PH. In: Ewing sarcoma. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. WHO classification of tumours of soft tissue and bone. Lyon: International Agency for Research on Cancer (IARC); 2013. p. 305–10. .
Folpe AL, Hill CE, Parham DM, O’Shea PA, Weiss SW. Immunohistochemical detection of FLI-1 protein expression: a study of 132 round cell tumors with emphasis on CD99-positive mimics of Ewing’s sarcoma/primitive neuroectodermal tumor. Am J Surg Pathol. 2000;24:1657–62.
Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–5.
Sorensen PH, Lessnick SL, Lopez-Terrada D, et al. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994;6:146–51.
Szuhai K, Ijszenga M, de Jong D, et al. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. 2009;15:2259–68.
Sumegi J, Nishio J, Nelson M, et al. A novel t(4;22)(q31; q12) produces an EWSR1-SMARCA5 fusion in extraskeletal Ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol. 2011;24:333–42.
Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35; q13) translocation. Hum Mol Genet. 2006;15:2125–37.
Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–6.
Italiano A, Sung YS, Zhang L, et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer. 2012;51:207–18.
Specht K, Zhang L, Sung YS, et al. Novel BCOR-MAML3 and ZC3H7B-BCOR gene fusions in undifferentiated small blue round cell sarcomas. Am J Surg Pathol. 2016;40:433–42.
Antonescu C. Round cell sarcomas beyond Ewing: emerging entities. Histopathology. 2014;64:26–37.
Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–32.
Straussman R, Nejman D, Roberts D, et al. Developmental programming of CpG island methylation profiles in the human genome. Nat Struct Mol Biol. 2009;16:564–71.
Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–92.
Johann PD, Erkek S, Zapatka M, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. 2016;29:379–93.
Koelsche C, Hovestadt V, Jones DT, et al. Melanotic tumors of the nervous system are characterized by distinct mutational, chromosomal and epigenomic profiles. Brain Pathol. 2015;25:202–8.
Mahoney SE, Yao Z, Keyes CC, Tapscott SJ, Diede SJ. Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics. 2012;7:400–8.
Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27:728–43.
Rohrich M, Koelsche C, Schrimpf D, et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016;131:877–87.
Schwalbe EC, Williamson D, Lindsey JC, et al. DNA methylation profiling of medulloblastoma allows robust subclassification and improved outcome prediction using formalin-fixed biopsies. Acta Neuropathol. 2013;125:359–71.
Seki M, Nishimura R, Yoshida K, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun. 2015;6:7557.
Sturm D, Orr BA, Toprak UH, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 2016;164:1060–72.
Sturm D, Witt H, Hovestadt V, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37.
Hovestadt V, Remke M, Kool M, et al. Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol. 2013;125:913–6.
Sahm F, Schrimpf D, Stichel D, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18:682–94.
Gaspar N, Hawkins DS, Dirksen U, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. 2015;33:3036–46.
Ladenstein R, Potschger U, Le Deley MC, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol. 2010;28:3284–91.
Le Deley MC, Paulussen M, Lewis I, et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol. 2014;32:2440–8.
Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72.
McPherson A, Hormozdiari F, Zayed A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138.
Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Braun M, Stomper J, Boehm D, et al. Improved method of detecting the ERG gene rearrangement in prostate cancer using combined dual-color chromogenic and silver in situ hybridization. J Mol Diagn. 2012;14:322–7.
Sahm F, Schrimpf D, Jones DT, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016;131:903–10.
Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer. 2012;51:127–39.
Arbajian E, Magnusson L, Brosjo O, et al. A benign vascular tumor with a new fusion gene: EWSR1-NFATC1 in hemangioma of the bone. Am J Surg Pathol. 2013;37:613–6.
Sadri N, Barroeta J, Pack SD, et al. Malignant round cell tumor of bone with EWSR1-NFATC2 gene fusion. Virchows Arch. 2014;465:233–9.
Kao YC, Sung YS, Zhang L, et al. BCOR overexpression is a highly sensitive marker in round cell sarcomas with BCOR genetic abnormalities. Am J Surg Pathol. 2016;40:1670–8.
Kao YC, Sung YS, Zhang L, et al. Recurrent BCOR internal tandem duplication and YWHAE-NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol. 2016;40:1009–20.
Ueno-Yokohata H, Okita H, Nakasato K, et al. Consistent in-frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet. 2015;47:861–3.
Argani P, Kao YC, Zhang L, et al. Primary renal sarcomas With BCOR-CCNB3 gene fusion: a report of 2 cases showing histologic overlap with clear cell sarcoma of kidney, suggesting further link between BCOR-related sarcomas of the kidney and soft tissues. Am J Surg Pathol. 2017;41:1702–12.
Yoshida Y, Nobusawa S, Nakata S, et al. CNS high-grade neuroepithelial tumor with BCOR internal tandem duplication: a comparison with its counterparts in the kidney and soft tissue. Brain Pathol. 2017. https://doi.org/10.1111/bpa.12585.
Huang SC, Zhang L, Sung YS, et al. Secondary EWSR1 gene abnormalities in SMARCB1-deficient tumors with 22q11-12 regional deletions: Potential pitfalls in interpreting EWSR1 FISH results. Genes Chromosomes Cancer. 2016;55:767–76.
Solomon DA, Brohl AS, Khan J, Miettinen M. Clinicopathologic features of a second patient with Ewing-like sarcoma harboring CIC-FOXO4 gene fusion. Am J Surg Pathol. 2014;38:1724–5.
Sugita S, Arai Y, Tonooka A, et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol. 2014;38:1571–6.
Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41:941–49.
Yoshida A, Arai Y, Kobayashi E, et al. CIC break-apart fluorescence in-situ hybridization misses a subset of CIC-DUX4 sarcomas: a clinicopathological and molecular study. Histopathology. 2017;71:461–9.
Kao YC, Sung YS, Chen CL, et al. ETV transcriptional upregulation is more reliable than RNA sequencing algorithms and FISH in diagnosing round cell sarcomas with CIC gene rearrangements. Genes Chromosomes Cancer. 2017;56:501–10.
Specht K, Sung YS, Zhang L, et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer. 2014;53:622–33.
Le Guellec S, Velasco V, Perot G, et al. ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol. 2016;29:1523–31.
Smith SC, Palanisamy N, Martin E, et al. The utility of ETV1, ETV4 and ETV5 RNA in-situ hybridization in the diagnosis of CIC-DUX sarcomas. Histopathology. 2017;70:657–63.
Kao YC, Sung YS, Zhang L, et al. BCOR upregulation in a poorly differentiated synovial sarcoma with SS18L1-SSX1 fusion-A pathologic and molecular pitfall. Genes Chromosomes Cancer. 2017;56:296–302.
Acknowledgements
We thank Simone Kocher, Jochen Meyer, Antje Habel, Petra Fischer, Birgit Schulte and Maria Wolters for excellent technical support. We also thank Dr. Meybrit Rasper and Regina Kloss from the CESS study group for helpful assistance. We thank the Tissue Bank of the National Center for Tumor Diseases Heidelberg for providing samples, and the Microarray unit of the Genomics and Proteomics Core Facility, German Cancer Research Center (DKFZ), for providing excellent methylation services. This study was supported by a German Cancer Aid grant 70112499 (to CK, AvD) and grant 108128 (to UD); ERA-Net-TRANSCAN consortium, project number 01KT1310, and Euro Ewing Consortium EEC, project number EU-FP7 602856, both funded under the European Commission Seventh Framework Program FP7-HEALTH (to UD).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Koelsche, C., Hartmann, W., Schrimpf, D. et al. Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod Pathol 31, 1246–1256 (2018). https://doi.org/10.1038/s41379-018-0045-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0045-3
This article is cited by
-
DNA methylation patterns suggest the involvement of DNMT3B and TET1 in osteosarcoma development
Molecular Genetics and Genomics (2023)
-
Sarcoma classification by DNA methylation profiling in clinical everyday life: the Charité experience
Clinical Epigenetics (2022)
-
Small round cell sarcomas
Nature Reviews Disease Primers (2022)
-
Genome-wide DNA methylation profiling is able to identify prefibrotic PMF cases at risk for progression to myelofibrosis
Clinical Epigenetics (2021)
-
Clinicopathologic and molecular analysis of embryonal rhabdomyosarcoma of the genitourinary tract: evidence for a distinct DICER1-associated subgroup
Modern Pathology (2021)
