
Abstract
White organic light-emitting diodes (WOLEDs) is a new generation of lighting technology and has stimulated wide-ranging studies. Despite the advantage of simple device structure, single-emitting-layer WOLEDs (SEL-WOLEDs) still face the challenges of difficult material screening and fine energy level regulation. Herein, we report efficient SEL-WOLEDs with a sky-blue emitting cerium(III) complex Ce-TBO2Et and an orange-red emitting europium(II) complex Eu(Tp2Et)2 as the emitters, showing a maximum external quantum efficiency of 15.9% and Commission Internationale de l’Eclairage coordinates of (0.33, 0.39) at various luminances. Most importantly, the electroluminescence mechanism of direct hole capture and hindered energy transfer between the two emitters facilitate a manageable weight doping concentration of 5% for Eu(Tp2Et)2, avoiding the low concentration (<1%) of the low-energy emitter in typical SEL-WOLEDs. Our results indicate that d-f transition emitters may circumvent fine energy level regulation and provide development potential for SEL-WOLEDs.
Similar content being viewed by others

Introduction
White organic light-emitting diodes (WOLEDs) have a great application prospect in daily lighting owing to their merits of low energy consumption, eye protection, and additional flexibility potential1,2,3,4,5,6. Compared with stacked WOLEDs7 or multiple-emitting-layer WOLEDs8,9, single-emitting-layer WOLEDs (SEL-WOLEDs) are favored for commercialization due to the drastically simplified device structure and reduced production costs10,11. However, a formidable challenge lies in the rational control of both singlet and triplet excitons between host materials and different color emitters in the single-emitting-layer12,13,14,15,16, making simultaneous efficiency improvement and color control a fundamental issue in SEL-WOLEDs17,18,19,20,21,22. Recently, considerable efforts have been devoted to molecular design and energy level regulation in host-guest multicomponent molecular systems22,23,24,25,26,27.
The observed white electroluminescence in typical SEL-WOLEDs usually involves complicated energy transfer processes from host material to emitters and between different color emitters. Even for the relatively simple binary system with blue and yellow/red emitters in the single-emitting-layer, the Förster resonance energy transfer, Dexter energy transfer, and the direct charge capture by luminescent materials all together induce the device design complexity28,29,30. Furthermore, efficient energy transfer between different color emitters makes the emission spectra correlate greatly to the doping concentration. Actually, rather low doping concentration (<1%) of the low-energy emitting material is generally needed22,31,32,33,34, which further increases the device fabrication difficulty.
In order to simplify device design and fabrication, reducing energy transfer channels between host material and emitters in the multicomponent single-emitting-layer is an intuitive method. Recently, we found that host materials are hardly involved in energy transfer process when luminescent lanthanide d-f transition complexes were used as the emitters35,36, which may be a solution for simple energy transfer regulation. In addition, we have demonstrated that d-f transition complexes have many advantages as emitters in OLEDs, such as theoretical high efficiency, short excited state lifetime, tunable emission color, and inexpensive cost due to the abundance of cerium in Earth’s crust is even slightly higher than that of copper35,36,37,38,39,40. Therefore, we could expect the exploration of d-f transition emitters in SEL-WOLEDs.
As a proof of concept, we synthesized a sky-blue emitting cerium(III) complex Ce-TBO2Et and an orange-red emitting europium(II) complex Eu(Tp2Et)2 with d-f transition characteristic, and fabricated their SEL-WOLEDs with a simple three-layered device structure. It is found that the energy transfer from host material to emitters is eliminated and that between two emitters is hindered, hence we obtained efficient and color-stable white electroluminescence with a controllable weight doping ratio of 10% Ce-TBO2Et and 5% Eu(Tp2Et)2 in N,N-dicarbazolyl-3,5-benzene (mCP).
Results
Synthesis and structure
Based on the previous work38,39,41, potassium hydrotris(3,5-diethylpyrazolyl)borate (KTp2Et)42 is chosen to synthesize luminescent lanthanide d-f transition complexes due to its rigid coordination skeleton and good protection for the metal center, as well as our prediction that the moderate ligand field would lead to sky-blue emitting Ce(III) complex Ce-TBO2Et and orange-red emitting Eu(II) complex Eu(Tp2Et)2 as depicted in Fig. 1a. The complexes were characterized by elemental analysis and X-ray single crystal diffraction (Supplementary Table S1). Due to the large steric hinderance of two ethyl groups in the Tp2Et ligand which weakens the B–N bond43, oxygen is easily introduced in Ce-TBO2Et as a bridging coordination atom by the addition of water in the reaction mixture. Thus the Ce(III) ion in Ce-TBO2Et is coordinated with five N atoms and two O atoms (Supplementary Fig. S1), resulting in a distorted single-capped-octahedral coordination geometry as depicted in Fig. 1b, and the average Ce−N and Ce−O bond distances are 2.609 Å and 2.333 Å, respectively. In the complex Eu(Tp2Et)2, the central Eu(II) ion is coordinated with six N atoms and encapsulated in a staggered manner with a trigonal antiprismatic coordination geometry (Fig. 1b, Supplementary Fig. S1), and the average Eu−N bond distance is 2.611 Å. The percentages of buried volume (%VBur)44, which measures the compactness of the first coordination sphere, are calculated to be 92% for Ce-TBO2Et and 83% for Eu(Tp2Et)2, and the steric maps are shown in Fig. 1c.

a Synthesis and structures of Ce-TBO2Et and Eu(Tp2Et)2. b Coordination geometry of Ce-TBO2Et (left) and Eu(Tp2Et)2 (right). Atom notation: Ce, yellow; Eu, cyan; N, blue; O, red. c Steric maps of Ce-TBO2Et (left) and Eu(Tp2Et)2 (right), hydrogen atoms are considered in the calculation, parameters adopted here are Bondi radii scaled by 1.17, sphere radius 3.5 Å, and mesh spacing value s = 0.10 Å
Photophysical properties
To systematically study the photophysical properties of the two complexes, steady-state and transient spectra were measured in dichloromethane solution as well as in solid powder state under nitrogen atmosphere. All the photoluminescence data are summarized in Table 1. When dispersed in dichloromethane solution, the broad and featureless absorption bands above 300 nm were detected, which are attributed to the 4f1 → 5d1 transition of Ce(III) ion and 4f7 → 4f65d1 transition of Eu(II) ion, respectively (Supplementary Fig. S2). Further time-dependent density functional theory (TD-DFT) calculation with hole-electron analysis (Fig. 2a, b) and natural transition orbital (NTO) analysis (Supplementary Fig. S3) confirmed the dominant f-d transition characteristics, and the predicted absorption bands are consistent with the experimental data (Table 1). The photoluminescence spectra of Ce-TBO2Et in dichloromethane solution and as solid powder both displayed sky-blue emissions with maximum emission wavelengths (λmax) of 469 nm and 474 nm, respectively (Fig. 2c). The short single-exponential fluorescence decay lifetimes (τ) of 66 ns and 55 ns for Ce-TBO2Et in dichloromethane solution and as solid powder (Fig. 2d) coincide with general lifetime span of d-f transition Ce(III) complexes35,45. Moreover, Ce-TBO2Et exhibited high photoluminescence quantum yield (PLQY, ΦPL) close to 100% both in dichloromethane solution and as solid powder (Table 1). The radiative rate constants (kr) and non-radiative rate constants (knr) of Ce-TBO2Et were calculated. The complex in dichloromethane solution and as solid powder showed similar high kr (1.8 × 107 s−1 vs. 1.5 × 107 s−1) as well as similar low knr (0.018 × 107 s−1 vs. 0.015 × 107 s−1), indicating a rigid coordination of the ligands to the central Ce(III) ion.

a Distributions of the hole and electron for the lowest energy transition (at 407 nm) for Ce-TBO2Et. b Distributions of the hole and electron for the lowest energy transition (at 423 nm) for Eu(Tp2Et)2. c Emission spectra of Ce-TBO2Et, inset is the photograph of solution under 365 nm radiation. d Transient photoluminescence decay curves of Ce-TBO2Et. e Emission spectra of Eu(Tp2Et)2, inset is the photograph of solution under 365 nm radiation. f Transient photoluminescence decay curves of Eu(Tp2Et)2. Solid represents the complex in solid powder state. Solution represents the complex in 1 mM dichloromethane solution
The photoluminescence spectra of Eu(Tp2Et)2 in dichloromethane solution and as solid powder both displayed orange-red emissions with maximum wavelengths of 600 nm and 604 nm (Fig. 2e), and the excited state lifetimes are 17 ns and 428 ns (Fig. 2f), respectively. The single-peak spectra and nanosecond excited state lifetimes of Eu(Tp2Et)2 are consistent with those of the reported d-f transition Eu(II) complexes37,38. In addition, the ΦPL values of Eu(Tp2Et)2 in dichloromethane solution and as solid powder are only 2% and 20%, respectively. The kr and knr of Eu(Tp2Et)2 in dichloromethane solution and as solid powder were also calculated, showing much higher knr than kr. However, the complex exhibited lower knr in solid powder state than that in solution, indicating that non-radiative channels are suppressed due to the more rigid environment in solid state, therefore, the ΦPL in solid powder is greatly improved. Compared with Ce-TBO2Et, the λmax of Eu(Tp2Et)2 is red shifted and the ΦPL is decreased. The former may arise mainly from higher energy Efd for the transition between the lowest 4fn5d0 and the lowest 4fn−15d1 states of free (gaseous) Ce(III) ion than that of Eu(II) ion (~6 eV vs. ~4 eV)46, while the latter may be the result of lower encapsulation of Eu(II) ion by Tp2Et ligands, i.e. smaller %VBur as calculated above (92% for Ce-TBO2Et and 83% for Eu(Tp2Et)2), leading to the nonnegligible quenching of luminescence.
We have also respectively doped Ce-TBO2Et and Eu(Tp2Et)2 in poly(methylmethacrylate) (PMMA) film with a doping concentration of 10 wt%. The photophysical properties of the doped films under nitrogen atmosphere are depicted in Supplementary Fig. S4 and summed in Table 1. The absorption spectra are consistent with that of dichloromethane solution (Supplementary Figs. S4a and S2). The emission peaks of Ce-TBO2Et and Eu(Tp2Et)2 doped in PMMA films are 475 nm and 593 nm (Supplementary Fig. S4b), respectively. The excited state lifetime of Ce-TBO2Et doped in PMMA film is 60 ns, comparable to that of solid powder and solution, and the ΦPL is 98% (Table 1). When doped in PMMA film, Eu(Tp2Et)2 exhibits longer excited state lifetimes (110 ns and 632 ns, Supplementary Fig. S4c) as well as a higher ΦPL of 28% compared to that of solid powder and solution (Table 1), which may result from the suppressed concentration and solvent quenching.
Thermal and electrochemical properties
During the synthesis, we found that both Ce-TBO2Et and Eu(Tp2Et)2 can be purified by thermal gradient sublimation, making them suitable candidates for OLEDs fabrication with the vacuum thermal evaporation method. The detailed thermal stabilities of Ce-TBO2Et and Eu(Tp2Et)2 were examined by thermogravimetric analysis and differential scanning calorimetry under nitrogen atmosphere (Supplementary Fig. S5). The complexes exhibited high decomposition temperatures (Td, corresponding to 5% weight loss) above 240 °C, high glass-transition temperature (Tg, 139 °C for Ce-TBO2Et) and high melting temperatures (Tm) above 160 °C, indicating good thermal stability for OLEDs application.
To understand the frontier energy levels of the two complexes, electrochemical properties of Ce-TBO2Et and Eu(Tp2Et)2 were measured by cyclic voltammogram (Supplementary Fig. S6). The oxidation onsets against ferrocenium/ferrocene (Fc+/Fc) were observed as 1.0 V and −0.6 V for Ce-TBO2Et and Eu(Tp2Et)2, respectively. Thus, the highest occupied molecular orbital (HOMO) energy levels are deduced to be −5.8 eV and −4.2 eV, respectively. This leads us to the conclusion that Eu(Tp2Et)2 has a much shallower HOMO energy level than that of Ce-TBO2Et. The lowest unoccupied molecular orbital (LUMO) energy levels were then calculated to be −3.0 eV and −1.9 eV for Ce-TBO2Et and Eu(Tp2Et)2, by considering the HOMO-LUMO energy gaps of 2.8 eV and 2.3 eV predicted from the absorption spectra, respectively (Supplementary Fig. S2).
Electroluminescence performance
Due to the important role of host material in OLEDs, serval host materials were evaluated by measuring the ΦPL of Ce-TBO2Et or Eu(Tp2Et)2 doped films. In particular, the ΦPL of Ce-TBO2Et doped in mCP film could be as high as ~100%, and the ΦPL of Eu(Tp2Et)2 doped in N,N’-di-[(1-naphthalenyl)-N,N’-diphenyl]-1,1’-biphenyl)-4,4’-diamine (NPB), 4,4’,4”-tris(N-3-methylphenyl-N-phenylamino)triphenylamine (m-MTDATA), and mCP films are 46%, 46%, and 57%, respectively (Supplementary Table S2, Supplementary Fig. S7). Thus, mCP is considered as a good host material for both Ce-TBO2Et and Eu(Tp2Et)2, and used as host material in the fabrication of the following OLEDs. It should be noted that the ΦPL of Eu(Tp2Et)2 doped in mCP film increased significantly when compared with that in solution or in solid powder state, indicating that solvent quenching and concentration quenching may be suppressed in the doped film.
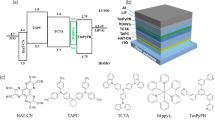
Prior to constructing SEL-WOLEDs, the electroluminescence properties of Ce-TBO2Et as a sky-blue emitter and Eu(Tp2Et)2 as an orange-red emitter were separately investigated by preparing the blue emitting device B1 and the orange-red emitting device O1 with a same device structure of indium tin oxide (ITO)/MoO3 (2 nm)/1-bis[4-[N,N’-di(4-tolyl)amino]phenyl]cyclohexane (TAPC, 60 nm)/mCP:Ce-TBO2Et (10 wt%, 30 nm) or mCP:Eu(Tp2Et)2 (12 wt%, 25 nm)/1,3,5-tri(m-pyrid-3-yl-phenyl)benzene (TmPyPB, 40 nm)/LiF (0.7 nm)/Al (100 nm). In the devices, MoO3 and TAPC serve as the hole injection layer and the hole transport layer, TmPyPB and LiF serve as the electron transport layer and the electron injection layer, respectively (see Supplementary Fig. S7 for the chemical structures of TAPC and TmPyPB). The two devices B1 and O1 showed only electroluminescence of emitters and no emission of host material was observed, which are different from the photoluminescence spectra of the emitting-layers with simultaneous emissions of the complex and mCP, respectively (Fig. 3a). The Ce-TBO2Et based device B1 showed a maximum luminance of 18,200 cd m−2 and a maximum EQE of 22.3%, while the Eu(Tp2Et)2 based device O1 showed a maximum luminance of 15,800 cd m−2 and a maximum EQE of 11.1% (Table 2, Fig. 3b, c). Noticeably, these performance surpass the best reported blue and red OLEDs with d-f transition lanthanide complexes (Supplementary Table S3)35,36,37,38,39,40,41,45,47,48.

a Photoluminescence spectra of 10 wt% Ce-TBO2Et doped mCP film and 12 wt% Eu(Tp2Et)2 doped mCP film (the excitation wavelength is 270 nm and 300 nm, respectively), and electroluminescence spectra of devices B1 and O1. b Current density-voltage-luminance traces of the devices B1, O1, and W1. c Power efficiency-luminance-EQE traces of the devices B1, O1 and W1. d Schematic device configuration and electroluminescence mechanism diagram of devices B1, O1 and W1. e Electroluminescence spectra of the device W1 at different luminances. f Electroluminescence spectra of devices Rx with different doping concentrations of Ir(bt)2acac
The differences between electroluminescence and photoluminescence spectra of Ce-TBO2Et or Eu(Tp2Et)2 in mCP suggest that hole and electron recombination dominantly occurs on d-f transition complexes rather than the host material, which avoids energy transfer from host molecule to the doped complex, so the emission of host material is no longer observed in the electroluminescence spectra. To clarify the electroluminescence mechanism, we fabricated two sets of hole-only devices with a device structure of ITO/MoO3 (2 nm)/mCP (40 nm)/mCP:Ce-TBO2Et or Eu(Tp2Et)2 (0, 5, 10 or 15 wt%, 25 nm)/mCP (40 nm)/MoO3 (2 nm)/Al. It is found that the current density of Ce-TBO2Et and Eu(Tp2Et)2 devices both reduced significantly first and raised then with the increased doping concentration of the complex, indicating that Ce-TBO2Et and Eu(Tp2Et)2 have the capability to capture holes (Supplementary Fig. S8). Furthermore, we could infer that Eu(Tp2Et)2 has priority to capture holes due to a much shallower HOMO energy level compared with that of Ce-TBO2Et. It is worth mentioning that although the excitons are not formed on mCP, the host material is important for electron transport. Furthermore, the high-energy triplet state (2.9 eV) of mCP is essential to prevent energy transfer from dopant to host material. Based on aforementioned studies, the electroluminescence mechanism of Ce-TBO2Et and Eu(Tp2Et)2 could be depicted in Fig. 3d. The holes transported by TAPC were captured by Ce-TBO2Et and Eu(Tp2Et)2, and then recombined with electrons transported by TmPyPB and mCP.
Encouraged by the results that both efficient sky-blue electroluminescence and orange-red electroluminescence were realized with the same simple three-layered device structure, we designed a SEL-WOLED by only changing the single emitting-layer with Ce-TBO2Et and Eu(Tp2Et)2 co-doped in mCP, and fabricated the device W1: ITO/MoO3 (2 nm)/TAPC (60 nm)/mCP:Ce-TBO2Et (10 wt%):Eu(Tp2Et)2 (5 wt%) (25 nm)/TmPyPB (40 nm)/LiF (0.7 nm)/Al (100 nm). The device showed a Commission Internationale de l´Eclairage (CIE) of (0.33, 0.39) at 1000 cd m−2, with emission wavelengths peaking at 490 nm and 580 nm (Fig. 3e), arising from Ce-TBO2Et and Eu(Tp2Et)2, respectively. And good color stability with CIE variation within (±0.01, ±0.01) from 10 to 10,000 cd m−2 was observed (Fig. 3e), meaning that the exciton allocations are basically independent on different exciton concentrations. The color temperature and color rendering index for the device W1 at 1000 cd m−2 are 5380 K and 76, respectively. Moreover, an intermediate efficiency between that of the devices B1 and O1 was obtained in the device W1, showing a maximum EQE of 15.9%, and the EQE is still up to 13.8% at a high luminance of 1000 cd m−2 (Fig. 3b, c, Table 2).
It is noticeable that the weight doping concentration of 5% for Eu(Tp2Et)2 is controllable, avoiding the low concentration (<1%) of the low-energy emitter in typical SEL-WOLEDs. To clarify the exceptionality, a series of reference devices Rx using a classic phosphorescence iridium complex bis(2-phenylbenzothiazolato)(acetylacetonate)iridium(III) (Ir(bt)2acac)49 as the orange-red emitter were fabricated with a device structure of ITO/MoO3 (2 nm)/TAPC (60 nm)/mCP:Ce-TBO2Et (10 wt%):Ir(bt)2acac (x wt%) (25 nm)/TmPyPB (40 nm)/LiF (0.7 nm)/Al (100 nm). It is found that the device with 5 wt% Ir(bt)2acac showed pure emission from Ir(bt)2acac, and no emission from Ce-TBO2Et was observed (Fig. 3f). A comparable emission from the sky-blue emitter Ce-TBO2Et to that of Ir(bt)2acac was measured until the doping concentration of Ir(bt)2acac is decreased to 0.1 wt%, resulting in a warm white emission with CIE coordinates of (0.33, 0.41) (Fig. 3f). The different optimal doping concentration of Eu(Tp2Et)2 in the device W1 compared with that of Ir(bt)2acac in the device Rx (5% vs. 0.1%) indicates there may be distinctive energy transfer between Ce-TBO2Et and Eu(Tp2Et)2.
In order to understand the energy transfer between Ce-TBO2Et and Eu(Tp2Et)2 in the single-emitting-layer, three doped films mCP:10 wt% Ce-TBO2Et, mCP:5 wt% Eu(Tp2Et)2, and mCP:10 wt% Ce-TBO2Et:5 wt% Eu(Tp2Et)2 with the same thickness (300 nm) were fabricated by vacuum thermal evaporation and their photophysical properties were measured (Fig. 4, Table 3). For the doped films mCP:10 wt% Ce-TBO2Et and mCP:5 wt% Eu(Tp2Et)2, weak f-d excitation peaks were both observed around 400 nm (Fig. 4a) with an absorption intensity (A) of 0.035 and 0.024 (Table 3), respectively. For the co-doped film mCP:10 wt% Ce-TBO2Et:5 wt% Eu(Tp2Et)2, the absorption intensity is measured to be 0.064 (Table 3), close to the sum of the above two values, suggesting independent excitation of the two emitters. The co-doped film mCP:10 wt% Ce-TBO2Et:5 wt% Eu(Tp2Et)2 exhibited two emission peaks at 490 and 600 nm, which is similar to the electroluminescence spectrum of the device W1 (Figs. 4a and 3e). The ratio of sky-blue photons to orange-red photons (r) in the co-doped film mCP:10 wt% Ce-TBO2Et:5 wt% Eu(Tp2Et)2 is ~1.4 as deduced from the peak splitting fitting of the spectrum (Supplementary Fig. S9), where the sky-blue and orange-red photons originate from Ce-TBO2Et and Eu(Tp2Et)2, respectively. Theoretically, r can be calculated by Eq. (1):
where A is absorption intensity and ΦET represents the energy transfer efficiency from the sky-blue emitter to the orange-red emitter, and the subscript B refers to the sky-blue emitter and the subscript O refers to the orange-red emitter. Therefore, the calculated ΦET from Ce-TBO2Et to Eu(Tp2Et)2 is around 20% (Table 3) and the energy diagram of the co-doped film mCP:10 wt% Ce-TBO2Et:5 wt% Eu(Tp2Et)2 is depicted in Fig. 4b. The energy transfer from Ce-TBO2Et to Eu(Tp2Et)2 is mostly inhibited, which accounts for the high doping concentration of Eu(Tp2Et)2 to achieve white emission in the device W1. As references, another two doped films mCP:5 wt% Ir(bt)2acac and mCP:10 wt% Ce-TBO2Et:5 wt% Ir(bt)2acac were also fabricated and their photophysical properties were studied (Fig. 4, Table 3). Though the absorption intensity of film mCP:10 wt% Ce-TBO2Et at 400 nm is non-ignorable as compared with that of the film mCP:5 wt% Ir(bt)2acac (0.035 vs. 0.126), the co-doped film mCP:10 wt% Ce-TBO2Et:5 wt% Ir(bt)2acac exhibited an emission spectrum that is basically consistent with that of the film mCP:5 wt% Ir(bt)2acac (Fig. 4c), indicating that the energy absorbed by Ce-TBO2Et is efficiently transferred to Ir(bt)2acac, and the ΦET from Ce-TBO2Et to Ir(bt)2acac is deduced to be nearly 100% (Table 3, Fig. 4d). Therefore, white emission can be obtained only when the doping concentration of Ir(bt)2acac is reduced to a very low level in the reference device Rx. In order to explore the different ΦET between Ce-TBO2Et and Eu(Tp2Et)2 or Ir(bt)2acac, we overlap the absorption spectrum of 10 wt% Eu(Tp2Et)2 or Ir(bt)2acac doped PMMA film with the normalized emission spectrum of 10 wt% Ce-TBO2Et doped PMMA film (Supplementary Fig. S10), and the overlapping area of Ce-TBO2Et and Eu(Tp2Et)2 is much smaller than that of Ce-TBO2Et and Ir(bt)2acac. This means the emission energy of Ce-TBO2Et is less efficiently absorbed by Eu(Tp2Et)2 than by Ir(bt)2acac. Therefore, the ΦET from Ce-TBO2Et to Eu(Tp2Et)2 is much lower than that from Ce-TBO2Et to Ir(bt)2acac.

a Excitation spectra (dashed lines) and emission spectra (solid lines) of mCP:10 wt% Ce-TBO2Et (blue lines), mCP:5 wt% Eu(Tp2Et)2 (orange lines), and mCP:10 wt% Ce-TBO2Et:5 wt% Eu(Tp2Et)2 (purple lines) films. b Energy diagram of the co-doped mCP:10 wt% Ce-TBO2Et:5 wt% Eu(Tp2Et)2 film. c Excitation spectra (dashed lines) and emission spectra (solid lines) of mCP:10 wt% Ce-TBO2Et (blue lines), mCP:5 wt% Ir(bt)2acac (orange lines), and mCP:10 wt% Ce-TBO2Et:5 wt% Ir(bt)2acac (purple lines) films. d Energy diagram of the co-doped mCP:10 wt% Ce-TBO2Et:5 wt% Ir(bt)2acac film. The excitation spectra are monitored at the maximum emission peak and the emission spectra are monitored at an excitation wavelength of 400 nm. ΦET represents the energy transfer efficiency from Ce-TBO2Et to Eu(Tp2Et)2 or Ir(bt)2acac
Discussion
In summary, a sky-blue emitting Ce(III) complex Ce-TBO2Et and an orange-red emitting Eu(II) complex Eu(Tp2Et)2 with effective d-f transition are synthesized and characterized. The maximum EQEs of the corresponding sky-blue OLEDs and orange-red OLEDs reached 22.3% and 11.1%, which are the highest record of d-f transition based blue and red OLEDs, respectively. Using the two complexes with complementary colors, we fabricated efficient and color-stable three-layered SEL-WOLEDs, showing a maximum EQE of 15.9% and CIE coordinates of (0.33, 0.39) at various luminances. It is found that excitons are directly formed on Ce-TBO2Et and Eu(Tp2Et)2 due to their hole capture capability in electroluminescent process, avoiding the energy transfer from host material to dopants. Moreover, hindered energy transfer between the two emitters provides an advantage on doping concentration optimization, i.e. avoiding the low concentration (<1%) of the low-energy emitter in typical SEL-WOLEDs. This work demonstrates that the application of d-f transition complexes is expected to bring a new perspective on SEL-WOLEDs construction with simplified energy level regulation and manufacturing process.
Materials and methods
General
All chemical reagents used in the synthesis process were commercially available and used as received unless otherwise mentioned. TAPC (99.5%) and TmPyPB (99%) are purchased from Xi’an Polymer Light Technology Corporation, and mCP (99.5%) is purchased from Jilin Oled Material Technology Corporation. K(Tp2Et) was synthesized as reported42. Synthesis of the Ce(III) and Eu(II) complexes were conducted in glove box. Ir(bt)2acac (99%) is purchased from Luminescence Technology Corporation and further purified by vacumm sublimation. Elemental analysis were performed on a VARIO EL analyzer (GmbH, Hanau, Germany).
Spectroscopic measurements
Single crystal X-ray diffraction data were measured on Rigaku XtaLAB PRO 007HF (Mo). UV absorption spectra were collected on a Shimadzu UV3600PLUS. Photoluminescence efficiencies and absorption intensity of doped films were measured using an absolute photoluminescence quantum yield measurement system on C9920-02, Hamamatsu Company. Steady-state and transient photoluminescence spectra were measured on an Edinburgh Analytical Instruments FLS980 spectrophotometer. All the compounds, no matter in solid powder or in solution state, were under inert atmosphere protection when carrying out photophysical characterizations.
Thermal stability measurements
Thermal gravimetric analysis (TGA) was carried out on TA Instruments SDT Q600. Differential scanning calorimetry (DSC) was measured on TA Instruments Q2000.
Cyclic voltammetry (CV) measurements
CV was carried out under inert atmosphere protection in dichloromethane (Ce-TBO2Et) or hexane (Eu(Tp2Et)2) solution at room temperature with a CHI voltammetric analyzer. Tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 M) was used as the supporting electrolyte. The conventional three-electrode configuration consists of a platinum working electrode, a platinum wire auxiliary electrode, and an Ag/AgCl wire pseudo-reference electrode with ferrocene as the external standard. Cyclic voltammogram was obtained at a scan rate of 100 mV s−1.
OLEDs fabrication and measurements
Indium tin oxide (ITO) patterned anode was commercially available with a sheet resistance of 14 Ω square−1 and a thickness of 80 nm. ITO substrates were cleaned with deionized water and ethanol. The organic and metal layers were deposited in different vacuum chambers with a base pressure better than 1 × 10−4 Pa. The active area for each device is 4 mm2. All electric testing and optical measurements were performed under ambient conditions with encapsulation of devices in a glove box. The electroluminescence spectra, current density-voltage-luminance curves, and EQE characteristics were measured by computer controlled Keithley 2400 source meter and absolute EQE measurement system (C9920-12) with photonic multichannel analyzer (PMA-12, Hamamatsu Photonics).
Synthesis of Ce-TBO2Et
Ce(CF3SO3)3 (5.87 g, 10.0 mmol), KTp2Et (8.41 g, 20.0 mmol), and H2O (0.180 g, 10.0 mmol) were added to a 100 mL round-bottomed flask with 50 mL tetrahydrofuran in a glove box. The mixture was stirred for 3 days at room temperature and then the solvent was removed under vacuum. The mixture was washed with hexane to obtain pale yellow solid powder. Pure product was obtained as crystalline powder after sublimation of the yield solid powder at 250 °C (0.560 g, 0.352 mmol, 7%). Anal. calcd for C70H114B4N20O2Ce2: C 52.84, H 7.22, N 17.60; found: C 52.97, H 7.34, N 17.82.
Synthesis of Eu(Tp2Et)2
EuI2 (0.406 g, 1.00 mmol), KTp2Et (0.880 g, 2.09 mmol) were added to a 100 mL round-bottomed flask with 50 mL tetrahydrofuran in a glove box. The mixture was stirred for 12 hours at room temperature and then the solvent was removed. Pure product was obtained as crystalline powder after sublimation at 180 °C (0.460 g, 0.503 mmol, 50%). Anal. calcd for C42H68B2N12Eu: C 55.15, H 7.49, N 18.38; found: C 55.07, H 7.53, N 18.78.
Data availability
[CCDC 2195888 (for Ce-TBO2Et) and 2195890 (for Eu(Tp2Et)2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.] The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
D’Andrade, B. W. & Forrest, S. R. White organic light-emitting devices for solid-state lighting. Adv. Mater. 16, 1585–1595 (2004).
Reineke, S. et al. White organic light-emitting diodes with fluorescent tube efficiency. Nature 459, 234–238 (2009).
Sun, Y. R. et al. Management of singlet and triplet excitons for efficient white organic light-emitting devices. Nature 440, 908–912 (2006).
Kamtekar, K. T., Monkman, A. P. & Bryce, M. R. Recent advances in white organic light-emitting materials and devices (WOLEDs). Adv. Mater. 22, 572–582 (2010).
Chen, Z. et al. Single-molecular white-light emitters and their potential WOLED applications. Adv. Mater. 32, 1903269 (2020).
Reineke, S. et al. White organic light-emitting diodes: status and perspective. Rev. Mod. Phys. 85, 1245–1293 (2013).
Parthasarathy, G., Gu, G. & Forrest, S. R. A full-color transparent metal-free stacked organic light emitting device with simplified pixel biasing. Adv. Mater. 11, 907–910 (1999).
Kido, J., Kimura, M. & Nagai, K. Multilayer white light-emitting organic electroluminescent device. Science 267, 1332–1334 (1995).
Luo, W. et al. High-performance organic light-emitting diodes with natural white emission based on thermally activated delayed fluorescence emitters. J. Mater. Chem. C. 8, 10431–10437 (2020).
Kido, J. et al. White light‐emitting organic electroluminescent devices using the poly(N‐vinylcarbazole) emitter layer doped with three fluorescent dyes. Appl. Phys. Lett. 64, 815–817 (1994).
Xiang, H. Y. et al. Research progress of full electroluminescent white light-emitting diodes based on a single emissive layer. Light Sci. Appl. 10, 206 (2021).
Li, Y. et al. Manipulating complementarity of binary white thermally activated delayed fluorescence systems for 100% exciton harvesting in OLEDs. Adv. Funct. Mater. 31, 2011169 (2021).
Cao, C. et al. Bipolar blue host emitter with unity quantum yield allows full exciton radiation in single-emissive-layer hybrid white organic light-emitting diodes. ACS Appl. Mater. Interfaces 11, 11691–11698 (2019).
Wang, Q. & Ma, D. G. Management of charges and excitons for high-performance white organic light-emitting diodes. Chem. Soc. Rev. 39, 2387–2398 (2010).
Tang, X. et al. All-fluorescence white organic light-emitting diodes exceeding 20% EQEs by rational manipulation of singlet and triplet excitons. Adv. Funct. Mater. 30, 1910633 (2020).
Sun, J. N. et al. Charge-transfer exciton manipulation based on hydrogen bond for efficient white thermally activated delayed fluorescence. Adv. Funct. Mater. 30, 1908568 (2020).
Zou, S. J. et al. The strategies for high-performance single-emissive-layer white organic light-emitting diodes. Laser Photonics Rev. 15, 2000474 (2021).
Zhu, X. Y. et al. Anthracene-based bipolar deep-blue emitters for efficient white OLEDs with ultra-high stabilities of emission color and efficiency. J. Mater. Chem. C. 9, 5198–5205 (2021).
Zhao, J. W. et al. White OLEDs with an EQE of 21% at 5000 cd m−2 and ultra high color stability based on exciplex host. Adv. Optical Mater. 6, 1800825 (2018).
Liu, H. et al. Achieving high electroluminescence efficiency and high color rendering index for all-fluorescent white OLEDs based on an out-of-phase sensitizing system. Adv. Funct. Mater. 31, 2103273 (2021).
Li, X. L. et al. High-efficiency WOLEDs with high color-rendering index based on a chromaticity-adjustable yellow thermally activated delayed fluorescence emitter. Adv. Mater. 28, 4614–4619 (2016).
Han, C. M. et al. Ladder-like energy-relaying exciplex enables 100% internal quantum efficiency of white TADF-based diodes in a single emissive layer. Nat. Commun. 12, 3640 (2021).
Zhang, C. et al. A π–D and π–A exciplex-forming host for high-efficiency and long-lifetime single-emissive-layer fluorescent white organic light-emitting diodes. Adv. Mater. 32, 2004040 (2020).
Ding, D. X. et al. Highly efficient and color-stable thermally activated delayed fluorescence white light-emitting diodes featured with single-doped single emissive layers. Adv. Mater. 32, 1906950 (2020).
Wu, Z. B. et al. Strategic-tuning of radiative excitons for efficient and stable fluorescent white organic light-emitting diodes. Nat. Commun. 10, 2380 (2019).
Wu, J. et al. Efficient excimer-based white OLEDs with reduced efficiency roll-off. Appl. Phys. Lett. 118, 073301 (2021).
Fleetham, T. et al. Efficient and stable single-doped white OLEDs using a palladium-based phosphorescent excimer. Chem. Sci. 8, 7983–7990 (2017).
Wei, P. C., Zhang, D. D. & Duan, L. Modulation of Förster and dexter interactions in single-emissive-layer all-fluorescent WOLEDs for improved efficiency and extended lifetime. Adv. Funct. Mater. 30, 1907083 (2020).
Wang, Q. et al. Manipulating charges and excitons within a single-host system to accomplish efficiency/CRI/color-stability trade-off for high-performance OWLEDs. Adv. Mater. 21, 2397–2401 (2009).
Wang, Q. et al. Harvesting excitons via two parallel channels for efficient white organic LEDs with nearly 100% internal quantum efficiency: fabrication and emission-mechanism analysis. Adv. Funct. Mater. 19, 84–95 (2009).
Bozkus, V., Aksoy, E. & Varlikli, C. Perylene based solution processed single layer WOLED with adjustable CCT and CRI. Electronics 10, 725 (2021).
Liu, X. K. et al. Remanagement of singlet and triplet excitons in single-emissive-layer hybrid white organic light-emitting devices using thermally activated delayed fluorescent blue exciplex. Adv. Mater. 27, 7079–7085 (2015).
Wu, Z. B. et al. Management of singlet and triplet excitons: a universal approach to high-efficiency all fluorescent WOLEDs with reduced efficiency roll-off using a conventional fluorescent emitter. Adv. Optical Mater. 4, 1067–1074 (2016).
Li, W. et al. Tri-spiral donor for high efficiency and versatile blue thermally activated delayed fluorescence materials. Angew. Chem. Int. Ed. 58, 11301–11305 (2019).
Fang, P. Y. et al. Lanthanide cerium(III) tris(pyrazolyl)borate complexes: efficient blue emitters for doublet organic light-emitting diodes. ACS Appl. Mater. Interfaces 13, 45686–45695 (2021).
Wang, L. D. et al. Deep-blue organic light-emitting diodes based on a doublet d–f transition cerium(III) complex with 100% exciton utilization efficiency. Light Sci. Appl. 9, 157 (2020).
Li, J. Y. et al. Highly efficient and air-stable Eu(II)-containing azacryptates ready for organic light-emitting diodes. Nat. Commun. 11, 5218 (2020).
Zhan, G. et al. Highly efficient and air-stable lanthanide EuII complex: new emitter in organic light emitting diodes. Angew. Chem. Int. Ed. 59, 19011–19015 (2020).
Zhao, Z. F. et al. Efficient rare earth cerium(III) complex with nanosecond d−f emission for blue organic light-emitting diodes. Natl Sci. Rev. 8, nwaa193 (2021).
Yan, W. C. et al. Highly efficient heteroleptic cerium(III) complexes with a substituted pyrazole ancillary ligand and their application in blue organic light-emitting diodes. Inorg. Chem. 60, 18103–18111 (2021).
Shipley, C. P. et al. Orange electroluminescence from a divalent europium complex. Adv. Mater. 11, 533–536 (1999).
Saly, M. J., Heeg, M. J. & Winter, C. H. Volatility, high thermal stability, and low melting points in heavier alkaline earth metal complexes containing tris(pyrazolyl)borate ligands. Inorg. Chem. 48, 5303–5312 (2009).
Kunrath, F. A. et al. Synthesis and reactivity in salt metathesis reactions oftrivalent [La(TpMe2)2X] (X=Cl, I) complexes: crystalstructures of [La(TpMe2)2Cl] and [La(TpMe2)2(κ2-pzMe2)]. Polyhedron 23, 2437–2445 (2004).
Falivene, L. et al. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 11, 872–879 (2019).
Zheng, X. L. et al. Bright blue-emitting Ce3+ complexes with encapsulating polybenzimidazole tripodal ligands as potential electroluminescent devices. Angew. Chem. Int. Ed. 46, 7399–7403 (2007).
Dorenbos, P. f → d transition energies of divalent lanthanides in inorganic compounds. J. Phys. Condens. Matter 15, 575–594 (2003).
Katkova, M. A. et al. Lanthanide imidodiphosphinate complexes: synthesis, structure and new aspects of electroluminescent properties. Synth. Met. 159, 1398–1402 (2009).
Yu, T. Z. et al. Ultraviolet electroluminescence from organic light-emitting diode with cerium(III)–crown ether complex. Solid State Electron. 51, 894–899 (2007).
Han, C. M. et al. A single phosphine oxide host for high-efficiency white organic light-emitting diodes with extremely low operating voltages and reduced efficiency roll-off. Adv. Mater. 23, 2491–2496 (2011).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22071003, 62104013, and 92156016), the National Key R&D Program of China (2021YFB3500400, 2021YFB3501800, and 2022YFB3503702), and the Beijing Natural Science Foundation (2202015). The authors acknowledge high-performance computing platform of Peking University and Beijing Super Computing Center (BSCC) for providing HPC resources for the quantum-chemical calculations.
Author information
Authors and Affiliations
Contributions
Z.L. proposed and designed this project. P.F. and P.H. conducted most of the experiments and wrote the manuscript. L.W. helped in designing device structure. Z.Z., G.Y., Y.H., Z.B., and Z.L. discussed the results and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, P., Huo, P., Wang, L. et al. Lanthanide complexes with d-f transition: new emitters for single-emitting-layer white organic light-emitting diodes. Light Sci Appl 12, 170 (2023). https://doi.org/10.1038/s41377-023-01211-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41377-023-01211-5
