
Abstract
Monosomy 7 and del(7q) are among the most common and poorly understood genetic alterations in myelodysplastic neoplasms and acute myeloid leukemia. Chromosome band 7q22 is a minimally deleted segment in myeloid malignancies with a del(7q). However, the rarity of “second hit” mutations supports the idea that del(7q22) represents a contiguous gene syndrome. We generated mice harboring a 1.5 Mb germline deletion of chromosome band 5G2 syntenic to human 7q22 that removes Cux1 and 27 additional genes. Hematopoiesis is perturbed in 5G2+/del mice but they do not spontaneously develop hematologic disease. Whereas alkylator exposure modestly accelerated tumor development, the 5G2 deletion did not cooperate with KrasG12D, NrasG12D, or the MOL4070LTR retrovirus in leukemogenesis. 5G2+/del mice are a novel platform for interrogating the role of hemopoietic stem cell attrition/stress, cooperating mutations, genotoxins, and inflammation in myeloid malignancies characterized by monosomy 7/del(7q).
Similar content being viewed by others
TO THE EDITOR:
Monosomy 7 (Mo7) and del(7q) [Mo7/del(7q22)] are highly prevalent chromosomal abnormalities in de novo pediatric and adult myelodysplastic neoplasm (MDS) and acute myeloid leukemia (AML) that are associated with an aggressive clinical course and therapeutic resistance [1, 2]. In addition, Mo7/del(7q) is highly enriched in myeloid malignancies that develop in patients with aplastic anemia or germline mutations in genes such as NF1, SAMD9/9L, and GATA2 or following treatment with radiation or alkylating agents [3, 4]. In patients with Mo7/del(7q), the transformation from MDS to AML is characterized by recurring cooperating mutations in NRAS/KRAS, SETBP1, RUNX1, and other genes [5]. The lack of accurate in vitro and in vivo models is a major barrier to understanding how Mo7/del(7q) contributes to leukemogenesis.
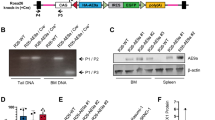
Chromosome band 7q22 is a minimally deleted segment in MDS and AML samples that is syntenic with mouse chromosome band 5A3 [6] (Supplementary Fig. 1). Based on the rarity of “second hit” mutations in any 7q gene in myeloid malignancies with Mo7/del(7q), loss of 7q22 likely represents a contiguous gene syndrome whereby haploinsufficiency for multiple genes contributes to leukemogenesis [2]. Utilizing chromosome engineering, we previously generated 5A3+/del mice harboring a heterozygous germline deletion corresponding to part of this minimally deleted segment, which is bounded by Fbxl13 and Srpk2 [7] (Supplementary Fig. 1). The 5A3 deletion impairs lymphoid repopulation and perturbs the hematopoietic stem cell (HSC) compartment without enhancing repopulating potential or initiating hematologic disease [7]. Studies of MDS and AML patient samples have implicated a second region of 7q22 flanked by EPO and UPK3BL in leukemogenesis. This interval contains CUX1 and 27 other genes and is syntenic to mouse chromosome band 5G2 (Supplementary Fig. 1). Here we report the generation and analysis of 5G2+/del mice harboring a heterozygous germline Epo-Upk3bl deletion (Supplementary Fig. 2).
The frequency of bone marrow (BM) c-kit+, lin-, Sca+ (KLS) cells is reduced in 5G2+/del mice compared to wild-type (WT) littermates, which is primarily due to a decrease in CD150neg multi-potent progenitors (Figs. 1a, b). Further analysis of the KLS CD48neg HSC population showed an increase in the myeloid-biased CD150hi cells (Fig. 1c). Interestingly, 5-bromo-2’-deoxyuridine (BrdU) labeling revealed a significant reduction in the proportion of 5G2+/del KLS CD48neg HSC in the S phase of the cell division cycle and an increase in the G0/G1 fraction (Fig. 1d). After backcrossing 5G2+/del mice to the C57BL/6 strain, we mixed WT or 5G2+/del BM cells (CD45.2) with WT CD45.1 competitors from congenic BoyJ mice at a 1:1 ratio and transplanted them into irradiated recipients. 5G2+/del cells showed a modest reduction in competitive fitness that did not achieve statistical significance (Fig. 1e). Consistent with RT-PCR analysis of individual 5G2 genes (Supplementary Fig. 2d), RNA sequencing revealed a ~50% reduction in the expression levels of genes within the deleted 5G2 interval (Fig. 1f) and showed that gene sets associated with interferon responses/inflammation were uniquely and significantly down-regulated in KLS, CD48-, CD150neg cells from 5G2+/del mice in comparison to WT controls (Supplementary Fig. 3).

a Frequency of c-kit+, lin-, Sca+ (KLS) cells per 106 nucleated BM cells in WT and 5G2+/del mice at 8–12 weeks of age (n = 5 per genotype). b, c Frequencies and percentages of CD150hi, CD150lo, and CD150neg cells within the KLS, CD48neg HSC population in WT and 5G2+/del mice. d BrdU staining showing the percentages of nucleated WT and 5G2+/del KLS, CD48neg HSC in the G0/G1, and S + M phases of the cell cycle. e Percentage of blood leukocytes derived from WT and 5G2+/del BM cells after transplantation with WT competitors at a 1:1 ratio into irradiated WT recipients. These data were pooled from three independent experiments (biologic replicates) with at least three recipients (technical replicates) in each experimental group. The procedures used for BM harvesting and processing, flow cytometry, cell sorting, competitive repopulation, RNA isolation, and TaqMan analysis have been described in detail [7]. Data from these experiments are presented as means ± SEM and statistical significance was determined by performing two-tailed Student’s t-tests unless stated otherwise. Asterisks denote significant differences between WT and 5G2+/del mice (*p < 0.05; **p < 0.01). f Transcriptome profiling of KLS, CD48neg, CD150neg HSCs from WT and 5G2+/del mice demonstrating reduced expression of genes in the G2 cytoband of mouse chromosome 5 ordered by location. Genes within the region that were filtered out of differential gene expression analysis due to low overall expression are represented by checkered bands. The bounds of the G2 region are denoted by black bands. The expression levels of the five genes highlighted in red font were verified by RT-PCR in the same KLS, CD48neg,CD150neg HSC population (see Supplementary Fig. 2d).
5G2+/del mice and their WT littermates remained well with normal hematologic parameters at >1 year of age (Supplementary Fig. 4). Likewise, thymus and spleen weights and BM cellularity were similar in 5G2+/del and WT mice at euthanasia (data not shown). Given the frequent occurrence of NRAS and KRAS mutations in myeloid malignancies with Mo7/del(7q), we generated cohorts of Mx1-Cre; KrasG12D/+ and Mx1-Cre; NrasG12D/+ mice either lacking or harboring the 5G2 deletion and induced Cre recombinase expression by injecting them with a single dose of polyI-polyC at weaning [8, 9]. As expected [8], Mx1-Cre; KrasG12D/+ mice developed a fully penetrant myeloproliferative disorder characterized by splenomegaly and leukocytosis that was similar in both 5G2 genotypes (Supplementary Figs. 5a–c). Additionally, the heterozygous 5G2 deletion did not modify survival or hematologic phenotypes in Mx1-Cre; NrasG12D/+ mice [9](Supplementary Fig. 5d). Furthermore, WT and 5G2+/del mice injected with the MOL4070LTR retrovirus [10] had similar survival and developed the same spectrum of hematologic malignancies (Supplementary Fig. 5e).
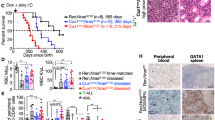
Mo7/del(7q) is strongly associated with therapy-induced MDS/AML following treatment with radiation and/or alkylating agents [4]. Accordingly, we injected 5G2+/del mice and WT littermates with the alkylating agent N-nitroso-N-ethylurea (ENU). This experiment revealed modest cooperativity between the 5G2 deletion and ENU exposure in tumorigenesis (Fig. 2a). To investigate if ENU treatment modulates competitive fitness, we mixed 5G2+/del or WT BM with WT competitor cells and transplanted them into irradiated syngeneic mice. Beginning five weeks after transplantation, these recipients received two doses of ENU or control vehicle separated by 7 days and were euthanized four weeks later to measure BM chimerism. The proportion of 5G2+/del cells was significantly reduced in recipient mice after ENU treatment in comparison to mice transplanted with WT competitors (Fig. 2b) and this difference was maintained over time (Supplementary Fig. 6). We next used s similar experimental design to assess the impact of the 5G2 deletion on the competitive fitness of HSC expressing NrasG12D from the endogenous locus. Specifically, mice were transplanted with CD45.2 BM cells from Mx1-Cre; NrasG12D/+;5G2+/del or control Mx1-Cre; NrasG12D/+ mice at a 1:1 ratio with WT CD45.1 competitors, and half of them were injected with ENU 5 and 6 weeks later (Fig. 2c). Recipient mice were monitored for 5 months after transplantation or until they became moribund and required euthanasia. In the absence of ENU treatment, the contribution of NrasG12D/+;5G2+/del double mutant cells to the HSC and myeloid compartments was reduced in recipient mice compared to control NrasG12D/+ cells while lymphoid repopulation was similar (Fig. 2d). By contrast, most of the recipients transplanted with either NrasG12D/+;5G2+/del or NrasG12D/+WT cells that received ENU died prematurely, primarily from lymphoid malignancies, which precluded assessing competitive fitness.

a Survival of ENU-treated WT (n = 18) and 5G2+/del (n = 13) littermates. Percent survival (time to euthanasia of moribund animals) is plotted vs time in days after ENU treatment. The cumulative probability of survival was computed by the Kaplan-Meier method and is significantly less in 5G2+/del mice (p = 0.0197 by log rank). Thymic lymphoma was the major cause of death in mice of both genotypes. b Chimeras were generated by transplanting WT or 5G2+/del BM with WT competitor cells at a ratio of 3:1 into irradiated congenic WT recipients. Recipient mice received control vehicle or ENU at a dose of 100 mg/kg intraperitoneally 5 and 6 weeks post-transplant. The fold change in the percentage of total leukocytes derived from WT or 5G2+/del cells 4 weeks after the second ENU treatment is shown. Data represent the mean ± standard error of the mean (s.e.m.) of 4-5 mice. Chimerism was analyzed using Student’s t-test. c Experimental design for assessing the effects of the 5G2 deletion on the competitive fitness of NrasG12D BM cells in the presence and absence of ENU treatment. Three-week-old Mx1-Cre; NrasG12D and Mx1-Cre; NrasG12D; 5G2+/del mice were injected with a single dose of pIpC to induce NrasG12D expression from the endogenous locus followed by bone marrow harvest at 8-12 weeks for competitive transplantation with WT competitor cells at a 1:1 ratio. Recipient mice were treated with a control vehicle or ENU at 5 and 6 weeks post-transplant as above and were monitored for 5 months post-transplant. d Contribution of donor NrasG12D (blue circles) and NrasG12D; 5G2+/del (red squares) cells to HSC, KLS, myeloid progenitor (MP), Myeloid, T cell and B cell populations in the BM of recipient mice that did not receive ENU post-transplant. Note that the 5G2 mutation impaired the ability of donor NrasG12D cells to repopulated the HSC, KLS, and MP compartments. Asterisks denote significant differences between NrasG12D (blue circles) and NrasG12D; 5G2+/del donor cells (*p < 0.05; ***p < 0.005 by Student’s t-test). Comparable competitive fitness data are not available from recipients that received ENU because most died prematurely from lymphoid malignancies.
Aly and colleagues identified 55 mutations and 6 microdeletions in the 7q22 gene CUX1 in 1480 adults with MDS, MPN, or AML (4.1%) [11]. Of these mutations, 85% were heterozygous and 75% encoded missense amino acid substitutions. Patients with MDS had the highest incidence of CUX1 mutations (~25%). Mo7/del(7q) was present in 81 additional cases (5.5%), including 5 with CUX1 mutations [11]. By contrast, a comprehensive molecular analysis of 77 pediatric MDS and MDS/MPN patient samples revealed Mo7/del(7q) in 40% and PTPN11, NRAS, and other Ras pathway mutations in 55% [12]. Germline mutations in the 7q21.2 genes SAMD9 and SAMD9L were identified in 17% of these pediatric cases and were strongly associated with loss of the chromosome 7 homolog harboring the mutant allele. The two CUX1 mutations occurred in samples with concurrent Ras pathway mutations and Mo7/del(7q) [12]. These data and other studies indicate that monosomy 7 is more common than CUX1 mutations in adult and pediatric myeloid malignancies, and demonstrate that CUX1 is only rarely mutated in patients with monosomy 7 [2].
Cux1 was independently investigated by two groups in mice using either an inducible knockdown approach [13, 14] or the Vav-iCre transgene to inactivate a conditional mutant allele [15]. In the first model, aged Cux1Mid mice with a 45% reduction in Cux1 protein levels developed an indolent MDS-like disorder characterized by normal survival and blood leukocyte counts, an increase in the percentage of myeloid cells, anemia, dysplasia, and modest splenomegaly. HSC from these mice showed impaired self-renewal and Cux1 knockdown cooperated with both ENU treatment and NrasG12D expression in leukemogenesis [13, 14]. In the second model, heterozygous Vav-iCre; Cux1+/- mice had normal blood leukocyte and erythrocyte numbers with macrocytosis at one year of age [15]. In agreement with these studies, we show that the heterozygous 5G2 deletion that removes Cux1 neither alters lifespan nor causes acute leukemia. However, our findings in Mx1-Cre; KrasG12D/+ and Mx1-Cre; NrasG12D/+ mice haploinsufficient for the 5G2 deletion contrast with previous studies showing that Flt3ITD or NrasG12D cooperated with reduced Cux1 expression in leukemogenesis [14, 15]. Haploinsufficiency for additional genes in the 5G2 interval - as commonly observed in human myeloid malignancies with Mo7/del(7q) - might account for these differences.
The HSC compartment of 5G2+/del mice is characterized by an increased percentage of CD150hi myeloid-biased cells, delayed/reduced entry of HSC into the S phase of the cell cycle, and impaired competitive fitness after ENU treatment and in the context of NrasG12D expression. The consistent absence of an in vivo growth advantage of 5A3del/+, 5G2+/del, and Cux1Mid HSCs under steady-state conditions is both intriguing and counter-intuitive as clonal outgrowth is a hallmark of myeloid malignancies. The discovery of germline SAMD9 and SAMD9L mutations as a cause of familial MDS and AML suggests an alternative explanation for how Mo7/del(7q) might contribute to leukemogenesis [3]. HSC upregulate SAMD9 and SAMD9L as an adaptive response to inflammatory signals in the BM microenvironment. The SAMD9 and SAMD9L mutations identified in pediatric MDS and AML encode biochemical gain-of-function proteins that inhibit Ras/mitogen-activated protein kinase (MAPK) signaling, suppress cell growth, perturb protein translation, and promote apoptosis, which favors the survival of Mo7/del(7q) clones that delete the mutant allele [3, 16, 17]. Knock-in mice harboring a conditional Samd9l mutant allele that models a mutation in familial MDS and AML develop BM aplasia with impaired HSC function that is exacerbated by inflammatory stress and associated with loss of the chromosomal segment harboring the mutant allele [18]. Our observation that gene sets associated with interferon signaling are down-regulated in 5G2+/del HSC provides a biologic rationale for why 7q22 deletions might be co-selected with loss of mutant SAMD9/9L alleles in response to inflammatory stress. Similarly, it is possible that haploinsufficiency for SAMD9, SAMD9L, and 7q22 cooperatively provide Mo7/del(7q) stem and progenitor cells with a survival – but not a proliferative - advantage in other disease settings characterized by chronic HSC stress/attrition [1, 19].
Together with a previous analysis of 5A3+/del mice [7], the studies of 5G2+/del mice reported here and observations in human patients suggest that Mo7/del(7q) functions as an “opportunistic” molecular abnormality in the context of HSC damage and dysfunction [1]. This idea is conceptually concordant with the outgrowth of TP53 mutant clones in therapy-induced AML [20]. The prevalent mutations in NRAS and other signaling genes in myeloid disorders with monosomy 7/del(7q) may partially overcome the fitness disadvantage associated with CUX1 haploinsufficiency and loss of the 7q22/5G2 interval by promoting cell cycle progression [14]. The distinct effects of the 5G2 and 5A3 deletions on HSC homeostasis is likely due to the fact that they independently removed 28 and 13 non-overlapping genes syntenic to different human 7q22 DNA segments. 5G2+/del mice are a genetically accurate model of the proposed 7q22 contiguous gene deletion syndrome for interrogating the role of Mo7/del(7q) in HSC homeostasis and for characterizing how a pro-inflammatory BM microenvironment shapes HSC survival, clonal evolution, and progression to MDS and AML. Future studies may also further elucidate the respective biologic and phenotypic consequences of haploinsufficiency for Cux1 and other individual 5G2 genes versus loss of the entire Upk3bl-Epo interval.
References
Inaba T, Honda H, Matsui H. The enigma of monosomy 7. Blood. 2018;131:2891–8.
Jotte MRM, McNerney ME. The significance of CUX1 and chromosome 7 in myeloid malignancies. Curr Opin Hematol. 2022;29:92–102.
Klco JM, Mullighan CG. Advances in germline predisposition to acute leukaemias and myeloid neoplasms. Nat Rev Cancer. 2021;21:122–37.
Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52.
Mori M, Kubota Y, Durmaz A, Gurnari C, Goodings C, Adema V, et al. Genomics of deletion 7 and 7q in myeloid neoplasm: from pathogenic culprits to potential synthetic lethal therapeutic targets. Leukemia. 2023;37:2082–93.
Le Beau MM, Espinosa R 3rd, Davis EM, Eisenbart JD, Larson RA, Green ED. Cytogenetic and molecular delineation of a region of chromosome 7 commonly deleted in malignant myeloid diseases. Blood 1996;88:1930–5.
Wong JC, Weinfurtner KM, Alzamora Mdel P, Kogan SC, Burgess MR, Zhang Y, et al. Functional evidence implicating chromosome 7q22 haploinsufficiency in myelodysplastic syndrome pathogenesis. Elife. 2015;https://doi.org/10.7554/eLife.07839.
Braun BS, Tuveson DA, Kong N, Le DT, Kogan SC, Rozmus J, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci USA. 2004;101:597–602.
Li Q, Haigis KM, McDaniel A, Harding-Theobald E, Kogan SC, Akagi K, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117:2022–32.
Wolff L, Koller R, Hu X, Anver MR. A Moloney murine leukemia virus-based retrovirus with 4070A long terminal repeat sequences induces a high incidence of myeloid as well as lymphoid neoplasms. J Virol. 2003;77:4965–71.
Aly M, Ramdzan ZM, Nagata Y, Balasubramanian SK, Hosono N, Makishima H, et al. Distinct clinical and biological implications of CUX1 in myeloid neoplasms. Blood Adv. 2019;3:2164–78.
Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant V, et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017;8:1557 https://doi.org/10.1038/s41467-017-01590-5
An N, Khan S, Imgruet MK, Gurbuxani SK, Konecki SN, Burgess MR, et al. Gene dosage effect of CUX1 in a murine model disrupts HSC homeostasis and controls the severity and mortality of MDS. Blood. 2018;131:2682–97.
An N, Khan S, Imgruet MK, Jueng L, Gurbuxani S, McNerney ME. Oncogenic RAS promotes leukemic transformation of CUX1-deficient cells. Oncogene. 2023;42:881–93.
Supper E, Rudat S, Iyer V, Droop A, Wong K, Spinella JF, et al. Cut-like homeobox 1 (CUX1) tumor suppressor gene haploinsufficiency induces apoptosis evasion to sustain myeloid leukemia. Nat Commun. 2021;12:2482 https://doi.org/10.1038/s41467-021-22750-8
Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48:792–7.
Thomas ME 3rd, Abdelhamed S, Hiltenbrand R, Schwartz JR, Sakurada SM, Walsh M, et al. Pediatric MDS and bone marrow failure-associated germline mutations in SAMD9 and SAMD9L impair multiple pathways in primary hematopoietic cells. Leukemia. 2021;35:3232–44.
Abdelhamed S, Thomas ME 3rd, Westover T, Umeda M, Xiong E, Rolle C, et al. Mutant Samd9l expression impairs hematopoiesis and induces bone marrow failure in mice. J Clin Invest. 2022;132:e158869 https://doi.org/10.1172/JCI158869
Wong JC, Bryant V, Lamprecht T, Ma J, Walsh M, Schwartz J, et al. Germline SAMD9 and SAMD9L mutations are associated with extensive genetic evolution and diverse hematologic outcomes. JCI Insight. 2018;3:e121086 https://doi.org/10.1172/jci.insight.121086
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–5.
Acknowledgements
This work was supported by NIH grants R50 CA211452 (to JCW), R01 HL144653 (to JMK), R01 CA216352 (to KS), K08CA256489 (to B.J.H.) and by the St. Jude Comprehensive Cancer Center (P30-CA21765). The content, however, does not necessarily represent the official views of the NIH and is solely the responsibility of the authors. The studies were also supported by the American Lebanese and Syrian Associated Charities of St. Jude Children’s Research Hospital and independent awards from the Edward P. Evans Foundation (to JMK and KS). We are grateful to Linda Wolff for providing the MOL4070LTR virus and to David Tuveson, Kevin Haigis and Tyler Jacks for providing KrasG12D/+ and NrasG12D/+ mice.
Author information
Authors and Affiliations
Contributions
JCW, JMK, and KS conceived the project and designed the experiments. JCW, KMW, TW, JK, EJL, MA, LM, and SA performed experiments and data analysis. BJM, MW, and JM performed computational and statistical analyses and produced the associated figure panels. JCW, JMK, and KS wrote the manuscript, which all of the authors edited, reviewed, and approved before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wong, J.C., Weinfurtner, K.M., Westover, T. et al. 5G2 mutant mice model loss of a commonly deleted segment of chromosome 7q22 in myeloid malignancies. Leukemia (2024). https://doi.org/10.1038/s41375-024-02205-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41375-024-02205-x
