
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a severe hyperinflammatory syndrome driven by pathologic activation of cytotoxic T-lymphocytes and macrophages. Despite advances in diagnostics and management, adult patients with lymphoma-associated HLH (LA-HLH) harbor particularly poor prognosis and optimal treatment remains challenging. As systematic data on LA-HLH are scarce, we aimed to synthesize research evidence by thorough analysis of the published literature in PubMed (MEDLINE-database) within the context of a scoping review. Of 595 search results, 132 articles providing information on 542 patients were reviewed and analyzed. Median patient age was 60 years (range, 18–98) with male predominance (62.7%). B- and T-NHL were equally represented (45.6% and 45.2%), Hodgkin’s lymphoma was reported in 8.9% of the cases. The majority of patients (91.6%) presented in Ann-Arbor-Stages III and IV, and bone marrow infiltration was observed in a significant proportion of patients (61.5%). Soluble CD25 levels were markedly elevated (median 10,000 U/ml), with levels beyond 10,000 U/ml indicating unfavorable prognosis for 30-day and overall survival. 66.8% of the patients died after median 5.1 months. LA-HLH remains a clinical challenge requiring specialized management. Timely diagnosis and appropriate lymphoma-specific treatment are of utmost importance to enhance patient outcomes.
Similar content being viewed by others

Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a severe hyperinflammatory syndrome driven by pathologic activation of cytotoxic T-lymphocytes and macrophages ultimately leading to cytokine storm and organ damage if not treated appropriately. First described in 1939 by Bodley Scott and Robb-Smith as histiocytic medullary reticulosis, HLH is now classified in the “H”-group of histiocytoses according to the Histiocyte Society [1, 2]. In the setting of autoimmune/autoinflammatory diseases, HLH is also referred to as macrophage activation syndrome (MAS or MAS-HLH).
Considering pathophysiology, HLH can be divided in a primary, genetically determined, and a secondary, acquired form. In primary HLH (also referred to as familial HLH, FHL), mutations in genes affecting lymphocyte toxicity or immune regulation prompt defective killing of antigen-presenting cells, thus resulting in persistent immune stimulation and overwhelming immune response. In secondary HLH, an interplay between preexisting immunosuppression, inflammation (i.e., autoinflammatory or rheumatic disorders), cytokine release within infections or malignant diseases as well as potential genetic predisposition reflects the complex pathophysiology leading to the clinical picture of hyperinflammation [3].
In contrast to primary HLH, which typically appears in infancy, secondary or acquired HLH more frequently occurs in adults. While in FHL, especially infections of viral origin represent the main trigger, the spectrum of underlying diseases in secondary HLH is diverse, with infections and malignancies being most common [4]. Of note, malignancy-associated HLH (M-HLH) may appear in the context of initial diagnosis reflecting paraneoplasia, during the disease course in case of progression or relapse, or during treatment as infection-triggered HLH. In addition, M-HLH may appear following cytokine release and T-cell activation within the context of novel therapies both in hematological and solid cancers, making the differentiation of treatment toxicity and malignancy-associated inflammation a particular challenge. As with other malignancies, lymphoma treatment increasingly incorporates immune-activating and cellular therapies (i.e., checkpoint inhibitors, T-cell engaging bispecific antibodies, chimeric antigen receptor modified T-cells), in which cytokine release is a well-known side effect with the potential to induce a life-threatening HLH-like clinical picture in extreme cases (also termed as HLH-like toxicity) [5, 6]. On the contrary, the programmed cell death protein-1 (PD-1) antibody Nivolumab demonstrated efficacy in the treatment of refractory or relapsed Epstein-Barr virus (EBV)-HLH in a pilot study, most likely due to expansion of PD-1 positive T-cells and restoring the defective anti-EBV response [7].
In the M-HLH group, lymphoma represent the most common trigger and typically exhibit the poorest prognosis [8]. This might be due to often-aggressive disease courses of certain lymphoma subtypes enriched in patients with M-HLH, but also due to distinct and unusual clinical features hindering lymphoma diagnosis. In particular, initial diagnosis may be delayed because of similarities between HLH and other inflammatory conditions such as sepsis. Moreover, histological diagnosis may be hindered by tissue infiltration with activated lymphocytes and macrophages that allow tumor cells to hide behind inflammatory infiltrates [9].
Clinically, HLH patients may present with a variety of symptoms according to the underlying disease, a triad consisting of fever, splenomegaly, and cytopenia is common though. Diagnosis of HLH is currently based on the HLH-2004 criteria established by the Histiocyte Society, whereby 5 of 8 criteria have to be fulfilled for HLH diagnosis [10]. During past few years, adaptation of these criteria has been proposed especially for adult patients with secondary HLH given the non-specificity of several of the HLH-2004 criteria, such as using higher ferritin or soluble CD25 (sCD25; synonym: soluble interleukin-2 receptor chain alpha, sIL-2Rα) cut-off values to best identify patients with HLH and distinguish from other inflammatory states like sepsis. Recently, the combination of elevated sCD25 of more than 3900 U/ml and ferritin greater 1000 µg/l showed best sensitivity and specificity in diagnosing M-HLH and might also aid in identifying patients with lymphoma-associated HLH in due time [11].
Despite these advances and a growing number of publications reporting on lymphoma-associated HLH (LA-HLH) in adults, systematic or multicenter data are still scarce given the rarity of LA-HLH. Within the context of a systematic scoping review, we therefore aimed to synthesize research evidence on LA-HLH by interrogation of the published literature in order to determine current evidence on epidemiology, LA-HLH subtypes and its clinical features, as well as providing an overview on applied therapies and prognosis.
Methods
Search strategy
On February 27th, 2020, a comprehensive literature search of the PubMed database was performed. To identify publications on LA-HLH, the following search terms and medical subject headings were used: “hemophagocytic lymphohistiocytosis” OR “haemophagocytic lymphohistiocytosis” AND “lymphoma-associated hemophagocytic lymphohistiocytosis” OR “hemophagocytic syndrome lymphoma” OR “haemophagocytic syndrome lymphoma” OR “Hodgkin hemophagocytic lymphohistiocytosis” OR “Hodgkin haemophagocytic lymphohistiocytosis” OR “T-cell non-Hodgkin lymphoma hemophagocytic lymphohistiocytosis” OR “T-cell non-Hodgkin haemophagocytic lymphohistiocytosis” OR “diffuse large B-cell lymphoma hemophagocytic lymphohistiocytosis” OR “diffuse large B-cell lymphoma haemophagocytic lymphohistiocytosis” OR “chronic lymphocytic leukemia hemophagocytic lymphohistiocytosis” OR “chronic lymphocytic leukemia haemophagocytic lymphohistiocytosis”.
Study selection and data collection
All case reports and studies referring to adult patients (≥18 years of age) with HLH triggered by lymphoma were screened. We considered patients to have HLH if they met at least five out of eight HLH-2004 diagnostic criteria or authors reported a diagnosis of HLH respectively. Only articles published in English language were included. We excluded articles focusing on pediatric patients, reporting insufficient data, or if lymphoma was not considered being the most likely HLH-triggering disease.
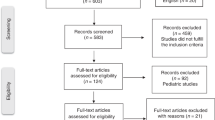
Study selection followed a three-step analysis process. First, based on inclusion and exclusion criteria, titles and abstracts of all 595 PubMed results were assessed for their relevance. Secondly, full-text content of articles was obtained if available. Of 73 articles with no available full text, the corresponding authors were requested to provide data. Subsequently, 67 articles were excluded from further analysis (64 articles without available full-text and three articles reporting on HLH cases not associated with lymphoma). In a third step, the remaining 297 full-text articles were reassessed using the abovementioned criteria and with consideration of data quality, resulting in 132 publications for detailed analysis. Figure 1 illustrates the process of study selection using a PRISMA flow diagram.

PRISMA flow diagram showing study selection process.
Data on lymphoma subtype (B-cell non-Hodgkin lymphoma, T-cell non-Hodgkin lymphoma (T-NHL), Hodgkin’s lymphoma, unspecified lymphoma), patient characteristics, clinical and laboratory parameters, treatment, and outcome were extracted depending on availability. Survival times if provided in the analyzed articles were extracted and recorded in months. In case the authors reported more than one associated lymphoma, the most likely HLH-triggering diagnosis was listed accordingly. To avoid analyzing duplicates (i.e., overlapping patient cohorts), author names and affiliations and year of publication were compared. Patient demographics were subsequently reviewed to identify cases published more than once.
Statistical analysis
Descriptive data are presented as median and corresponding ranges or frequencies (%). We used Chi-Square test to compare differences regarding categorial variables and Mann-Whitney U test for continuous variables. Survival times were recorded as provided in the analyzed articles and visualized using Kaplan-Meier method, the log-rank test was performed for comparison of subgroups. Further survival analysis was carried out using Kaplan-Meier method and Cox’s proportional hazards model. Variables with a p < 0.05 in univariate analysis were included in multivariate analysis using Cox’s proportional hazards model with a backward stepwise selection procedure. All statistical tests were two-sided. A p < 0.05 was considered statistically significant. All statistical analyses were carried out using IBM SPSS Statistics Version 27 (IBM Corp., Armonk, N.Y., US).
Results
Epidemiology
Of 595 search results, 132 articles (N = 113 case reports [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124]; N = 19 single or multicenter studies [125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143]) providing data on 542 LA-HLH patients were eligible for further analysis and included in this study. Three hundred forty patients were male (62.7%) and median age was 60 years (range, 18–98 years), patients with T-NHL had a lower median age compared to those with B-cell non-Hodgkin lymphoma (B-NHL) (49 vs. 64 years, p < 0.001). As expected, given the growing diagnostic vigilance, the number of publications concerning LA-HLH showed a steady increase during the past years (Supplementary Fig. 1).
The majority of reported patients came from Asia (N = 323/542, 59.6%), followed by Europe and North America (28.6% and 11.1%, respectively). In more detail, most of the published patients (pts) were from Japan and China (174 and 138 pts, respectively), followed by France (93 pts). Figure 2 provides an overview on the geographic distribution and shows corresponding lymphoma subgroups, Supplementary Table 1 shows countries of origin and the corresponding lymphoma subtypes. Of note, T-NHL was reported more frequently in publications originating from China compared to all other countries, with particular emphasis on natural killer/T-cell lymphoma (NKTCL).

Abbreviations: B-NHL B-cell non-Hodgkin lymphoma, T-NHL T-cell non-Hodgkin lymphoma, HL Hodgkin’s lymphoma.
Lymphoma subtypes
There was an equal distribution of T-NHL and B-NHL-triggered HLH (45.2% and 45.6% of all patients, respectively). Hodgkin’s lymphoma-associated HLH was reported in 8.9% of all cases. The most common T-NHL subgroups according to 2016 World Health Organization classification of lymphoid neoplasms [144] were peripheral T-cell lymphoma, not otherwise specified (21.2% of all T-NHL-HLH cases) and extranodal NKTCL, nasal type (29.8%). Moreover, subcutaneous panniculitis-like T-cell lymphoma (SPTCL) and angioimmunoblastic T-NHL (AITL) were frequently reported (8.6% and 6.1%, respectively). In the B-NHL group diffuse large B-cell lymphoma represents the main etiology (50.6% of all B-NHL-associated HLH cases), followed by intravascular large B-cell lymphoma (23.1%). Figure 3 shows a graphic impression of different LA-HLH subgroups, and Supplementary Table 2 gives a detailed listing of lymphoma subtypes with their percentage distributions.

Abbreviations: B-NHL B-cell non-Hodgkin lymphoma, T-NHL T-cell non-Hodgkin lymphoma, HL Hodgkin’s lymphoma, PTCL, NOS Peripheral T-cell lymphoma, not otherwise specified, SPTCL Subcutaneous panniculitis-like T-cell lymphoma, ENKTCL, NT Extranodal natural killer/T-cell lymphoma, nasal type, NKTCL Natural killer/T-cell lymphoma, AITL Angioimmunoblastic T-cell lymphoma, ALCL Anaplastic large-cell lymphoma, DLBCL Diffuse large B-cell lymphoma, BCL, NOS B-cell lymphoma, not otherwise specified, MZL Marginal zone lymphoma, CLL Chronic lymphocytic leukemia, LPL Lymphoplasmacytic lymphoma, HGBCL High-grade B-cell lymphoma, MCL Mantle cell lymphoma.
Patient characteristics, clinical features
Besides typical symptoms like fever, or enlarged liver and spleen, there was a wide spectrum of symptoms caused by involvement of liver, lung, or the gastrointestinal tract. Of note, there was a high percentage of bone marrow infiltration due to lymphoma throughout all subgroups, which was highest in the B-NHL subgroup with almost 80%. The vast majority of patients (91.6%) presented in Ann-Arbor stages III and IV. Half of the patients with T-NHL exhibited skin changes. Table 1 comprises clinical features of the total lymphoma cohort, and patients grouped by subentities.
Laboratory features
Single or more extensive laboratory values were available for 225 patients. About 60% of the patients presented with cytopenia of at least two lineages; thrombocytopenia less than 100 × 109/l was observed most frequently (85.6%), followed by anemia and neutropenia (61.4 and 44.2%, respectively). Ferritin as a hallmark of HLH was increased in 90% of the patients, median ferritin level was 4808 µg/l (range, 12–526,259), and 30% of the patients presented with extremely elevated values beyond 15,000 µg/l. Comparing different lymphoma subgroups, patients with T-NHL showed significantly higher median ferritin levels than the B-NHL group (7604 vs. 2785 µg/l, p = 0.045) and more often had values of above 10,000 µg/l (47.8 vs. 23.6%, p = 0.003). Elevated triglycerides were observed in 62% of the patients, while low fibrinogen was present in 42%. Virtually all patients had elevated soluble CD25 (sCD25) levels, with a median of 10,380 U/ml (range, 1194–108,640) for the entire cohort. Comparing sCD25 levels between lymphoma subgroups, there was no significant difference between the B-NHL and T-NHL group (median 9585 U/ml [range, 1194–71,834] vs. 14,000 U/ml [range, 3588–108,640], p = 0.392). However, the sCD25/ferritin ratio was higher in patients with HLH triggered by B-NHL. Patients with Hodgkin’s lymphoma-HLH showed the highest sCD25 levels with a median level of 24,414 U/ml (range, 4250–96,415). Lactate dehydrogenase levels were elevated in nearly all patients (97.3%), with a significant increase of more than five times the upper limit of normal in one third (34.4% of the patients). Patients with T-NHL tended to have lower platelet counts and lower fibrinogen than B-NHL patients. Table 2 shows detailed laboratory features of all patients with available data and grouped into B- and T-NHL associated HLH, Supplementary Table 6 provides laboratory features of Hodgkin’s lymphoma-HLH patients.
EBV status was available for 131 patients. Of these, 29 (22.1%) had past infection and 49 (37.4%) presented with EBV reactivation defined as active EBV replication. Comparing lymphoma subgroups, patients with T-NHL showed a higher percentage of EBV replication than B-NHL (41.7 vs. 18.8%), while virtually all Hodgkin’s lymphoma patients exhibited EBV replication (89.5%). Data on EBV status are comprised in Supplementary Table 3.
Treatment
Detailed information on HLH treatment and lymphoma-specific therapy (or if no such treatment was administered) was available for 184/542 patients (33.9%) and 280/542 patients (51.7%), respectively. 94.3% (264/280) of the patients with available information received treatment for either HLH, lymphoma, or both. Forty patients (14.3%) were treated for HLH only, while 222 patients (79.3%) received lymphoma-specific treatment (alone or in combination) and 90 patients (32.1%) were treated for HLH and lymphoma. Sixteen patients (5.7%) received neither HLH- nor lymphoma-specific treatment. 29 of 222 patients with information on lymphoma treatment (13.1%) received hematopoietic stem cell transplantation (HSCT) (allogeneic or autologous). Table 3 provides an overview on HLH and lymphoma-specific treatment strategies in more detail.
Treatment of the triggering disease (i.e., lymphoma-directed therapy or combined treatment directed against HLH and lymphoma) resulted in lower mortality (57.7%; vs. 100% mortality for patients who had received HLH-directed therapy only or no treatment at all). Without lymphoma-specific treatment, median estimated survival time was less than 1 month (Supplementary Table 4).
Prognostic factors, outcome
Information on outcome and corresponding follow-up times were available for 271 patients, of whom 181 (66.8%) died after a median estimated survival time of 5.1 months (Fig. 4a). 75 of 271 patients (27.7%) died within the first month after HLH diagnosis, while 48 of these 75 patients (64%) had T-NHL (N = 25 NK/T-cell lymphoma, N = 23 other T-NHL). Comparing lymphoma subgroups, patients with NK/T-cell lymphoma-HLH had shorter estimated survival time than patients with B-NHL, T-NHL, and Hodgkin’s lymphoma-HLH (1.0 months vs. 12.0, 3.2, and 31.0 months, respectively; p < 0.001) (Fig. 4b). Of 222 patients who received lymphoma-specific therapy, 208 had available follow-up data. Median estimated survival time in these patients was 11.0 months, with a particular poor prognosis for patients with T-NHL and NK/T-cell lymphoma (median survival 6.0 and 3.4 months, Fig. 4c). Even though HSCT was carried out only in minority of patients, those receiving HSCT showed considerably longer estimated survival (B-NHL: 36.0 vs. 12.0 months, p = 0.004; T-NHL: 93.0 vs. 3.2 months, p < 0.001, respectively; Supplementary Fig. 2a, b).

Kaplan-Meier plots displaying estimated survival. Plot (a) shows information for the entire cohort, (b) for lymphoma subgroups, and (c) for patients who had received any lymphoma-specific treatment depending on lymphoma subgroup. Abbreviations: LA-HLH lymphoma-associated hemophagocytic lymphohistiocytosis, B-NHL B-cell non-Hodgkin lymphoma, T-NHL T-cell non-Hodgkin lymphoma, HL Hodgkin’s lymphoma, NKTCL natural killer/T-cell lymphoma.
For patients with sufficient data, univariate Cox regression analyses for both overall and 30-day mortality were conducted. Results are summarized in Supplementary Table 5. Age over 60 years, sCD25 levels exceeding 10,000 U/ml, and ferritin levels above 15,000 µg/l were associated with poorer overall survival (OS) in univariate analysis; while sCD25 levels above 10,000 U/ml and platelet counts below 50 × 109/l were associated with poorer 30-day survival. Parameters, which showed statistical significance in univariate analysis were included in a multivariate Cox regression analysis. For 30-day mortality, we also included ferritin above 15,000 µg/l, which shows a tendency towards poorer survival in univariate analysis and is an essential parameter in HLH. The results of the multivariate Cox regression analysis are presented in Fig. 5 as Forest plots displaying hazard ratios and corresponding confidence intervals. Both ferritin levels above 15,000 µg/l and sCD25 levels above 10,000 U/ml independently predicted poorer OS, while the latter one also predicted poorer 30-day survival.

Multivariate Cox regression analysis for possible prognostic factors. Results are presented as Forest plots with Hazard ratios and confidence intervals, plots provide information on (a) overall mortality and (b) 30-day mortality (death within 30 days). Abbreviations: HR hazard ratio, CI confidence interval, sCD25 soluble CD25.
Discussion
Adult hemophagocytic lymphohistiocytosis (aHLH) is a clinical syndrome characterized by pathologic hyperinflammation caused by aberrant immune activation resulting in a cytokine storm with often fatal organ failure [145, 146]. In most patients, triggering diseases such as infections or malignancies can be identified. With increasing patient age, HLH associated with lymphoma (LA-HLH) is the leading HLH-subtype, typically exhibiting a poorer prognosis compared to HLH following infections or autoimmune diseases [127]. Despite a growing number of published cases or case series during the past years (Supplementary Fig. 1), systematic data on LA-HLH are limited. Within the framework of a scoping review, and to the best of our knowledge, we here present the largest systematic analysis on LA-HLH, comprising data of 132 articles reporting on a total of 542 patients.
The increasing frequency of publications on adult HLH might be due to several reasons. On one hand, we assume higher awareness and diagnostic vigilance, which might be driven mainly by efforts towards a more standardized work-up in HLH during past years (resulting in several published national and international guidelines [9, 147, 148]). On the other hand, there are hints for a rising incidence rate, as reported by a German study [149]. The authors outline a growing number of HLH cases between 2014 and 2020, mainly due to an increase in older HLH patients, for whom there is a higher prevalence of HLH triggers like malignancies and autoimmune diseases. Beyond, this study reports increasing numbers of inpatient deaths related to HLH, possibly attributed to higher age, disease severity and higher lymphoma incidence, harboring especially poor prognosis.
In line with latest epidemiological data on malignant lymphoma and a recent nationwide Swedish study reporting on malignancy-associated HLH, we found a male predominance with 62.7% of all reported LA-HLH cases [150, 151]. Median age at diagnosis in the present cohort was 60 years, while patients with T-NHL-associated HLH were significantly younger compared to those with B-NHL-HLH (49 vs. 64 years, p < 0.001). This difference might reflect the significant proportion of NK/T-cell lymphoma within the T-NHL group – a lymphoma subtype with an often particularly young age of onset [152]. Besides NK/T-cell lymphoma, other common subtypes comprised angioimmunoblastic T-cell lymphoma, subcutaneous panniculitis-like T-NHL (SPTCL) and intravascular large B-cell lymphoma (displayed and summarized also in Fig. 3 and Supplementary Table 2). Some of these rare lymphoma subtypes seem to be enriched in LA-HLH patients. Mechanistically, this might be explained by aberrant immune activity of the tumor cells and the tumor microenvironment causing hyperinflammation with cytokine storm, the association of lymphoma pathophysiology with EBV tropism, and/or modulating genetic factors leading to increased risk for HLH development [153, 154]. An example is loss-of-function mutations involving the Hepatitis A Virus-Cellular Receptor 2 gene (HAVCR2), which are enriched in SPTCL and result in dysregulated interferon-γ-inducible inflammatory host response as predisposing factor for HLH [155,156,157]. Of note, germline HAVCR2 mutations have been reported in idiopathic HLH/HLH-like systemic disease, supporting molecular testing in addition to deep skin biopsies in these patients [158, 159]. The discovery of HAVCR2 mutations in inflammatory conditions is a prime example how better understanding of the molecular basis of HLH can lead to treatment optimization, as shown by promising responses following immunomodulatory treatment with the JAK1/2 inhibitor ruxolitinib in SPTCL and patients with lupus panniculitis [159, 160].
Besides more or less disease-specific factors, preexisting iatrogenic or acquired immunosuppression (i.e., infections with human immunodeficiency virus, drugs like steroids or azathioprine) is frequently observed in HLH patients. Consequently, adult HLH may exhibit with variable and occasionally atypical clinical presentation, reflecting the complex pathophysiology and interaction of the aforementioned factors. On the other hand, the multifaceted clinical picture not infrequently leads to delayed or misdiagnosis of HLH, especially if lymphoma-associated.
In this analysis, typical symptoms comprised fever and organomegaly, and nearly all patients with LA-HLH were diagnosed in Ann Arbor stages III and IV. Interestingly, more than half of the patients had lymphoma infiltration in the bone marrow. Moreover, lymphoma infiltration in liver, spleen, and skin was frequently reported. Besides typically aggressive courses and advanced disease stages of lymphoma presenting with HLH, possible explanations might be higher rates of extranodal manifestations in certain lymphoma subtypes enriched in our cohort or preceding immunosuppression, which was shown to significantly increase the risk for aggressive lymphoma (and likewise extranodal manifestations) [161,162,163,164]. Even though the presented data have to be interpreted with caution given potential publication bias with a tendency to publish illustrative cases (i.e., with bone marrow infiltration and/or liver/spleen involvement) more frequently, several implications on diagnosis and management of LA-HLH may be drawn. Keeping in mind the especially poor prognosis as demonstrated in this cohort and published studies on LA-HLH [126, 165,166,167], and even worse survival of less than 1 month if no lymphoma-specific therapy or only HLH-directed therapy is applied (Supplementary Table 4), a comprehensive and vigorous diagnostic work-up with the aim of timely trigger identification is justified. This includes imaging (preferably using positron-emission tomography guidance [168]) and (repetitive) biopsy of suspicious tissue or lymph node excision, especially in patients with no obvious signs of a triggering disease or suspected but yet not proven lymphoma [9]. A recent study from French intensive care specialists demonstrated a high rate of trigger identification in 95% of the examined patients using an aggressive diagnostic approach including tissue biopsies of lymph node, skin, bone marrow, and liver. Of note, it was possible to apply timely trigger specific therapy in 68% of the patients in the Intensive Care Unit [169].
Laboratory hallmarks of HLH comprise cytopenia and in particular highly elevated ferritin and sCD25 levels [170, 171]. In our analysis with special emphasis on LA-HLH, nearly all patients fulfilled the HLH-2004 criteria for ferritin and sCD25 (ferritin >500 µg/l: 90% of all patients, sCD25 ≥ 2400 U/ml: 98% of all patients). Patients with T-NHL-associated HLH had significantly higher ferritin levels compared to those with B-NHL (7645 vs. 2785 µg/l, p = 0.033) and presented with lower median platelet counts and fibrinogen, indicating a more profound inflammatory response. Aberrant immune activity and inflammation is a well-known phenomenon in certain T-NHL subtypes, for example NK/T-cell lymphoma, which represent a major group in this report and often go along with EBV-infection [172]. Besides, we observed a particular high rate of EBV replication in the Hodgkin’s lymphoma group (Supplementary Table 3). A strong association of EBV and Hodgkin’s lymphoma-associated HLH was shown in the past, highlighting the role of EBV in the pathogenesis of HLH in this lymphoma subgroup [173]. Active EBV replication acting as a co-trigger should thus be considered in patients with HLH features and suspected malignancy, and there are case reports showing positive EBV-PCR before lymphoma-diagnosis was established [174].
Soluble CD25 (sCD25; synonym: soluble interleukin-2 receptor alpha chain, sIL-2Rα) is a surrogate parameter of T-cell activation and was shown to be a sensitive test in diagnosing adult HLH as well as for monitoring disease activity [171, 175, 176]. In addition, a high sCD25/ferritin ratio was proposed as a marker for the diagnosis of LA-HLH [140]. In our analysis, we found highly elevated median sCD25 levels of about 10,000 U/ml, with highest values in patients with Hodgkin’s lymphoma-associated HLH. Although elevated sCD25 levels are also observed in inflammation, infection, or solid tumors, the values in hematological malignancies (i.e., lymphoma) are typically higher [177]. In an attempt to simplify (and perhaps accelerate) the diagnosis of HLH in hematologic malignancies, Zoref-Lorenz et al. developed a novel combination of ferritin and sCD25, termed optimized HLH inflammatory index (OHI). Using defined cut-offs for ferritin (>1000 µg/l) and sCD25 (>3900 U/ml), patients with malignancy-associated HLH were best identified [11]. In a Japanese study comparing 57 LA-HLH patients to 53 patients with HLH due to benign diseases, a sCD25 cut-off of 5000 U/ml showed best sensitivity and specificity for a diagnosis of LA-HLH [178]. In synopsis of the published literature and the data of our analysis, significantly elevated sCD25 levels in patients with clinical signs of hyperinflammation should prompt diagnostics for a yet undetected lymphoma. Besides the importance in recognizing and diagnosing HLH, sCD25 was recently shown to have also prognostic value in adult HLH [179]. In the present analysis, we demonstrated an adverse prognostic impact for sCD25 levels beyond 10,000 U/ml both for OS and 30-day survival, which might be explained by advanced lymphoma disease and pronounced inflammation leading to organ failure. Given the limited sample size (only 64 and 62 patients eligible for this analysis) and retrospective design of our study, this finding should be interpreted with caution though, and potential consequences (i.e., treatment intensification) should be discussed thoroughly on an individual case basis considering disease severity and potentially harmful treatment effects.
The particular poor prognosis of LA-HLH (this cohort: median estimated survival 5.1 months overall and 11.0 months if lymphoma-specific therapy was applied) warrants further treatment optimization. In the present study, a detailed analysis for example on the early use of etoposide was not feasible due to limited data availability. However, the dismal prognosis of patients not receiving any lymphoma-specific therapy and a patient proportion of almost 30% dying within the first month after HLH diagnosis indicates the need of combined anti-inflammatory (HLH-directed) and lymphoma-specific therapy as soon as possible. LA-HLH patients often present with severe disease characterized by imminent or manifest organ failure clinically and highly elevated ferritin and sCD25 levels as laboratory surrogate parameters. In these severely ill patients, profound immunosuppression beyond steroids and polyvalent immunoglobulins is typically necessary in order to effectively suppress hyperinflammation, thus allowing for further diagnostics and to bridge the time gap between HLH diagnosis and begin of lymphoma-specific therapy. In this situation, current guidelines recommend adding dose-adjusted etoposide (i.e., 50–100 mg/m2 once or twice weekly) [8, 9, 180] given its activity against inflammation and lymphoma. This approach is supported by several studies showing beneficial effects for early use of etoposide in patients with LA-HLH [126, 181]. However, large prospective studies are lacking with respect to rarity of LA-HLH and often dramatic clinical disease courses. Thus, we strongly encourage international collaborations to study aspects of optimal treatment systematically in a larger cohort of well-characterized lymphoma-HLH patients, in particular with the advent of more targeted (and maybe less harmful) therapies like ruxolitinib [182].
Beneficial effects for incorporating etoposide in subsequent lymphoma-therapy have also been suggested, a recent Chinese study demonstrated high overall response rates and promising survival data in patients with B-NHL-HLH (5-year overall survival 73%) treated with dose-adjusted EPOCH-R regimen in first-line [183]. This observation is in line with previous reports on high-risk patients with aggressive B-NHL, showing 4-year OS of 75% [184] and 10-year OS of 72% for patients treated with R-CHOEP-14 [185]. Based on these data, inclusion of etoposide in first line therapy seems reasonable in LA-HLH patients, especially in younger patients with B-NHL triggered HLH.
As demonstrated within this cohort, lymphoma presenting with HLH typically exhibit aggressive disease courses and poor prognosis even if initial cytokine storm is controlled and lymphoma-specific therapy is applied (median survival 11 months for these patients in the present analysis). For that reason, primary consolidation therapy using high-dose chemotherapy and subsequent stem cell transplantation is proposed in guidelines, even though these considerations are mainly derived from a study on peripheral T-NHL [186]. Data regarding this special issue are very limited and primarily case reports. In the present analysis, 29 patients had HSCT (SCT; N = 21 autologous, N = 8 allogeneic) and exhibited a considerably better prognosis. Of note, all patients in the B-NHL group (N = 14) underwent autologous SCT. In line with these findings, a recent study by Song et al. reported encouraging OS and similar prognosis for LA-HLH patients undergoing autologous SCT when compared to lymphoma patients without HLH undergoing autologous SCT [187]. In this study, all patients received conditioning regimen with cyclophosphamide, BCNU, and etoposide. Obviously, these data are to some extent preliminary and have to be interpreted with caution. However, in the absence of more sophisticated data, consolidating high-dose chemotherapy and autologous SCT (or initiation of donor search for allogeneic SCT in patients with disseminated NK/T-cell lymphoma) should be evaluated depending on the individual patient’s constitution in B-NHL- and T-NHL-HLH.
Our study has several limitations: first of all, retrospective design and limited data availability depending on published information prevented several more detailed analyses. Moreover, a publication bias with a tendency to publish notably illustrative cases (as discussed above) cannot be ruled out. Despite these restrictions, we here to the best of our knowledge present the largest analysis on LA-HLH, providing a comprehensive overview especially regarding to lymphoma subtypes, clinical presentation, and laboratory features. The especially poor prognosis in LA-HLH patients was confirmed even if lymphoma-specific therapy is applied, justifying an aggressive diagnostic approach and underlining the need of larger and prospective studies to determine and refine optimal management in lymphoma-associated HLH.
References
Bodley Scott R, Robb-Smith AHT. Histiocytic medullary reticulosis. Lancet. 1939;234:194–8.
Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672–81.
Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515.
Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383:1503–16.
Hines MR, Keenan C, Maron Alfaro G, Cheng C, Zhou Y, Sharma A, et al. Hemophagocytic lymphohistiocytosis-like toxicity (carHLH) after CD19-specific CAR T-cell therapy. Br J Haematol. 2021;194:701–7.
Liu LL, Skribek M, Harmenberg U, Gerling M. Systemic inflammatory syndromes as life-threatening side effects of immune checkpoint inhibitors: case report and systematic review of the literature. J Immunother cancer. 2023;11:e005841.
Liu P, Pan X, Chen C, Niu T, Shuai X, Wang J, et al. Nivolumab treatment of relapsed/refractory Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults. Blood. 2020;135:826–33.
Daver N, McClain K, Allen CE, Parikh SA, Otrock Z, Rojas-Hernandez C, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017;123:3229–40.
La Rosée P, Horne A, Hines M, von Bahr Greenwood T, Machowicz R, Berliner N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. 2019;133:2465–77.
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
Zoref-Lorenz A, Murakami J, Hofstetter L, Iyer S, Alotaibi AS, Mohamed SF, et al. An improved index for diagnosis and mortality prediction in malignancy-associated hemophagocytic lymphohistiocytosis. Blood. 2022;139:1098–110.
Adler NR, Sia CS, Polchleb C, Jane S, Aung AK. Intravascular large B cell lymphoma with haemophagocytic syndrome: a double lethal masquerade. Intern Med J. 2015;45:1310–2.
Alaoua A, Gilbert G, Ghannouchi N, Houchlef M, Letaief A, Bahri F. Primary bilateral adrenal lymphoma revealed by hemophagocytic syndrome. Ann Endocrinol. 2011;72:247–50.
Aljitawi OS, Boone JM. Lymphoma-associated hemophagocytic lymphohistiocytosis. Blood. 2012;120:932.
Anghel G, Petrinato G, Severino A, Remotti D, Insabato L, De Renzo A, et al. Intravascular B-cell lymphoma: report of two cases with different clinical presentation but rapid central nervous system involvement. Leuk Lymphoma. 2003;44:1353–9.
Bailey C, Dearden C, Ardeshna K. Haemophagocytic lymphohistiocytosis as a consequence of untreated B-cell chronic lymphocytic leukaemia. BMJ Case Rep. 2017;2017:bcr2016219057.
Bains A, Mamone L, Aneja A, Bromberg M. Lymphoid malignancy-associated hemophagocytic lymphohistiocytosis: search for the hidden source. Ann Diagn Pathol. 2017;28:37–42.
Baselga E, Pujol RM, Costa I, Bordas R, De Moragas JM. Subcutaneous angiocentric T-cell lymphoma associated with fatal hemophagocytic syndrome. Int J Dermatol. 1997;36:363–7.
Bhagwati NS, Oiseth SJ, Abebe LS, Wiernik PH. Intravascular lymphoma associated with hemophagocytic syndrome: a rare but aggressive clinical entity. Ann Hematol. 2004;83:247–50.
Blom A, Beylot-Barry M, D’Incan M, Laroche L. Lymphoma-associated hemophagocytic syndrome (LAHS) in advanced-stage mycosis fungoides/Sezary syndrome cutaneous T-cell lymphoma. J Am Acad Dermatol. 2011;65:404–10.
Boland PJ, Hegerova LT, Williams SJ, McKenna RW, Bachanova V, Eckfeldt CE. Successful treatment of two cases of classical Hodgkin lymphoma-associated hemophagocytic lymphohistiocyosis with R-CEPP. Leuk Lymphoma. 2017;58:478–81.
Brown NA, Ross CW, Gudjonsson JE, Wale D, Pawarode A, Maillard I, et al. Subcutaneous panniculitis-like T-cell lymphoma with bone marrow involvement. Am J Clin Pathol. 2015;143:265–73.
Chan EY, Pi D, Chan GT, Todd D, Ho FC. Peripheral T-cell lymphoma presenting as hemophagocytic syndrome. Hematol Oncol. 1989;7:275–85.
Cheng FY, Tsui WM, Yeung WT, Ip LS, Ng CS. Intravascular lymphomatosis: a case presenting with encephalomyelitis and reactive haemophagocytic syndrome diagnosed by renal biopsy. Histopathology. 1997;31:552–4.
Chhabra S, Strair RK, Rubin AD. Haemophagocytic lymphohistiocytosis and primary central nervous system lymphoma. Intern Med J. 2013;43:463–4.
Cho SG, Koh YB, Chang HS, Park G, Kang CS, Park JW, et al. Successful treatment with splenectomy and interferon alpha against recurred hemophagocytic syndrome in remission state of anaplastic large cell lymphoma following high-dose therapy and autologous peripheral blood stem cell transplantation. Eur J Haematol. 2005;74:259–62.
Chubachi A, Miura I, Hatano Y, Ohshima A, Nishinari T, Miura AB. Syndrome of inappropriate secretion of antidiuretic hormone in patients with lymphoma-associated hemophagocytic syndrome. Ann Hematol. 1995;70:53–55.
Ciaudo M, Chauvenet L, Audouin J, Rossert J, Favier R, Horellou MH, et al. Peripheral-T-cell lymphoma with hemophagocytic histiocytosis localised to the bone marrow associated with inappropriate secretion of antidiuretic hormone. Leuk Lymphoma. 1995;19:511–4.
Cuttelod M, Pascual A, Baur Chaubert AS, Cometta A, Osih R, Duchosal MA, et al. Hemophagocytic syndrome after highly active antiretroviral therapy initiation: a life-threatening event related to immune restoration inflammatory syndrome? Aids. 2008;22:549–51.
Davidson-Moncada JK, McDuffee E, Roschewski M. CD5+ diffuse large B-cell lymphoma with hemophagocytosis. J Clin Oncol. 2013;31:e76–79.
Dawson L, den Ottolander GJ, Kluin PM, Leeksma O. Reactive hemophagocytic syndrome as a presenting feature of Hodgkin’s disease. Ann Hematol. 2000;79:322–6.
Demirkan F, Vural F, Ozsan GH, Ozcan MA, Ozkal S, Undar B. Hemophagocytic syndrome associated with inappropriate secretion of antidiuretic hormone in lymphoma and acute myeloblastic leukemia: report of two cases. Leuk Lymphoma. 2001;42:1401–4.
Dominguez-Munoz MA, Morales-Camacho RM, Prats-Martin C, Avila R, Vargas MT, Burillo S, et al. Unusual co-occurrence of Hodgkin lymphoma and hemophagocytic lymphohistiocytosis in a bone marrow aspirate. Ann Hematol. 2016;95:1019–21.
Dufau JP, Le Tourneau A, Molina T, Le Houcq M, Claessens YE, Rio B, et al. Intravascular large B-cell lymphoma with bone marrow involvement at presentation and haemophagocytic syndrome: two Western cases in favour of a specific variant. Histopathology. 2000;37:509–12.
Epperla N, Harrington AM, Hemauer K, Shah NN. Extracavitary primary effusion lymphoma associated with hemophagocytic lymphohistiocytosis. Am J Hematol. 2016;91:1161–4.
Eser B, Altuntas F, Er O, Kontas O, Ferahbas A, Cetin M, et al. A case of subcutaneous panniculitis-like T-cell lymphoma with haemophagocytosis developing secondary to chemotherapy. J Eur Acad Dermatol Venereol. 2004;18:713–5.
Flew SJ, Radcliffe KW. Haemophagocytic lymphohistiocytosis complicating Hodgkin’s lymphoma in an HIV-positive individual. Int J STD AIDS. 2010;21:601–3.
Fretwell TB, Hanna M. An incidental finding of severe hyperferritinaemia: a lesson to be learned. J R Coll Physicians Edinb. 2018;48:30–32.
Fung KM, Chakrabarty JH, Kern WF, Magharyous H, Gehrs BC, Li S. Intravascular large B-cell lymphoma with hemophagocytic syndrome (Asian variant) in a Caucasian patient. Int J Clin Exp Pathol. 2012;5:448–54.
Gerard L, Oksenhendler E. Hodgkin’s lymphoma as a cause of fever of unknown origin in HIV infection. AIDS Patient Care STDS. 2003;17:495–9.
Ghose A, Yellu M, Wise-Draper T, Sharma D, Qualtieri J, Latif T, et al. Lymphoma presenting as secondary HLH: a review with a tale of two cases. Clin Lymphoma Myeloma Leuk. 2014;14:e187–193.
Greil C, Roether F, La Rosee P, Grimbacher B, Duerschmied D, Warnatz K. Rescue of cytokine storm due to HLH by hemoadsorption in a CTLA4-deficient patient. J Clin Immunol. 2017;37:273–6.
Hagihara M, Inoue M, Hua J, Iwaki Y. Lymphocyte-depleted Hodgkin lymphoma complicating hemophagocytic lymphohistiocytosis as an initial manifestation: a case report and review of the literature. Intern Med. 2012;51:3067–72.
Han SM, Teng CL, Hwang GY, Chou G, Tsai CA. Primary splenic lymphoma associated with hemophagocytic lymphohistiocytosis complicated with splenic rupture. J Chin Med Assoc. 2008;71:210–3.
Hanaoka M, Tsukimori K, Hojo S, Abe Y, Mutou T, Muta K, et al. B-cell lymphoma during pregnancy associated with hemophagocytic syndrome and placental involvement. Clin Lymphoma Myeloma. 2007;7:486–90.
Harada S, Shinohara T, Naruse K, Machida H. Diffuse 18F-fluorodeoxyglucose accumulation in the bone marrow of a patient with haemophagocytic lymphohistiocytosis due to Hodgkin lymphoma. BMJ Case Rep. 2016;2016:bcr2016217555.
Harvey Y, Wordsworth H, Sia H. Epstein-Barr virus-negative Hodgkin lymphoma presenting as haemophagocytic lymphohistiocytosis. Br J Haematol. 2015;169:2.
He M, Jia J, Zhang J, Beejadhursing R, Mwamaka Sharifu L, Yu J, et al. Pregnancy-associated hemophagocytic lymphohistiocytosis secondary to NK/T cells lymphoma: a case report and literature review. Medicine. 2017;96:e8628.
Hirai H, Shimazaki C, Hatsuse M, Okano A, Ashihara E, Inaba T, et al. Autologous peripheral blood stem cell transplantation for adult patients with B-cell lymphoma-associated hemophagocytic syndrome. Leukemia. 2001;15:311–2.
Hrudka J, Eis V, Herman J, Prouzova Z, Rosenwald A, Duska F. Panniculitis-like T-cell-lymphoma in the mesentery associated with hemophagocytic syndrome: autopsy case report. Diagn Pathol. 2019;14:80.
Hu S, Bansal P, Lynch D, Rojas Hernandez CM, Dayao Z. Rituximab, etoposide, methylprednisolone, high-dose cytarabine, and cisplatin in the treatment of secondary hemophagocytic lymphohistiocytosis with classical Hodgkin lymphoma: a case report and review of the literature. J Med Case Rep. 2016;10:365.
Hussain S, Hallam S, Beltran L, Haroon A, Majumdar K, Shamash J, et al. Intravascular large B-cell lymphoma presenting as a pituitary mass with bilateral adrenal enlargement and haemophagocytic lymphohistiocytosis. Br J Haematol. 2018;181:851–2. Jun
Inagaki N, Sugimoto K, Hosone M, Isobe Y, Yamamoto Y, Sasaki M, et al. Disseminated Mucor infection and thrombotic microangiopathy in lymphoma-associated hemophagocytic syndrome. Int J Hematol. 2008;88:355–6.
Isotani H, Kameoka K. Hemophagocytic syndrome associated with B cell lymphoma in a patient with mitochondrial diabetes. Ann Hematol. 2001;80:187–8.
Jamil A, Nadzri N, Harun N, Ong CL. Primary cutaneous diffuse large B-cell lymphoma leg type presenting with hemophagocytic syndrome. J Am Acad Dermatol. 2012;67:e222–3.
Jang KA, Choi JH, Sung KJ, Moon KC, Koh JK, Kwon YM, et al. Primary CD56 + nasal-type T/natural killer-cell subcutaneous panniculitic lymphoma: presentation as haemophagocytic syndrome. Br J Dermatol. 1999;141:706–9.
Jassal DS, Kasper K, Morales C, Rubinger M. Autologous peripheral stem cell transplantation for aggressive hemophagocytic syndrome associated with T-cell lymphoma: case study and review. Am J Hematol. 2002;69:64–66.
Jung B, Zoric L, Chanques G, Konate A, Nocca D, Jaber S. Acute abdomen and severe lactic acidosis can lead to a surprising diagnosis. Intensive Care Med. 2010;36:169–70.
Karkouche R, Ingen-Housz-Oro S, Le Gouvello S, Charlotte F, Thomas M, Zehou O, et al. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma with KIR3DL2 and NKp46 expression in a human immunodeficiency virus carrier. J Cutan Pathol. 2015;42:199–205.
Kato T, Tanabe J, Kanemoto M, Kobayashi C, Morita S, Karahashi T. A case of extranodal NK/T-cell lymphoma, nasal type mimicking typical manifestations of adult-onset Still’s disease (AOSD) with hemophagocytic syndrome: diagnostic consideration between malignant lymphoma without lymphadenopathy and AOSD. Mod Rheumatol. 2009;19:675–80.
Kim JE, Kim CW, Park SH, Chi JG. Hemophagocytic syndrome associated with occult B-cell lymphoma: an autopsy case. J Korean Med Sci. 1998;13:77–80.
Kim MS, Cho YU, Jang S, Seo EJ, Lee JH, Park CJ. A case of primary bone marrow diffuse large B-cell lymphoma presenting with fibrillar projections and hemophagocytic lymphohistiocytosis. Ann Lab Med. 2017;37:544–6.
Kobayashi T, Ohno H. Intravascular large B-cell lymphoma associated with t(14;19)(q32;q13) translocation. Intern Med. 2011;50:2007–10.
Koduri PR, Carandang G, DeMarais P, Patel AR. Hyperferritinemia in reactive hemophagocytic syndrome report of four adult cases. Am J Hematol. 1995;49:247–9.
Kojima H, Takei N, Mukai Y, Hasegawa Y, Suzukawa K, Nagata M, et al. Hemophagocytic syndrome as the primary clinical symptom of Hodgkin’s disease. Ann Hematol. 2003;82:53–56.
Krishna R, Byrne E, Burbridge J, Salooja N, Naresh KN. The Hammersmith hospital hematopathology case of the month: hemophagocytic lymphohistiocytosis secondary to Epstein-Barr virus-associated T-cell lymphoma. Leuk Lymphoma. 2011;52:1127–32.
Kueck BD, Hanson CA, Weissman DE, Bayliss K. Primary lymph node presentation of angiocentric lymphoma associated with features of a hemophagocytic syndrome. Am J Hematol. 1989;30:104–11.
Kuo CY, Yeh ST, Huang CT, Lin SF. Diffuse large B-cell lymphoma presenting with type B lactic acidosis and hemophagocytic syndrome. Kaohsiung J Med Sci. 2014;30:428–9.
Kwon SY, Lee JJ, Chung IJ, Kim HJ, Park MR, Kim HS, et al. Hepatosplenic B-cell lymphoma associated with hemophagocytic syndrome: a case report. J Korean Med Sci. 1999;14:671–4.
Lee SY, Wu CW, Chang WH, Ku HC. Fever and jaundice caused by hemophagocytic syndrome. J Formos Med Assoc. 2019;118:649–50.
Ma L, Katz Y, Sharan KP, Schwarting R, Kim AS. Epstein-Barr virus positive anaplastic large cell lymphoma: myth or reality? Int J Clin Exp Pathol. 2010;4:100–10.
Machaczka M, Nahi H, Karbach H, Klimkowska M, Hagglund H. Successful treatment of recurrent malignancy-associated hemophagocytic lymphohistiocytosis with a modified HLH-94 immunochemotherapy and allogeneic stem cell transplantation. Med Oncol. 2012;29:1231–6.
Machaczka M, Vaktnas J. Haemophagocytic syndrome associated with Hodgkin lymphoma and Pneumocystis jiroveci pneumonitis. Br J Haematol. 2007;138:672.
Maejima H, Tanei R, Morioka T, Miyakoshi S. Haemophagocytosis-related intravascular large B-cell lymphoma associated with skin eruption. Acta Derm Venereol. 2011;91:339–40.
Matsumura Y, Kuroda J, Shimura Y, Kiyota M, Yamamoto-Sugitani M, Kobayashi T, et al. Cyclosporine A and reduced-intensity conditioning allogeneic stem cell transplantation for relapsed angioimmunoblastic T cell lymphoma with hemophagocytic syndrome. Intern Med. 2012;51:2785–7.
Mayson E, Saverimuttu J, Warburton P. Two-faced haemophagocytic lymphohistiocytosis: comparative review of two cases of adult haemophagocytic lymphohistiocytosis. Intern Med J. 2014;44:198–201.
Miura T, Kawakami Y, Sato M, Ohtsuka M, Yamamoto T. Hemophagocytic syndrome occurred in a patient with subcutaneous panniculitis-like T-cell lymphoma without overt skin lesion: successful treatment with steroid pulse therapy. J Dermatol. 2011;38:1113–5.
Miyahara M, Sano M, Shibata K, Matsuzaki M, Ibaraki K, Shimamoto Y, et al. B-cell lymphoma-associated hemophagocytic syndrome: clinicopathological characteristics. Ann Hematol. 2000;79:378–88.
Mizutani S, Kuroda J, Shimura Y, Kobayashi T, Tsutsumi Y, Yamashita M, et al. Cyclosporine A for chemotherapy-resistant subcutaneous panniculitis-like T cell lymphoma with hemophagocytic syndrome. Acta Haematol. 2011;126:8–12.
Morita Y, Kenzaka T, Yoshimoto H, Ohno N. Hodgkin’s lymphoma preceded by haemophagocytic lymphohistiocytosis. BMJ Case Rep. 2013;2013:bcr2013010129.
Motegi S, Nishizaki Y, Muramatsu C, Nakamura H, Kobayashi F, Shiozawa H, et al. Hemophagocytic syndrome in ileum-origin B-cell lymphoma. J Gastroenterol. 2003;38:995–9.
Murase T, Nakamura S, Tashiro K, Suchi T, Hiraga J, Hayasaki N, et al. Malignant histiocytosis-like B-cell lymphoma, a distinct pathologic variant of intravascular lymphomatosis: a report of five cases and review of the literature. Br J Haematol. 1997;99:656–64.
Nakayama S, Morita Y, Espinoza JL, Rai S, Taniguchi Y, Taniguchi T, et al. Multiple cytokine-producing aggressive EBV-positive diffuse large B cell lymphoma, not otherwise specified with hemophagocytic syndrome. Ann Hematol. 2020;99:381–3.
Narimatsu H, Morishita Y, Saito S, Shimada K, Ozeki K, Kohno A, et al. Usefulness of bone marrow aspiration for definite diagnosis of Asian variant of intravascular lymphoma: four autopsied cases. Leuk Lymphoma. 2004;45:1611–6.
Neistadt B, Carrubba A, Zaretsky MV. Natural killer/T-cell lymphoma and secondary haemophagocytic lymphohistiocytosis in pregnancy. BMJ Case Rep. 2018;2018:bcr2018224832.
Ng CS, Chan JK, Cheng PN, Szeto SC. Nasal T-cell lymphoma associated with hemophagocytic syndrome. Cancer. 1986;58:67–71.
Noguchi M, Kawano Y, Sato N, Oshimi K. T-cell lymphoma of CD3+CD4+CD56+granular lymphocytes with hemophagocytic syndrome. Leuk Lymphoma. 1997;26:349–58.
Nosari A, Oreste PL, Biondi A, Costantini MC, Santoleri L, Intropido L, et al. Hepato-splenic gammadelta T-cell lymphoma: a rare entity mimicking the hemophagocytic syndrome. Am J Hematol. 1999;60:61–65.
Notaro E, Shustov A, Chen X, Shinohara MM. Kikuchi-Fujimoto disease associated with subcutaneous panniculitis-like T-Cell lymphoma. Am J Dermatopathol. 2016;38:e77–80.
Obama K, Tara M, Niina K. L-asparaginase-Based induction therapy for advanced extranodal NK/T-cell lymphoma. Int J Hematol. 2003;78:248–50.
Ohno H, Takimoto K. Gastric mucosa-associated lymphoid tissue lymphoma complicated with hemophagocytic syndrome in an elderly woman. Ann Hematol. 2010;89:1175–6.
Pasvolsky O, Zoref-Lorenz A, Abadi U, Geiger KR, Hayman L, Vaxman I, et al. Hemophagocytic lymphohistiocytosis as a harbinger of aggressive lymphoma: a case series. Int J Hematol. 2019;109:553–62.
Peeters P, Sennesael J, De Raeve H, De Waele M, Verbeelen D. Hemophagocytic syndrome and T-cell lymphoma after kidney transplantation: a case report. Transpl Int. 1997;10:471–4.
Pongpairoj K, Rerknimitr P, Wititsuwannakul J, Asawanonda P. Eruptive telangiectasia in a patient with fever and haemophagocytic syndrome. Clin Exp Dermatol. 2016;41:696–8.
Real E, Gomez A, Alcaraz MJ, Saez AI, Pastor E, Grau E. Fulminant hemophagocytic syndrome as presenting feature of T-cell lymphoma and Epstein-Barr virus infection. Haematologica. 2000;85:439–40.
Rivera XI, McGhan LJ, Schatz JH, Puvvada SD. Double hit lymphoma presenting as haemophagocytic lymphohistiocytosis. BMJ Case Rep. 2017;2017:bcr2017220401.
Romero Fernandez E, Pardo JR, Doyle A, Albendea MC, de la Rua AR. Hemophagocytic syndrome associated NK/T nasal type lymphoma presenting as hypereosinophilic syndrome: a case report and literature review. Leuk Res. 2011;35:e97–9.
Sano T, Sakai H, Takimoto K, Ohno H. Rituximab alone was effective for the treatment of a diffuse large B-cell lymphoma associated with hemophagocytic syndrome. Int J Clin Oncol. 2007;12:59–62.
Sasaki K, Yamato M, Yasuda K, Rakugi H, Isaka Y. Rhabdomyolysis caused by peripheral T-cell lymphoma in skeletal muscle. Am J Emerg Med. 2013;31:1537 e1533–1535.
Sato T, Kogawa K, Iyama S, Kobayashi D, Sato Y, Kuribayashi K, et al. Successful treatment of advanced peripheral T-cell lymphoma with an angiocentric growth pattern complicated with hemophagocytic syndrome by high-dose chemotherapy and autologous peripheral blood stem cell transplantation. Ann Hematol. 2002;81:739–43.
Shimizu Y, Tanae K, Takahashi N, Kohri M, Arai E, Bessho M, et al. Primary cutaneous anaplastic large-cell lymphoma presenting with hemophagocytic syndrome: a case report and review of the literature. Leuk Res. 2010;34:263–6.
Soiza RL, Ghosh S, McAlpine JK, Vickers MA. Non-fatal haemophagocytic syndrome in an elderly patient. Age Ageing. 2005;34:522–4.
Suvorava N, Richmond S, Patel N, Bell B, Mesa H. Between a rock and a hard place. Am J Hematol. 2016;91:351–3.
Tabata R, Tabata C, Kimura T, Nagai T, Yasumizu R. Prominent granulomas in bone marrow in disseminated lymphoma with hemophagocytic syndrome. Ann Hematol. 2011;90:1365–7.
Takahashi E, Kajimoto K, Fukatsu T, Yoshida M, Eimoto T, Nakamura S. Intravascular large T-cell lymphoma: a case report of CD30-positive and ALK-negative anaplastic type with cytotoxic molecule expression. Virchows Arch. 2005;447:1000–6.
Takahashi T, Kanda Y, Mori M, Saito T, Chiba S, Mitani K, et al. B cell lymphoma-associated hemophagocytic syndrome after PBSCT. Bone Marrow Transpl. 1998;21:623–5.
Takami A, Nakao S, Ueda M, Miura Y, Matsuda T, Kawamura Y. Successful treatment of B-cell lymphoma associated with hemophagocytic syndrome using autologous peripheral blood CD34 positive cell transplantation followed by induction of autologous graft-versus-host disease. Ann Hematol. 2000;79:389–91.
Tan B, Abdelmalek C, O’Donnell JE, Toltaku T, Chaudhry R, Wang JC, et al. A case report of primary nasal natural killer (NK)/T-cell lymphoma in an African American patient presenting with hemophagocytic syndrome. Am J Case Rep. 2017;18:160–5.
Tatara R, Sato M, Fujiwara S, Oh I, Muroi K, Ozawa K, et al. Hemoperfusion for Hodgkin lymphoma-associated hemophagocytic lymphohistiocytosis. Intern Med. 2014;53:2365–8.
Terrier B, Aouba A, Vasiliu V, Charlier C, Delarue R, Buzyn A, et al. Intravascular lymphoma associated with haemophagocytic syndrome: a very rare entity in Western countries. Eur J Haematol. 2005;75:341–5.
Tsai AS, Ko CW, Yeh HZ, Chang CS, Wang RC. Peripheral T-cell lymphoma of the colon associated with hemophagocytic lymphohistiocytosis. J Chin Med Assoc. 2013;76:169–72.
Tsukamoto Y, Katsunobu Y, Omura Y, Maeda I, Hirai M, Teshima H, et al. Subcutaneous panniculitis-like T-cell lymphoma: successful initial treatment with prednisolone and cyclosporin A. Intern Med. 2006;45:21–4.
Tzeng HE, Teng CL, Yang Y, Young JH, Chou G. Occult subcutaneous panniculitis-like T-cell lymphoma with initial presentations of cellulitis-like skin lesion and fulminant hemophagocytosis. J Formos Med Assoc. 2007;106:S55–9.
Uehara T, Yokota A, Onoda M, Yamamoto K, Terano T. Successful autologous peripheral blood stem cell transplantation for a patient with primary adrenal lymphoma with hemophagocytic syndrome. Clin Lymphoma Myeloma. 2008;8:184–7.
Uni M, Yoshimi A, Maki H, Maeda D, Nakazaki K, Nakamura F, et al. Successful treatment with recombinant thrombomodulin for B-cell lymphoma-associated hemophagocytic syndrome complicated by disseminated intravascular coagulation. Int J Clin Exp Pathol. 2013;6:1190–4.
Varghese D, Haseer Koya H, Cherian SV, Mead K, Sharma A, Sharma N, et al. Hemophagocytic lymphohistiocytosis: an uncommon presentation of enteropathy-associated T-cell lymphoma. J Clin Oncol. 2013;31:e226–230.
Watabe R, Shibata K, Hirase N, Kodera T, Muta K, Nishimura J, et al. Angiotropic B-cell lymphoma with hemophagocytic syndrome associated with syndrome of inappropriate secretion of antidiuretic hormone. Ann Hematol. 2000;79:581–4.
Xu Z, Burns BF. Hemophagocytosis due to bone marrow ALCL, ALK. Blood. 2014;124:478.
Yamamoto K, Nagata K, Hamaguchi H. Translocation (11;14)(q13;q32) in CD5-positive B-cell lymphoma associated with haemophagocytic syndrome. Br J Haematol. 1999;106:1069–70.
Yamamoto K, Nakamura Y, Arai H, Aoyagi M, Saito K, Furusawa S, et al. Translocation (14;19)(q32;q13) detected by spectral karyotyping and lack of BCL3 rearrangement in CD5-positive B-cell lymphoma associated with hemophagocytic syndrome. Cancer Genet Cytogenet. 2001;130:38–41.
Yu JT, Hwang WL, Wang RC, Teng CL. Reduced intensity conditioning allogeneic hematopoietic stem cell transplant could be beneficial to angioimmunoblastic T-cell lymphoma patients with hemophagocytic lymphohistiocytosis. Ann Hematol. 2012;91:805–7.
Yun S, Taverna JA, Puvvada SD, Anwer F. NK/T-cell non-Hodgkin’s lymphoma with secondary haemophagocytic lymphohistiocytosis treated with matched unrelated donor allogeneic stem cell transplant. BMJ Case Rep. 2014;2014:bcr2014205602.
Zabernigg A, Fend F, Thaler J, Gattringer C. An unusual case of a splenic gamma/delta T-cell lymphoma with angiocentric tendency and haemophagocytic syndrome. Leuk Lymphoma. 1996;23:631–4.
Zhan Y, Teruya-Feldstein J. Immune dysregulation: EBV(+) DLBCL and HLH in a patient with T-LGL. Blood. 2019;133:1695.
Apodaca E, Rodriguez-Rodriguez S, Tuna-Aguilar EJ, Demichelis-Gomez R. Prognostic factors and outcomes in adults with secondary hemophagocytic lymphohistiocytosis: a single-center experience. Clin Lymphoma Myeloma Leuk. 2018;18:e373–80.
Bigenwald C, Fardet L, Coppo P, Meignin V, Lazure T, Fabiani B, et al. A comprehensive analysis of Lymphoma-associated haemophagocytic syndrome in a large French multicentre cohort detects some clues to improve prognosis. Br J Haematol. 2018;183:68–75.
Birndt S, Schenk T, Heinevetter B, Brunkhorst FM, Maschmeyer G, Rothmann F, et al. Hemophagocytic lymphohistiocytosis in adults: collaborative analysis of 137 cases of a nationwide German registry. J Cancer Res Clin Oncol. 2020;146:1065–77.
Jia J, Song Y, Lin N, Liu W, Ping L, Zheng W, et al. Clinical features and survival of extranodal natural killer/T cell lymphoma with and without hemophagocytic syndrome. Ann Hematol. 2016;95:2023–31.
Jiang T, Ding X, Lu W. The prognostic significance of beta2 microglobulin in patients with hemophagocytic lymphohistiocytosis. Dis Markers. 2016;2016:1523959.
Lecronier M, Prendki V, Gerin M, Schneerson M, Renvoise A, Larroche C, et al. Q fever and Mediterranean spotted fever associated with hemophagocytic syndrome: case study and literature review. Int J Infect Dis. 2013;17:e629–633.
Lin TA, Yang CF, Liu YC, Liu JH, Chiou TJ, Hsiao LT, et al. Hematopoietic stem cell transplantation for subcutaneous panniculitis-like T-cell lymphoma: single center experience in an Asian population. Int J Hematol. 2019;109:187–96.
Ohno T, Ueda Y, Nagai K, Takahashi T, Konaka Y, Takamatsu T, et al. The serum cytokine profiles of lymphoma-associated hemophagocytic syndrome: a comparative analysis of B-cell and T-cell/natural killer cell lymphomas. Int J Hematol. 2003;77:286–94.
Okamoto M, Yamaguchi H, Isobe Y, Yokose N, Mizuki T, Tajika K, et al. Analysis of triglyceride value in the diagnosis and treatment response of secondary hemophagocytic syndrome. Intern Med. 2009;48:775–81.
Rutnin S, Porntharukcharoen S. Boonsakan P. Clinicopathologic, immunophenotypic, and molecular analysis of subcutaneous panniculitis-like T-cell lymphoma: a retrospective study in a tertiary care center. J Cutan Pathol. 2019;46:44–51.
Sano H, Kobayashi R, Tanaka J, Hashino S, Ota S, Torimoto Y, et al. Risk factor analysis of non-Hodgkin lymphoma-associated haemophagocytic syndromes: a multicentre study. Br J Haematol. 2014;165:786–92.
Shimazaki C, Inaba T, Okano A, Hatsuse M, Takahashi R, Hirai H, et al. Clinical characteristics of B-cell lymphoma-associated hemophagocytic syndrome (B-LAHS): comparison of CD5+ with CD5- B-LAHS. Intern Med. 2001;40:878–82.
Shimazaki C, Inaba T, Shimura K, Okamoto A, Takahashi R, Hirai H, et al. B-cell lymphoma associated with haemophagocytic syndrome: a clinical, immunological and cytogenetic study. Br J Haematol. 1999;104:672–9.
Shimizu I, Ichikawa N, Yotsumoto M, Sumi M, Ueno M, Kobayashi H. Asian variant of intravascular lymphoma: aspects of diagnosis and the role of rituximab. Intern Med. 2007;46:1381–6.
Takahashi N, Miura I, Chubachi A, Miura AB, Nakamura S. A clinicopathological study of 20 patients with T/natural killer (NK)-cell lymphoma-associated hemophagocytic syndrome with special reference to nasal and nasal-type NK/T-cell lymphoma. Int J Hematol. 2001;74:303–8.
Tsuji T, Hirano T, Yamasaki H, Tsuji M, Tsuda H. A high sIL-2R/ferritin ratio is a useful marker for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann Hematol. 2014;93:821–6.
Wang J, Ding W, Gao L, Yao W, Chen M, Zhao S, et al. High frequency of bone marrow involvement in intravascular large B-cell lymphoma. Int J Surg Pathol. 2017;25:118–26.
Wang Y, Huang W, Hu L, Cen X, Li L, Wang J, et al. Multicenter study of combination DEP regimen as a salvage therapy for adult refractory hemophagocytic lymphohistiocytosis. Blood. 2015;126:2186–92.
Yu JT, Wang CY, Yang Y, Wang RC, Chang KH, Hwang WL, et al. Lymphoma-associated hemophagocytic lymphohistiocytosis: experience in adults from a single institution. Ann Hematol. 2013;92:1529–36.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90.
Fajgenbaum DC, June CH. Cytokine Storm. N. Engl J Med. 2020;383:2255–73.
Setiadi A, Zoref-Lorenz A, Lee CY, Jordan MB, Chen LYC. Malignancy-associated haemophagocytic lymphohistiocytosis. Lancet Haematol. 2022;9:e217–27.
Hines MR, von Bahr Greenwood T, Beutel G, Beutel K, Hays JA, Horne A, et al. Consensus-based guidelines for the recognition, diagnosis, and management of hemophagocytic lymphohistiocytosis in critically Ill children and adults. Crit Care Med. 2022;50:860–72.
Lehmberg K, Nichols KE, Henter JI, Girschikofsky M, Greenwood T, Jordan M, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica. 2015;100:997–1004.
Kuron D, Voran JC, von Samson-Himmelstjerna FA, Baldus C, Kunzendorf U, Schulte K, et al. Epidemiology of haemophagocytic lymphohistiocytosis at the population level in Germany. Br J Haematol. 2023;201:285–9.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Lofstedt AH, Jadersten M, Meeths M, Henter JI. Malignancy-associated hemophagocytic lymphohistiocytosis in Sweden: incidence, clinical characteristics and survival. Blood 2023 Aug 18. https://doi.org/10.1182/blood.2023020715.
Au WY, Ma SY, Chim CS, Choy C, Loong F, Lie AK, et al. Clinicopathologic features and treatment outcome of mature T-cell and natural killer-cell lymphomas diagnosed according to the World Health Organization classification scheme: a single center experience of 10 years. Ann Oncol. 2005;16:206–14.
Chiba S, Sakata-Yanagimoto M. Advances in understanding of angioimmunoblastic T-cell lymphoma. Leukemia. 2020;34:2592–606.
Marsh RA. Epstein-Barr virus and hemophagocytic lymphohistiocytosis. Front Immunol. 2017;8:1902.
Gayden T, Sepulveda FE, Khuong-Quang DA, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet. 2018;50:1650–7.
Polprasert C, Takeuchi Y, Kakiuchi N, Yoshida K, Assanasen T, Sitthi W, et al. Frequent germline mutations of HAVCR2 in sporadic subcutaneous panniculitis-like T-cell lymphoma. Blood Adv. 2019;3:588–95.
Sonigo G, Battistella M, Beylot-Barry M, Ingen-Housz-Oro S, Franck N, Barete S, et al. HAVCR2 mutations are associated with severe hemophagocytic syndrome in subcutaneous panniculitis-like T-cell lymphoma. Blood. 2020;135:1058–61.
Moonla C, Polprasert C, Komvilaisak P, Rattanathammethee T, Kongkiatkamon S, Wudhikarn K, et al. Germline HAVCR2 mutations and their relation to the clinical spectrum of subcutaneous panniculitis-like T-cell lymphoma and hemophagocytic lymphohistiocytosis: results from a multicenter study and meta-analysis. Haematologica. 2023;108:2743–52.
Zhang Q, Zhou CJ, Li DH, Cui L, Li WJ, Ma HH, et al. Efficacy of ruxolitinib for HAVCR2 mutation-associated hemophagocytic lymphohistiocytosis and panniculitis manifestations in children. Br J Haematol. 2023;202:135–46.
Levy R, Fusaro M, Guerin F, Chetouani A, Moshous D, Fischer A, et al. Efficacy of ruxolitinib in subcutaneous panniculitis-like T-cell lymphoma and hemophagocytic lymphohistiocytosis. Blood Adv. 2020;4:1383–7.
Ferreri AJ, Campo E, Seymour JF, Willemze R, Ilariucci F, Ambrosetti A, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127:173–83.
Lunning MA, Vose JM. Angioimmunoblastic T-cell lymphoma: the many-faced lymphoma. Blood. 2017;129:1095–102.
Clarke CA, Morton LM, Lynch C, Pfeiffer RM, Hall EC, Gibson TM, et al. Risk of lymphoma subtypes after solid organ transplantation in the United States. Br J Cancer. 2013;109:280–8.
Grogg KL, Miller RF, Dogan A. HIV infection and lymphoma. J Clin Pathol. 2007;60:1365–72.
Han AR, Lee HR, Park BB, Hwang IG, Park S, Lee SC, et al. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol. 2007;86:493–8.
Li N, Jiang M, Wu WC, Zhou HJ, Zou LQ. Lymphoma-associated hemophagocytic syndrome: a retrospective study from a single center. Hematology. 2022;27:909–16.
Tamamyan GN, Kantarjian HM, Ning J, Jain P, Sasaki K, McClain KL, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: Relation to hemophagocytosis, characteristics, and outcomes. Cancer. 2016;122:2857–66.
Wang J, Wang D, Zhang Q, Duan L, Tian T, Zhang X, et al. The significance of pre-therapeutic F-18-FDG PET-CT in lymphoma-associated hemophagocytic lymphohistiocytosis when pathological evidence is unavailable. J Cancer Res Clin Oncol. 2016;142:859–71.
Tudesq JJ, Valade S, Galicier L, Zafrani L, Boutboul D, de Bazelaire C, et al. Diagnostic strategy for trigger identification in severe reactive hemophagocytic lymphohistiocytosis: a diagnostic accuracy study. Hematol Oncol. 2021;39:114–22.
Machowicz R, Kröger N, Krieger T, Zborowska H, Rejowski S, Gorka M, et al. Hyperferritinemia in adult HLH: the best what we have, so far [published e-Letter response 23 April 2015]. Blood. 2015;125:1548–1552.
Hayden A, Lin M, Park S, Pudek M, Schneider M, Jordan MB, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood Adv. 2017;1:2529–34.
Liapis K, Apostolidis J, Delimpasis S. EBV-associated hemophagocytic syndrome. Am J Hematol. 2011;86:422.
Menard F, Besson C, Rince P, Lambotte O, Lazure T, Canioni D, et al. Hodgkin lymphoma-associated hemophagocytic syndrome: a disorder strongly correlated with Epstein-Barr virus. Clin Infect Dis. 2008;47:531–4.
Komisarof J, McGann K, Huston A, Katerji H, Morgan MA. An atypical presentation of hemophagocytic lymphohistiocytosis (HLH) secondary to occult Hodgkin lymphoma. Case Rep. Hematol. 2021;2021:6672257.
Lin M, Park S, Hayden A, Giustini D, Trinkaus M, Pudek M, et al. Clinical utility of soluble interleukin-2 receptor in hemophagocytic syndromes: a systematic scoping review. Ann Hematol. 2017;96:1241–51.
Damoiseaux J. The IL-2 - IL-2 receptor pathway in health and disease: The role of the soluble IL-2 receptor. Clin Immunol. 2020;218:108515.
Nakase K, Tsuji K, Tamaki S, Tanigawa M, Ikeda T, Miyanishi E, et al. Elevated levels of soluble interleukin-2 receptor in serum of patients with hematological or non-hematological malignancies. Cancer Detect Prev. 2005;29:256–9.
Tabata C, Tabata R. Possible prediction of underlying lymphoma by high sIL-2R/ferritin ratio in hemophagocytic syndrome. Ann Hematol. 2012;91:63–71.
Wimmer T, Mattes R, Stemmler HJ, Hauck F, Schulze-Koops H, Stecher SS, et al. sCD25 as an independent adverse prognostic factor in adult patients with HLH: results of a multicenter retrospective study. Blood Adv. 2023;7:832–44.
La Rosée P. Treatment of hemophagocytic lymphohistiocytosis in adults. Hematol Am Soc Hematol Educ Program. 2015;2015:190–6.
Song Y, Wang J, Wang Y, Wu L, Wang Z. Requirement for containing etoposide in the initial treatment of lymphoma associated hemophagocytic lymphohistiocytosis. Cancer Biol Ther. 2021;22:598–606.
Ahmed A, Merrill SA, Alsawah F, Bockenstedt P, Campagnaro E, Devata S, et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol. 2019;6:e630–e637.
Liang JH, Wang L, Zhu HY, Qian J, Liao H, Wu JZ, et al. Dose-adjusted EPOCH regimen as first-line treatment for non-Hodgkin lymphoma-associated hemophagocytic lymphohistiocytosis: a single-arm, open-label, phase II trial. Haematologica. 2020;105:e29–e32.
Gang AO, Strom C, Pedersen M, d’Amore F, Pedersen LM, Bukh A, et al. R-CHOEP-14 improves overall survival in young high-risk patients with diffuse large B-cell lymphoma compared with R-CHOP-14. A population-based investigation from the Danish Lymphoma Group. Ann Oncol. 2012;23:147–53.
Frontzek F, Ziepert M, Nickelsen M, Altmann B, Glass B, Haenel M, et al. Rituximab plus high-dose chemotherapy (MegaCHOEP) or conventional chemotherapy (CHOEP-14) in young, high-risk patients with aggressive B-cell lymphoma: 10-year follow-up of a randomised, open-label, phase 3 trial. Lancet Haematol. 2021;8:e267–e277.
Wilhelm M, Smetak M, Reimer P, Geissinger E, Ruediger T, Metzner B, et al. First-line therapy of peripheral T-cell lymphoma: extension and long-term follow-up of a study investigating the role of autologous stem cell transplantation. Blood Cancer J. 2016;6:e452.
Song Y, Yin Q, Wang J, Wang Z. Autologous hematopoietic stem cell transplantation for patients with lymphoma-associated hemophagocytic lymphohistiocytosis. Cell Transpl. 2021;30:9636897211057077.
Acknowledgements
This analysis was presented in part at the 2021 and 2022 annual meetings of the German, Austrian, and Swiss Societies of Hematology and Medical Oncology.
Funding
SB is supported by a clinician scientist grant from the Jena Interdisciplinary Center of Clinical Research (IZKF). Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
JK performed the literature search, extracted and collected the data, which were reviewed by TS, PLR, and SB. JK and SB assembled the data and performed data analysis. JK and SB wrote the initial draft of the manuscript. TS, TE, US, PLR, and AH reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Knauft, J., Schenk, T., Ernst, T. et al. Lymphoma-associated hemophagocytic lymphohistiocytosis (LA-HLH): a scoping review unveils clinical and diagnostic patterns of a lymphoma subgroup with poor prognosis. Leukemia 38, 235–249 (2024). https://doi.org/10.1038/s41375-024-02135-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-024-02135-8
