
Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is a malignant hematologic disease caused by gene mutations in T-cell progenitors. As an important epigenetic regulator, PHF6 mutations frequently coexist with JAK3 mutations in T-ALL patients. However, the role(s) of PHF6 mutations in JAK3-driven leukemia remain unclear. Here, the cooperation between JAK3 activation and PHF6 inactivation is examined in leukemia patients and in mice models. We found that the average survival time is shorter in patients with JAK/STAT and PHF6 comutation than that in other patients, suggesting a potential role of PHF6 in leukemia progression. We subsequently found that Phf6 deficiency promotes JAK3M511I-induced T-ALL progression in mice by inhibiting the Bai1-Mdm2-P53 signaling pathway, which is independent of the JAK3/STAT5 signaling pathway. Furthermore, combination therapy with a JAK3 inhibitor (tofacitinib) and a MDM2 inhibitor (idasanutlin) reduces the Phf6 KO and JAK3M511I leukemia burden in vivo. Taken together, our study suggests that combined treatment with JAK3 and MDM2 inhibitors may potentially increase the drug benefit for T-ALL patients with PHF6 and JAK3 comutation.
Similar content being viewed by others


Introduction
T-cell acute lymphoblastic leukemia (T-ALL) is one of the most common hematologic malignancies, resulted from gene mutations and genomic rearrangements in T-cell progenitors [1, 2]. The 5-year survival rate of adult T-ALL patients is less than 50%, and the mortality rate of relapsed adult T-ALL can be as high as 90% [3]. Recent studies have shown that T-ALL results from multistep transformation processes that involve the accumulation of genetic defects, including activating mutations of NOTCH1 or JAK-STAT, super-enhancer generating mutations of TAL1, deep deletion of cell cycle-related genes (such as CDKN2, RB1, and CDKN1B), and inactivating mutations of WT1, LEF1, GATA3, and PHF6 [4,5,6]. The close interrelationship between key regulators of early T-cell development and T-ALL oncogenic signals is best illustrated by the prominent roles of the JAK-STAT signal pathway in T-ALL [5, 7, 8]. More than 25% of T-ALL patients carry JAK-STAT mutations [9,10,11], of which JAK3 mutation is the most frequent in T-ALL cases [8, 9, 12, 13]. The continuous activation of JAK-STAT signaling has been shown to play an essential role in T-ALL initiation and progression [7, 8, 12, 13].
JAK family kinases are nonreceptor tyrosine kinases that function as signal transducers to activate STAT protein to support the differentiation, proliferation and survival of early T-cell progenitors [10]. Activated STAT proteins translocate to nucleus and act as transcription factors to regulate gene expression and/or induce novel epigenetic changes [11]. Aberrant JAK signaling has been linked with T-ALL development. JAK1 mutations can be found in 10–15% of T-ALL and 1–2% of acute myeloid leukemia (AML) patients [14, 15]. JAK2V617F mutation is mainly associated with myeloproliferative neoplasms [16]. JAK3 mutations can be identified in 16.1% of T-ALL cases. JAK3 plays a key role in regulating T and B cell development [9, 13, 17], while the rate of the most common JAK3 mutation (M511I) is 34.7% in all JAK3 mutations [8, 9, 13]. Transplantation of mice with bone marrow (BM) progenitor cells expressing the active JAK3M511I mutant allele induces a lymphoproliferative disorder followed by a T-ALL-like disease [12]. Moreover, the JAK3M511I mutation induces phosphorylation and activation of STAT5, which subsequently activates the oncogenes to drive leukemia cell proliferation [8, 12].
It has been reported that JAK3 mutations could act as “driver” mutations in T-ALL. Interestingly, JAK3 mutations are frequently accompanied by a high number of genetic changes in T-ALL, such as changes in epigenetic regulatory genes (ASXL1, DNMT3A, EED, EZH2, PHF6, and SUZ12) [5, 13, 17, 18]. PHF6 is one of the most common mutated epigenetic regulatory gene coexisting with JAK3 in T-ALL patients [8, 17, 19]. Studies have found that Phf6-deficient HSCs had higher proliferation and reconstitution capacity than the wild-type HSCs, although loss of Phf6 alone is not sufficient to induce aberrant hematopoietic transformation [20,21,22,23]. Nevertheless, Phf6 loss could significantly accelerate leukemia development driven by aberrant expression of TLX3 or lower the threshold of NOTCH1-induced T-ALL development. Phf6 deficiency could also activate leukemia stem cell transcriptional programs and enhance T-ALL leukemia-initiating cell (LIC) activity [20,21,22]. While these studies provide mechanistic insight into the importance of PHF6 in regulating early T-cell development, the mechanism of how PHF6 and JAK3 co-occurring events drive leukemogenesis still needs to be functionally dissected and elucidated.
Here we examined the role of cooperatively PHF6 mutations in the context of JAK/STAT mutant-induced leukemia in patients and further functionally dissect their biological events using JAK3M511I T-ALL mouse model. We found that PHF6 mutations frequently coexist with JAK3 mutations in T-ALL patients, and Phf6 comutation with JAK3 can drive aggressive leukemia in mice.
Materials and methods
Generation of leukemia mice models
Phf6 conditional deletion mice were generated using the homologous recombination technique and mated with Vav1-Cre or Mx1-Cre mice to delete Phf6 in hematopoietic cells. The mice, including donors and recipients used in all our experiments were male mice of 8 weeks. The MSCV-JAK3M511I-IRES-GFP retroviral vector was kindly provided by Dr. Jan Cools [8, 12]. Lineage-negative (Lin−) cells were transfected with JAK3M511I-GFP virus and transplanted into male mice to establish Vav1-Cre;Phf6fl/y + JAK3M511I and Mx1-Cre;Phf6fl/y + JAK3M511I mice models. Detailed mouse lines used in this study have been listed in supplementary Table 1 (Table S1). This research was approved by the Institute Ethics Committee and the Institutional Animal Care and Use Committee (IACUC), Institute of Hematology and Blood Diseases Hospital, CAMS/PUMC, KT2020052-EC-1.
Extreme limiting dilution analysis of LICs
The recipient mice were sublethally irradiated and received 500,000, 50,000, 5000, 500, or 100 GFP+ BM cells. The mice were monitored for 4 months. The extreme limiting dilution LIC data were analyzed with ELDA software online [24].
Inhibitor experiments
For the in vivo experiment, mice were treated through oral gavage with tofacitinib (40 mg/kg per day), idasanutlin (30 mg/kg per day) (MedChemExpress, New Jersey, USA), or vehicle (0.2% dimethylsulfoxide [DMSO]). The mice received medication daily until death. For the in vitro experiment, MOLT-4 cells were treated with tofacitinib (2.0 μM), idasanutlin (0.5 μM) or 0.2% DMSO in culture medium for 48 h and then proliferation and apoptosis were examined.
RNA-Seq and ChIP-Seq assay
For RNA-Seq analysis of Phf6 WT/KO + JAK3M511I cells, 1 µg RNA per sample was used for RNA sample preparations. Transcriptome sequencing was performed on Illumina NovaSeq 6000 platform (Illumina, CA, USA) to a total target depth of 10 million 150 bp paired end reads. Differential expression analysis was performed by DESeq2 R package (1.16.1).
The ChIP assay was performed with the ChIP assay kit (Cell Signaling Technology, Boston, MA, USA) according to manufacturer’s recommendations. Chromatin from cross-linked HA-PHF6-overexpressed K562 or control cells was sheared using an ultrasonicator (Covaris, S220, ABI, New York, USA) to obtain DNA (100–400 bp). Immunoprecipitation was conducted with ChIP-grade HA-tag antibody (Abcam, ab9110, Cambridge, UK), PHF6 antibody (Sigma-Aldrich, HPA001023, Missouri, USA) or normal IgG antibody (Cell Signaling Technology, CST2729, Boston, MA, USA).
RNA-Seq and ChIP-Seq data are available at GEO under accession number GSE159444 and GSE159549.
Results
PHF6 mutation frequently co-exists with JAK3 mutation in acute leukemia patients
We analyzed the genetic data of 449 T-ALL cases from different clinical centers, among which JAK/STAT mutations occurred in 98 samples (21.83%) [13, 17, 25]. JAK/STAT and PHF6 comutation accounted for 7.80% (35/449) of all T-ALL cases. Interestingly, JAK3 and PHF6 comutation accounted for 4.90% of the 449 cases (Fig. 1A, B). PHF6 mutation occurred in 44.90% of patients with JAK3 mutation (P < 0.001, Fig. 1C, left). JAK3 mutation occurred in 25.00% of PHF6-mutated patients and was significantly associated with PHF6 mutation (P < 0.001, Fig. 1C, right).

A, B The percentage of JAK3 or PHF6 mutations in 449 T-ALL patients from three independent clinical centers. C Left panel, the frequency of PHF6 mutations (orange) in T-ALL patients with JAK3 mutations vs the frequency of PHF6 mutations in T-ALL patients with WT JAK3. Right panel, the frequency of JAK3 mutations (orange) in T-ALL patients with PHF6 mutations vs the frequency of JAK3 mutations in T-ALL patients with WT PHF6. Fisher’s exact test was applied to calculate the P values to analyze the significance of the positive association between JAK3 mutations and PHF6 mutations. D Frameshift (fs), nonsense (non), and missense (mis) of PHF6 or JAK3 found in 44 T/M MPAL cases from Alexander et al. [19]. Each type of mutation is indicated by a unique color. Gender are also shown for each patient. E Total case number, median age, gender ratio, mutation type and average survival time of 44 T/M MPAL patients from Alexander et al. [19].
We analyzed PHF6 and JAK/STAT mutations in 44 T/myeloid mixed phenotype acute leukemia cases (T/M MPAL) from 102 MPAL patients in Alexander et al. [19]. We found that PHF6 and JAK/STAT comutation accounted for 6.82% (3/44) in total cases (Fig. 1D, E and Table S2). We assigned the 44 T/myeloid MPAL patients into four groups including PHF6 and JAK/STAT comutation, single JAK/STAT or PHF6 mutation, and non-PHF6/JAK/STAT mutation (others), and found that the survival time of the PHF6 and JAK/STAT comutation group was significantly shorter than the single JAK/STAT mutation group (P < 0.0001) or the non-PHF6/JAK/STAT mutation group (P < 0.0001, Figs. 1E and S1A). The combined clinical data suggested that PHF6 mutations may play a synergetic role with JAK/STAT mutations in leukemia development.
PHF6 mutation acts as an early event to accelerate JAK3 M511I hematopoietic progenitor cell transformation
To evaluate the potential role of PHF6 mutation in leukemia-initiating events, we generated Phf6 knockout (Vav1-Cre;Phf6fl/y) and wild-type (Phf6fl/y) mice. We sorted Lin− cells from BM of Vav1-Cre;Phf6fl/y or Phf6fl/y male mice and transfected with JAK3M511I-GFP virus. Equal number of GFP+ cells were transplanted into male recipients to establish Vav1-Cre;Phf6fl/y + JAK3M511I (presented as VC Phf6 + JAK3M511I) and Phf6fl/y + JAK3M511I (as Phf6 WT + JAK3M511I) mouse lines (Figs. 2A and S1B) [8, 12]. While all mice succumbed to leukemia, the survival time of VC Phf6 + JAK3M511I mice was significantly shorter than Phf6 WT + JAK3M511I mice (Fig. 2B). The percentage of GFP+ leukemia cells was higher in the peripheral blood (PB) of VC Phf6 + JAK3M511I mice than Phf6 WT + JAK3M511I mice (Figs. 2C and S1C). Although GFP+ leukemia cells in the two groups were mainly CD3+ or CD8+, the percentages of myeloid and B cells were significantly increased in the PB of VC Phf6 + JAK3M511I mice when compared with Phf6 WT + JAK3M511I mice (Fig. S1D, E). The VC Phf6 + JAK3M511I mice showed more aggressive leukemia phenotypes than Phf6 WT + JAK3M511I mice, including higher white blood cells (WBCs), neutrophils (Neu), and lymphocytes (Lym) in PB (Figs. 2D, E and S1F). The weights of spleen, liver, and thymus were slightly increased in VC Phf6 + JAK3M511I mice when compared with Phf6 WT + JAK3M511I mice (Fig. S1G–J). The percentage of GFP+ leukemia cells and the degree of extramedullary infiltration in spleen, liver, lung, and brain, were increased in VC Phf6 + JAK3M511I mice than Phf6 WT + JAK3M511I mice, (Fig. 2F, G). These results demonstrated that Phf6 deficiency accelerated the initiation of JAK3M511I hematopoietic progenitor cell transformation.

A Scheme of constructing VC Phf6 + JAK3M511I and Phf6 WT + JAK3M511I T-ALL mouse models. B Kaplan–Meier survival curves of VC Phf6 + JAK3M511I T-ALL mice and Phf6 WT + JAK3M511I T-ALL mice (log-rank test P < 0.0001, n = 15 per group). VC Phf6 + JAK3M511I T-ALL mice (median survival time = 85 days), Phf6 WT + JAK3M511I T-ALL mice (median survival time = 143 days). C Percentage of GFP+ leukemia cells in PB at different time points. D The counts of WBCs, lymphocytes (Lym), and neutrophils (Neu) in PB by routine blood tests. E Wright–Giemsa staining of peripheral blood cells. F Percentage of GFP+ cells in spleen, liver, and thymus. G Hematoxylin and eosin (HE) staining of BM, spleen, liver, lung, and brain. A red triangle or white arrow indicates the leukemia infiltration area. H Percentage of GFP+ leukemia cells in BM at 21 weeks after transplantation. I, J Percentages of T cells, B cells, and myeloid cells in the GFP+ population in BM at 21 weeks after transplantation. K Wright–Giemsa staining of BM cells.
To further investigate the role of Phf6 deletion in the transformation of hematopoietic progenitors with JAK3 mutations, we quantified the biological characteristics of VC Phf6 + JAK3M511I cells in BM with various surface markers. The percentage of GFP+ cells was higher in BM of VC Phf6 + JAK3M511I mice (44.1%) than the Phf6 WT + JAK3M511I mice (7.8%) (Fig. 2H, I). The percentages of myeloid and B cells in the BM of VC Phf6 + JAK3M511I mice were 20.9% and 25.7% respectively, and were higher than the controls (Fig. 2I, J). Wright–Giemsa staining revealed the development of a complex hematolymphoid neoplasm characterized by the coexistence of different populations of atypical cells displaying both lymphoid and myeloid differentiation in VC Phf6 + JAK3M511I mice when compared to the controls (Fig. 2K). These results suggested that Phf6 deficiency induced the polyclonal expansion of JAK3M511I hematopoietic progenitors.
Phf6 deletion promotes JAK3 M511I-induced T-ALL progression
Since JAK3 mutations could act as “driver” mutations in leukemia patients, we investigated whether PHF6 mutations act as subsequent events to promote JAK3M511I-induced leukemia development by generating Mx1-Cre;Phf6fl/y (MC Phf6fl/y) and Mx1-Cre;Phf6+/y (MC) mice, and constructed MC Phf6fl/y + JAK3M511I and MC + JAK3M511I mice (Fig. 3A). MC Phf6fl/y or MC Lin− cells were transfected with retrovirus containing JAK3M511I/GFP ex vivo, and equal number of GFP+ cells were injected into lethally irradiated mice. pIpC was injected at 3 weeks post-transplantation to delete Phf6 when CD3+ cells in GFP+ cells were above 70% in JAK3M511I-induced T-cell leukemia (Fig. 3A, B). Phf6 deletion was confirmed by western blotting of BM cells from MC Phf6fl/y + JAK3M511I and MC + JAK3M511I mice treated with pIpC (presented as MC Phf6 KO + JAK3M511I or MC Phf6 WT + JAK3M511I) (Fig. 3C). We sorted GFP+ cells from BM of MC Phf6 WT + JAK3M511I and MC Phf6 KO + JAK3M511I mice, and performed Wright–Giemsa staining to discern the morphological characteristics of GFP+ cells. We found that most of the GFP+ cells in BM of the two mouse groups were abnormal lymphocytes with larger cell size, irregular nuclear contours, and prominent nucleoli (Fig. S1K). Interestingly, both MC Phf6 WT + JAK3M511I and MC Phf6 KO + JAK3M511I mice developed CD8+ T-ALL without myeloid expansion (Fig. 3D, E). Furthermore, the percentages of GFP+ cells in PB, BM, spleen, liver, and thymus were higher in MC Phf6 KO + JAK3M511I than in MC Phf6 WT + JAK3M511I mice (Fig. 3F, G). The WBC and lymphocyte counts were also increased, while the platelet count was decreased in the PB of MC Phf6 KO + JAK3M511I mice (Fig. 3H). The weights of spleens and livers of MC Phf6 KO + JAK3M511I were much higher than MC Phf6 WT + JAK3M511I mice (Fig. 3I). Importantly, the survival time of MC Phf6 KO + JAK3M511I mice was significantly shorter than MC Phf6 WT + JAK3M511I mice (Fig. 3J). These data indicated that Phf6 mutation acquired after JAK/STAT mutation could promote JAK3M511I-induced T-ALL progression.

A Schematic representation of Phf6 deletion after JAK3M511I-induced T-ALL in a mouse model. B Left panel, percentage of GFP+ leukemia cells in the PB of mice at 3 weeks after transplantation. Right panel, percentage of CD3+ cells in GFP+ leukemia cells in the PB of mice at 3 weeks after transplantation. C The Phf6 protein expression level in GFP+ cells of T-ALL mice treated with pIpC. D, E Percentage of T cells, B cells and myeloid cells in GFP+ cells in the BM of T-ALL mice treated with pIpC. F Percentage of GFP+ cells in the PB and BM of T-ALL mice treated with pIpC. G Percentage of GFP+ cells in the spleen, liver, and thymus of T-ALL mice treated with pIpC. H The counts of WBCs, lymphocytes and platelets in the PB of T-ALL mice treated with pIpC by routine blood tests. I The weights of the spleen, liver, and thymus of T-ALL mice treated with pIpC. J Kaplan–Meier survival curves of MC Phf6 KO + JAK3M511I T-ALL mice (n = 16) and MC Phf6 WT + JAK3M511I T-ALL mice (n = 12) after pIpC injection (log-rank test P < 0.0001).
Phf6 deficiency increases the activity of leukemia-initiating cells in T-ALL
To determine whether Phf6 deficiency promotes leukemia initiation by increasing LIC number or activity, GFP+ primary Phf6 WT + JAK3M511I and VC Phf6 + JAK3M511I cells were sorted and transplanted into secondary recipients (Fig. 4A). The mice transplanted with VC Phf6 + JAK3M511I cells had more leukemic cells in the PB, BM and extramedullary organs than the controls (Figs. 4B, C and S2A), with significantly shorter survival time than the controls (Fig. 4D). Importantly, secondary leukemia cells arising from VC Phf6 + JAK3M511I primary cells in the BM were mainly lymphoid cells (96.3%) (Figs. 4E and S2B), in contrast to the primary VC Phf6 + JAK3M511I cells showed much greater heterogeneity (Fig. 2I). Wright–Giemsa staining showed that most GFP+ cells of the two groups were abnormal lymphocytes with typical lymphoblastic characteristics (Fig. S2C). These results suggested that the GFP+ cells were mainly T leukemic cells based on immunophenotypes and morphological characteristics and the GFP+ CD8+ T-cell subclone had a dominant growth advantage in VC Phf6 + JAK3M511I cell populations.

A Schematic representation of secondary transplantation. B, C Percentage of GFP+ leukemia cells in the PB and BM of mice at 8 weeks after secondary transplantation. D Kaplan–Meier survival curves of VC Phf6 + JAK3M511I T-ALL mice and Phf6 WT + JAK3M511I T-ALL mice in the secondary transplantation assay (log-rank test P < 0.0001, n = 10 per group). E Percentage of T cells, B cells and myeloid cells in GFP+ leukemia cells in the BM of mice at 8 weeks after secondary transplantation. F Percentage of GFP+ CD25+ cells in GFP+ leukemia cells in the BM of Phf6 KO + JAK3M511I mice and Phf6 WT + JAK3M511I mice. G The expression of TCR-β in GFP+CD8+cells of Phf6 KO + JAK3M511I mice and Phf6 WT + JAK3M511I mice. H, I Phf6 KO + JAK3M511I mice had more LICs (P < 0.0001) according to the extreme limiting dilution assay (n = 10 per dose). J LIC analysis in mice transplanted with Phf6 knockout and wild-type leukemia cells. Confidence intervals showing 1/ (stem cell frequency) based on H.
To further identify the properties of VC Phf6 + JAK3M511I leukemia cells, we analyzed T-cell surface markers of GFP+ cells (CD4, CD8, CD25, and CD127). The CD25 marker is highly expressed in T cells during embryonic development [26], and we found that the percentage of GFP+ CD25+ cells was increased in VC Phf6 + JAK3M511I mice than the controls, while no obvious CD127 expression was observed (Fig. 4F). Notably, the expression of TCR-β was significantly decreased in Phf6-deficient leukemia cells, suggesting that VC Phf6 + JAK3M511I cells were more immature (Fig. 4G). Consistent with the presence of more immature T cells in the VC Phf6 + JAK3M511I populations, extreme limiting dilution assay demonstrated a marked increase in LICs activity in Phf6 KO T-ALL cells when compared with Phf6 WT T-ALL cells, indicating that loss of Phf6 promoted LICs self-renewal and proliferation (Figs. 4H–J and S2D).
Loss of Phf6 enhances leukemia cell proliferation through acceleration of the cell cycle
To identify the definitive role of Phf6 deficiency in leukemia cell over-proliferation, we investigated cell cycle and apoptosis in VC Phf6 + JAK3M511I cells. We found more VC Phf6 + JAK3M511I cells were in G1/S and G2/M stages than the control cells (Fig. 5A). However, the cell apoptosis rate was similar in both groups in vivo (Fig. S3A).

A Representative FACS plots showing the cell cycle of GFP+ cells in BM (upper panel). Percentage of GFP+ cells at each cell cycle stage (lower panel). B Gene interaction analysis showing the significantly altered expression pattern in genes that regulate the cell cycle, lymphocyte differentiation, cell adhesion and apoptosis in VC Phf6 + JAK3M511I T-ALL cells compared with Phf6 WT + JAK3M511I T-ALL cells. C Heatmap of cell cycle-related and apoptosis-related genes. D Validation of the expression of cell cycle-related genes in GFP+ T-ALL cells. E Validation of the expression of apoptosis-related genes in GFP+ T-ALL cells. F Validation of the expression of P53 signaling pathway-targeted genes in GFP+ T-ALL cells. G–I Gene set enrichment analysis (GSEA) of VC Phf6 + JAK3M511I T-ALL cells versus Phf6 WT + JAK3M511I T-ALL cells.
To explore the underlying molecular mechanisms of Phf6 loss in accelerating T-ALL cell proliferation, we analyzed the transcriptional profiles of VC Phf6 + JAK3M511I and Phf6 WT + JAK3M511I T-ALL cells. It revealed a distinct gene expression signature in VC Phf6 + JAK3M511I cells (2377 genes upregulated and 3751 genes downregulated; P < 0.05) (Fig. S3B) and these differentially expressed genes were significantly enriched for activities associated with cell cycle, apoptosis, adhesion and lymphocyte differentiation (Fig. 5B). We identified 35 cell cycle-related genes were upregulated, and 39 apoptosis-related genes were downregulated (P < 0.05) (Fig. 5C). We further validated that members of Mcm and Cdc gene families, considered to enhance cell proliferation, were upregulated, while genes related to apoptosis, such as Caspase family genes, were downregulated in VC Phf6 + JAK3M511I cells (Fig. 5D, E). Notably, the analysis showed Phf6 loss inhibited the P53 signaling pathway, as this was validated by the decreased mRNA expression of Gadd45a, Gadd45b and Fasl in Phf6 KO T-ALL cells when compared with the control cells (Fig. 5F).
GO and KEGG analysis revealed active nuclear division and cell cycle in VC Phf6 + JAK3M511I cells (Fig. S3C, D), while GSEA showed upregulation of cell cycle progression and DNA replication gene expression in VC Phf6 + JAK3M511I cells (Fig. 5G). T-cell maturation might be altered since naive CD8+ T-cell-related genes were enriched in VC Phf6 + JAK3M511I when compared with the controls (Figs. 5H and S3E). Meanwhile, embryonic stem cell related genes were enriched in VC Phf6 + JAK3M511I cells (Fig. 5I). These data suggested that Phf6 loss promoted JAK3M511I induced T-ALL progression by accelerating the cell cycle and increasing immature T cells.
Phf6 deficiency increases Bai1-mediated P53 degradation
To determine the underlying molecular mechanism driving the enhanced oncogenic potential of JAK3M511I in the absence of Phf6, we examined the phosphorylation level of Stat5 in Phf6 WT + JAK3M511I, VC Phf6 + JAK3M511I, and WT cells. The p-Stat5 was increased in both Phf6 WT + JAK3M511I and VC Phf6 + JAK3M511I when compared with WT cells, while Phf6 loss did not further enhance p-Stat5 in VC Phf6 + JAK3M511I when compared with Phf6 WT + JAK3M511I cells (Fig. S4A). We further probed that how Phf6 downregulates P53 signaling pathway, and found that Phf6 deficiency decreased P53 protein expression but not P53 mRNA expression in mouse leukemia cells (Fig. 6A left panel, B left panel). To investigate if PHF6 regulates P53 expression independent of the JAK3-STAT5 signaling pathway, we examined P53 protein levels in PHF6 knockdown (KD) U2OS cells treated with X-ray (35 Gy). We found that the protein expression of P53 was decreased, while the mRNA expression of P53 was unchanged in PHF6 KD U2OS cells when compared with the controls (Fig. 6A right panel, B right panel). The ubiquitination of P53 was significantly increased in PHF6 KD U2OS cells and PHF6 KD MOLT-4 T-ALL cells when compared with the controls (Fig. 6C). These results indicated that PHF6 regulates the ubiquitination of P53.

A Left panel, the protein expression of P53, Mdm2 and Phf6 in Phf6 WT + JAK3M511I and VC Phf6 + JAK3M511I leukemia cells. Right panel, the protein levels of P53, MDM2, and PHF6 in PHF6 KD U2OS cells and control cells treated with X-rays (35 Gy). B Left panel, the mRNA expression of P53 and Phf6 in Phf6 WT + JAK3M511I and VC Phf6 + JAK3M511I leukemia cells. Right panel, the mRNA expression of P53 and PHF6 in PHF6 KD U2OS cells and control cells. C Co-IP was performed with P53 antibody. P53 ubiquitination was determined by WB with anti-ubiquitin antibody in PHF6 KD/Con U2OS cells treated with X-rays (35 Gy) (upper panel) and PHF6 KD/Con MOLT-4 cells treated with γ-rays (7.5 Gy) (lower panel). D Left panel, Bai1 and Phf6 mRNA expression in Phf6 WT + JAK3M511I and VC Phf6 + JAK3M511I leukemia cells. Right panel, BAI1 and PHF6 mRNA expression in PHF6 KD U2OS cells and control cells. E Left panel, protein levels of Phf6 and Bai1 in Phf6 WT + JAK3M511I and VC Phf6 + JAK3M511I leukemia cells. Right panel, protein levels of PHF6 and BAI1 in PHF6 KD U2OS cells and control cells treated with X-rays (35 Gy). F The protein level of HA-PHF6 in PHF6 OE K562 cells. G PHF6 binding at the ADGRB1 (BAI1) gene locus in PHF6 OE K562 cells. H The relative amount of immunoprecipitated DNA quantified by ChIP-qPCR is given as the percentage of input DNA. I Quantification of luciferase activity from K562 cells (PHF6 KD or OE) co-transfected with a luciferase reporter containing the BAI1 sequence (or control sequence) and Renilla luciferase. J Co-IP was performed with P53 antibody. P53 ubiquitination was examined in BAI1 KD/Con U2OS cells treated with X-rays (35 Gy). K The protein levels of BAI1, P53, MDM2, and PHF6 in BAI1 KD/Con U2OS cells. L The protein levels of Flag-BAI1, P53, MDM2, and PHF6 in PHF6 KD + BAI1 OE U2OS cell (lane 3), PHF6 KD + Empty Vector U2OS cell (lane 2) and PHF6 Con U2OS cell (lane 1). M The protein levels of Flag-BAI1, P53, MDM2, and PHF6 in PHF6 KD + BAI1 OE MOLT-4 cell (lane 3), PHF6 KD + Empty Vector MOLT-4 cell (lane 2) and PHF6 Con MOLT-4 cell (lane 1).
It has been reported that adhesion G protein-coupled receptor B1 (ADGRB1), also known as brain-specific angiogenesis inhibitor 1 (BAI1), prevents MDM2-mediated P53 ubiquitination, and loss of BAI1 reduces P53 level [27]. We thus examined Bai1/BAI1 expression in VC Phf6 + JAK3M511I cells and PHF6 KD U2OS cells, and found that the mRNA and protein expression of Bai1/BAI1 were significantly decreased in both cells in comparison with Phf6 WT + JAK3M511I or PHF6 WT U2OS cells, respectively (Fig. 6D, E). Chromatin immunoprecipitation sequencing (ChIP-seq) analysis of HA-PHF6-overexpressing (PHF6 OE) K562 cells (Fig. 6F) showed that PHF6 directly bound to BAI1 gene (Fig. 6G). We used PHF6 or HA antibody to enrich PHF6 protein in PHF6-OE K562 cells and further verified the binding of PHF6 protein to the BAI1 DNA sequence by ChIP-qPCR (Figs. 6H and S4B). K562 cells were then co-transfected with a reporter vector containing the BAI1 sequence (or the control) inserted upstream of firefly luciferase and Renilla luciferase vector. We found that PHF6 KD reduced BAI1 luciferase activity, while PHF6 OE increased BAI1 luciferase activity (Fig. 6I). We further knocked-down BAI1 in U2OS cells and observed that BAI1 KD significantly reduced the P53 level and increased the P53 ubiquitination (Fig. 6j, k). To further investigate whether PHF6 regulates P53 expression via BAI1, the expression of BAI1 was restored in PHF6 KD U2OS cells and PHF6 KD MOLT-4 cells. We found that the expression of P53 was significantly increased in PHF6 KD U2OS cells and PHF6 KD MOLT-4 cells when BAI1 was rescued (Figs. 6L, M and S4C). We further investigated the interaction between PHF6 and BAI1-P53 in the Kasumi-1 cells without irradiation. We found that the mRNA and protein expressions of BAI1 were significantly decreased in PHF6 KD Kasumi-1 cells when compared with that of controls (Fig. S4D, E). The protein level of P53 was also decreased in PHF6 KD Kasumi-1 cells (Fig. S4E) and could be partially rescued when BAI1 was overexpressed in PHF6 KD Kasumi-1 cells (Fig. S4F). Taken together, our data suggested that Phf6 loss inhibited the Bai1-Mdm2-P53 signaling pathway rather than by activating the Jak3/Stat5 signaling pathway.
Combined treatment with idasanutlin and tofacitinib shows greater antitumor effects in Phf6 KO + JAK3 M511I T-ALL
Since P53 signaling pathway plays a central role in the pathogenesis of Phf6 KO + JAK3M511I T-ALL, we sought to determine whether dual activation of P53 and inactivation of JAK3 would be beneficial in Phf6 KO + JAK3M511I T-ALL mice. Tofacitinib is a JAK3-specific inhibitor [12]. Idasanutlin is a selective MDM2 antagonist that can activate P53 [28, 29]. VC Phf6 + JAK3M511I T-ALL mice were treated with placebo, single tofacitinib, single idasanutlin, or combined tofacitinib + idasanutlin (To + Id) (Fig. 7A). We measured p-Stat5 and P53 in VC Phf6 + JAK3M511I BM cells from treated or nontreated mice. We found that p-Stat5 was decreased (Figs. 7B and S5A), and P53 was increased in mice treated with To + Id (Fig. 7C). The survival time of mice treated with To + Id was significantly longer than mice treated with placebo, single tofacitinib or idasanutlin (Fig. 7D). The counts of hemoglobin and PLT were increased, while the count of leukemia cells was decreased in the PB of mice treated with To + Id when compared with mice in other groups (Fig. 7E). Additionally, the percentages of GFP+ leukemia cells in the PB, BM, spleen, liver, and thymus were decreased in mice treated with To + Id when compared with other groups (Figs. 7F and S5B–E). The weights of spleen, liver, and thymus were significantly decreased in mice treated with To + Id in comparison with other groups (Fig. S5F, G). HE staining showed that the degree of leukemia cell infiltration in the spleen, liver, brain, thymus, and BM was reduced in mice treated with To + Id when compared with other groups (Fig. 7G). Immunohistochemical staining of Ki67, a marker of cell proliferation, showed significantly reduced staining in the To + Id-treated group when compared with the other groups (Fig. S5H), while TUNEL (TdT-mediated dUTP nick-end labeling) staining showed a significant increase in apoptotic cells in mice treated with To + Id than other groups (Fig. 7G).

A Schematic representation of different drug treatments in VC Phf6 + JAK3M5111 T-ALL mice. B Phosphoflow cytometry was used to measure the phosphorylation level of Stat5 in leukemia cells from VC Phf6 + JAK3M5111 T-ALL mice treated with placebo, single tofacitinib, single idasanutlin, or combined tofacitinib and idasanutlin through oral gavage. C Western blotting was used to assess the expression of P53 in leukemia cells from VC Phf6 + JAK3M5111 T-ALL mice treated with different drugs. D Kaplan–Meier survival curves of VC Phf6 + JAK3M5111 T-ALL mice treated with different drugs (n = 10 per group). Mice treated with combined tofacitinib and idasanutlin VS placebo (log-rank test P < 0.0001), single tofacitinib (log-rank test P = 0.0019) or single idasanutlin (log-rank test P = 0.0034). E The counts of HGB, PLT, and WBC in PB by routine blood tests. F The percentage of GFP+ leukemia cells in BM from VC Phf6 + JAK3M5111 T-ALL mice treated with different drugs. G Immunohistochemical staining for hematoxylin and eosin (HE) (magnification, ×40) and TUNEL staining (green represents TUNEL, blue represents DAPI, magnification, ×60) in BM, spleen, liver, brain, and thymus from VC Phf6 + JAK3M5111 T-ALL mice treated with different drugs. H Construction of PHF6 KD + JAK3M511I and PHF6 Con464 + JAK3M511I MOLT-4 cells and their WB verification. I The proliferation of MOLT-4 PHF6 KD + JAK3M511I cells was much faster than MOLT-4 PHF6 Con464 + JAK3M511I, MOLT-4 PHF6 KD, MOLT-4 Con464 or MOLT-4 WT cells respectively. J, K Proliferation and apoptosis of MOLT-4 PHF6 KD + JAK3M511I cells were examined in vitro with various drug treatment of 0.2% DMSO, single tofacitinib (2.0 μM), single idasanutlin (0.5 μM) or combined tofacitinib + idasanutlin for 48 h.
Also, we constructed JAK3M511I-overexpression MOLT-4 cells that were treated with shPHF6 (PHF6 KD) or sh-Control (PHF6 Con464) respectively, and confirmed by WB along with MOLT-4 cells (WT) (Fig. 7H). We found that the proliferation of PHF6 KD + JAK3M511I cells was much faster than PHF6 Con464 + JAK3M511I or other control cells (Fig. 7I). Furthermore, the proliferation of PHF6 KD + JAK3M511I MOLT-4 cells was significantly decreased while their percentage of apoptosis was increased after treated with To + Id when compared with single drug or control treatment groups (Fig. 7J, K). Our result thus demonstrated that combination therapy with tofacitinib and idasanutlin can reduce leukemia burden better than single drug treatment in Phf6 KO + JAK3M511I mice, which was also validated by the result in human MOLT-4 cells.
Discussion
Understanding the mechanism of how epigenetic regulatory genes target chromatin and redirect gene transcription and activation in leukemogenesis is imperative for developing novel therapies. As a chromatin remodeling-related gene, PHF6 is frequently mutated in T-ALL patients [13, 17, 30]. Here, we demonstrated that PHF6 mutations more commonly coexisted with JAK/STAT mutations in T-ALL patients (Fig. 1A). The average survival time of patients with JAK/STAT and PHF6 comutation was shorter than that of patients without this comutation (Figs. 1E and S1A). Phf6 deletion led to rapid development of JAK3M511I-induced T-ALL by inhibiting the Mdm2-P53 signaling pathway. Leukemia progression can be contained better by specific inhibition of JAK3 and Mdm2-P53 in Phf6-deficient and JAK3M511I T-ALL cells (Fig. 7D). Furthermore, we revealed that the progression of JAK3M511I-induced T-ALL from low to high malignancy is triggered by the coexistence of PHF6 mutation, and that provided a potential therapeutic window for the modulation of P53 and JAK3 activity in the treatment of T-ALL patients with PHF6 and JAK3 comutation.
It has been reported that Phf6 is essential for HSC homeostasis and T-ALL initiation, although Phf6 appears to play a modest role in normal T-cell differentiation and proliferation [20]. Loss of Phf6 slightly reduced number of T cells in PB and BM, but did not lead to spontaneous hematological malignant transformation in mice [20]. Earlier studies theorized that PHF6 may play important roles in lineage-specification during leukemogenesis. For example, lack of Phf6 promoted Notch-induced T-ALL initiation and MLL-AF9-induced AML progression, while decelerated the development of BCR-ABL1-induced B-ALL [31]. However, Thomas and colleagues suggested that PHF6 might not act in a strictly lineage-dependent manner. They found that Phf6 knockout accelerated TLX1/TLX3-induced B-ALL [22], and that is different from the oncogenic role of Phf6 in BCR-ABL1-induced B-ALL. It indicated that the role(s) of PHF6 may depend on the combination of oncogenic mutations and specific molecular pathways that drive leukemia. In our studies, we found that primary Phf6 KO + JAK3M511I progenitor cells developed a complex hematolymphoid neoplasm characterized by the coexistence of different populations of atypical cells (Fig. 2I, J). However, when we depleted Phf6 in MC Phf6 + JAK3M511I mice with pIpC, leukemia cells were mainly lymphoid cells (Fig. 3D, E). These results suggested that if PHF6 mutation and JAK3 mutation occurred in hematopoietic progenitors, it may induce a complex hematolymphoid neoplasm. When PHF6 mutation acted as “a secondary strike” in JAK3M511I-induced T-ALL, it might also promote the T-ALL progression. Based on our observations, we reasoned that PHF6 might function as a tumor suppressor, while PHF6 loss or mutations favors T-ALL initiation by lowering the threshold for subsequent oncogenic transformation in hematopoietic progenitors, and postulated the role in promoting T-ALL development.
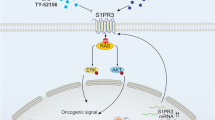
We further demonstrated how PHF6 functions in JAK3M511I-induced T-ALL independent of the Jak3-Stat5 signaling pathway. As expected, we found that Stat5 was activated in both Phf6 KO/WT + JAK3M511I cells (Fig. S4A) [12]. However, the Mdm2-P53 signaling pathway was inhibited only in Phf6 KO + JAK3M511I but not in Phf6 WT + JAK3M511I leukemia cells (Fig. 6A). This suggested that T-ALL progression due to Phf6 loss is dependent on additional signaling pathways. BAI1 belongs to the adhesion subgroup of GPCRs, which functions at preventing MDM2-mediated P53 ubiquitination, and its loss could substantially reduce the P53 level [27]. Consistent with prior findings showing that P53 loss increased penetrance and accelerated progression of leukemia [28, 32], we showed here that PHF6 loss increased JAK3M511I-induced T-ALL initiation by downregulating BAI1 expression, thereby decreasing BAI1 and MDM2 interaction, and destabilizing P53 (modeled in Fig. 8). Thus, our results defined a PHF6-BAI1-P53 signaling axis and linked it with JAK3M511I-induced T-ALL genesis. It will be interesting to assess whether T-ALL patients with JAK3 and PHF6 comutation could further benefit from combined therapy with MDM2 and JAK3 inhibitors.

In JAK3M511I-induced leukemia, PHF6 could bind to ADGRB1 (BAI1) gene, increase its expression, upregulate BAI1 level, prevent MDM2-mediated P53 ubiquitination, stabilize the P53 protein, and suppress leukemia development (Left panel). In the same leukemia model, loss of PHF6 could downregulate the expression of BAI1, lead to increased MDM2-P53 binding and P53 degradation, and accelerate JAK3M511I-induced T-ALL progression (Right panel).
In conclusion, we revealed that PHF6 and JAK3 mutations cooperatively drive T-ALL progression probably via inhibiting the BAI1-MDM2-P53 signaling pathway, in addition to activating the JAK3/STAT5 signaling pathway. We further demonstrated that combination therapy with tofacitinib and idasanutlin reduced the Phf6 KO + JAK3M511I leukemia burden in vivo. Our study suggested that the combined usage of JAK3 and MDM2 inhibitors should increase the drug benefit for T-ALL patients with PHF6 and JAK3 comutation.
References
Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016;16:494–507.
Juliusson G, Hough R. Leukemia. Prog Tumor Res. 2016;43:87–100.
Marks DI, Rowntree C. Management of adults with T-cell lymphoblastic leukemia. Blood 2017;129:1134–42.
Albertí-Servera L, Demeyer S, Govaerts I, Swings T, Cools J. Single-cell DNA amplicon sequencing reveals clonal heterogeneity and evolution in T-cell acute lymphoblastic leukemia. Blood 2020;137:801–11.
Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Investig. 2012;122:3398–406.
Yeh T-C, Liang D-C, Liu H-C, Jaing T-H, Chen S-H, Hou J-Y, et al. Clinical and biological relevance of genetic alterations in pediatric T-cell acute lymphoblastic leukemia in Taiwan. Pediatr Blood Cancer. 2019;66:e27496.
Dorritie KA, McCubrey JA, Johnson DE. STAT transcription factors in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia 2014;28:248–57.
de Bock CE, Demeyer S, Degryse S, Verbeke D, Sweron B, Gielen O, et al. HOXA9 Cooperates with Activated JAK/STAT Signaling to Drive Leukemia Development. Cancer Discov. 2018;8:616–31.
Xiao W, Bharadwaj M, Levine M, Farnhoud N, Pastore F, Getta BM, et al. PHF6 and DNMT3A mutations are enriched in distinct subgroups of mixed phenotype acute leukemia with T-lineage differentiation. Blood Adv. 2018;2:3526–39.
Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205:751–8.
Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene. 2013;32:2601–13.
Degryse S, de Bock CE, Cox L, Demeyer S, Gielen O, Mentens N, et al. JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model. Blood. 2014;124:3092–100.
Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica. 2015;100:1301–10.
Asnafi V, Noir SL, Lhermitte L, Gardin C, Legrand F, Vallantin X, et al. JAK1 mutations are not frequent events in adult T-ALL: a GRAALL study. Br J Haematol. 2010;148:178–79.
Lin PH, Li HY, Fan SC, Yuan TH, Chen M, Hsu YH, et al. A targeted next-generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: implications for clinical practice. Cancer Med. 2017;6:349–60.
Tashi T, Swierczek S, Prchal JT. Familial MPN predisposition. Curr Hematol Malig Rep. 2017;12:442–47.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–18.
Zhou X, Gu Y, Han Q, Soliman M, Song C, Ge Z. Coexistence of EZH2, NOTCH1, IL7R, and PHF6 mutations in adult T-cell acute lymphoblastic leukemia. Turkish J Haematol. 2017;34:366–68.
Alexander TB, Gu Z, Iacobucci I, Dickerson K, Choi JK, Xu B, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature. 2018;562:373–79.
Hsu YC, Chen TC, Lin CC, Yuan CT, Hsu CL, Hou HA, et al. Phf6-null hematopoietic stem cells have enhanced self-renewal capacity and oncogenic potentials. Blood Adv. 2019;3:2355–67.
Wendorff AA, Quinn SA, Rashkovan M, Madubata CJ, Ambesi-Impiombato A, Litzow MR, et al. Phf6 loss enhances HSC self-renewal driving tumor initiation and leukemia stem cell activity in T-ALL. Cancer Discov. 2019;9:436–51.
McRae HM, Garnham AL, Hu Y, Witkowski MT, Corbett MA, Dixon MP, et al. PHF6 regulates hematopoietic stem and progenitor cells and its loss synergizes with expression of TLX3 to cause leukemia. Blood. 2019;133:1729–41.
Miyagi S, Sroczynska P, Kato Y, Nakajima-Takagi Y, Oshima M, Rizq O, et al. The chromatin-binding protein Phf6 restricts the self-renewal of hematopoietic stem cells. Blood. 2019;133:2495–506.
Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8.
Spinella JF, Cassart P, Richer C, Saillour V, Ouimet M, Langlois S, et al. Genomic characterization of pediatric T-cell acute lymphoblastic leukemia reveals novel recurrent driver mutations. Oncotarget. 2016;7:65485–503.
Habu S, Okumura K, Diamantstein T, Shevach EM. Expression of interleukin 2 receptor on murine fetal thymocytes. Eur J Immunol. 1985;15:456–60.
Dan Z, Satoru O, Zhaobin Z, Reichert ZR, Liquan Y, Yonehiro K, et al. BAI1 suppresses medulloblastoma formation by protecting p53 from Mdm2-mediated degradation. Cancer Cell. 2018;33:1004–16.
Ciardullo C, Aptullahoglu E, Woodhouse L, Lin WY, Wallis JP, Marr H, et al. Non-genotoxic MDM2 inhibition selectively induces a pro-apoptotic p53 gene signature in chronic lymphocytic leukemia cells. Haematologica. 2019;104:2429–42.
Lehmann C, Friess T, Birzele F, Kiialainen A, Dangl M. Superior anti-tumor activity of the MDM2 antagonist idasanutlin and the Bcl-2 inhibitor venetoclax in p53 wild-type acute myeloid leukemia models. J Hematol Oncol. 2016;9:50.
Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42:338–42.
Soto-Feliciano YM, Bartlebaugh JME, Liu Y, Sanchez-Rivera FJ, Bhutkar A, Weintraub AS, et al. PHF6 regulates phenotypic plasticity through chromatin organization within lineage-specific genes. Genes Dev. 2017;31:973–89.
Zhang L, McGraw KL, Sallman DA, List AF. The role of p53 in myelodysplastic syndromes and acute myeloid leukemia: molecular aspects and clinical implications. Leuk Lymphoma. 2017;58:1777–90.
Acknowledgements
This work was supported by funds from the Ministry of Science and Technology of China (2018YFA0107801 and 2017YFA0103402 to WPY); the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences, CIFMS (2017-I2M-3-015 to WPY and XMW and 2016-I2M-1-017 to TC and YJC); and the National Natural Science Foundation of China (81629001 to WPY, 82070169 and 81870088 to XMW, 81970149 and 81770105 to WPY).
Author information
Authors and Affiliations
Contributions
WPY and XMW conceived the project, supervised the research and revised the paper. SNY, SBH and XMW performed most of the experiments and analyzed the data. TXG, SY, YJL, FZ, JG and YXW assisted with the mouse experiments, flow cytometry analysis and data processing. XMW and SNY wrote the manuscript. YJC, JS and TC contributed to the data analyses and paper discussion. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, S., Wang, X., Hou, S. et al. PHF6 and JAK3 mutations cooperate to drive T-cell acute lymphoblastic leukemia progression. Leukemia 36, 370–382 (2022). https://doi.org/10.1038/s41375-021-01392-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-021-01392-1
This article is cited by
-
PHF6 loss reduces leukemia stem cell activity in an acute myeloid leukemia mouse model
Cancer Cell International (2024)
-
Idasanutlin and navitoclax induce synergistic apoptotic cell death in T-cell acute lymphoblastic leukemia
Leukemia (2023)
-
PHF6 maintains acute myeloid leukemia via regulating NF-κB signaling pathway
Leukemia (2023)
-
Impact of genetic patterns on sorafenib efficacy in patients with FLT3-ITD acute myeloid leukemia undergoing allogeneic hematopoietic stem cell transplantation: a multi-center, cohort study
Signal Transduction and Targeted Therapy (2023)
-
Oncogenetic landscape of T-cell lymphoblastic lymphomas compared to T-cell acute lymphoblastic leukemia
Modern Pathology (2022)
