
Abstract
Despite recent advances in therapeutic approaches, patients with MLL-rearranged leukemia still have poor outcomes. Here, we find that the RNA-binding protein IGF2BP3, which is overexpressed in MLL-translocated leukemia, strongly amplifies MLL-Af4-mediated leukemogenesis. Deletion of Igf2bp3 significantly increases the survival of mice with MLL-Af4-driven leukemia and greatly attenuates disease, with a minimal impact on baseline hematopoiesis. At the cellular level, MLL-Af4 leukemia-initiating cells require Igf2bp3 for their function in leukemogenesis. At the molecular level, IGF2BP3 regulates a complex posttranscriptional operon governing leukemia cell survival and proliferation. IGF2BP3-targeted mRNA transcripts include important MLL-Af4-induced genes, such as those in the Hoxa locus, and the Ras signaling pathway. Targeting of transcripts by IGF2BP3 regulates both steady-state mRNA levels and, unexpectedly, pre-mRNA splicing. Together, our findings show that IGF2BP3 represents an attractive therapeutic target in this disease, providing important insights into mechanisms of posttranscriptional regulation in leukemia.
Similar content being viewed by others


Introduction
Chromosomal rearrangements of the mixed-lineage leukemia (MLL, KMT2A) gene are recurrently found in a subset of acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), and acute leukemia of ambiguous lineage [1]. Despite recent advances in therapeutic approaches, patients with MLL-rearranged leukemia have poor outcomes, high risk of relapse, and show resistance to novel targeted therapies [2, 3]. MLL encodes an H3K4 methyltransferase required for hematopoietic stem cell (HSC) development during both embryonic and adult hematopoiesis [4,5,6,7]. Many translocation partners for MLL, including AF4 (AFF1), encode proteins that regulate transcriptional elongation [8,9,10,11,12,13,14]. Of more than 90 translocation fusion partner genes, MLL-AF4 (KMT2A-AFF1) is the most common MLL fusion protein in patients [15]. Biologically, MLL-AF4-driven leukemia is a distinct entity compared to non-MLL-rearranged leukemias, with a unique gene expression profile showing significant overlap with stem cell programs [16,17,18].
At the posttranscriptional level, emerging evidence suggests a role for microRNAs, RNA-binding proteins (RBP), and other RNA-based mechanisms in regulating gene expression during leukemogenesis [19,20,21]. We recently identified the oncofetal RBP Insulin like growth factor 2 mRNA binding protein 3 (IGF2BP3) as an important regulator of gene expression in MLL-rearranged B-ALL [22]. IGF2BP3 is expressed during embryogenesis, lowly expressed in healthy adult tissues, and strongly reexpressed in cancer cells [23]. Elevated levels of IGF2BP3 expression are correlated with diminished patient survival in many cancers and may be a marker of disease aggressiveness in B-ALL [24,25,26]. Previously, we determined that overexpression of IGF2BP3 in bone marrow (BM) of mice led to a pathologic expansion of hematopoietic stem and progenitor cells (HSPC). IGF2BP3 interacted with and upregulated oncogenic transcripts (e.g., MYC, CDK6) via the 3′UTR, contributing to the pathologic proliferative phenotype [22]. Together, these studies illuminated a novel role for posttranscriptional gene regulation in the pathologic proliferation of HSPCs.
Experimentally, MLL-AF4-driven leukemogenesis has been studied using a range of in vitro and in vivo models leading to significant progress in our understanding of MLL-rearranged leukemia [16, 27,28,29,30,31]. Here, we explicitly tested the requirement for Igf2bp3 in a bona-fide model of MLL-Af4-driven leukemogenesis [32]. Deletion of Igf2bp3 significantly increased survival of MLL-Af4 transplanted mice and decreased the numbers and self-renewal capacity of MLL-Af4 leukemia-initiating cells (LICs). Mechanistically, we found that IGF2BP3 targets and modulates the expression of transcripts encoding regulators of leukemogenesis, through multiple posttranscriptional mechanisms. Together, our findings show that IGF2BP3 is a critical regulator of MLL-AF4-mediated leukemogenesis and a potential therapeutic target in this disease.
Methods
Molecular biology assays
ChIP-PCR on RS4;11 and SEM cells were performed as previously described [33]. IGF2BP3 ChIP primer sequences were kindly provided by Dr. James Mulloy (University of Cincinnati) [32]. Protein and mRNA extracts were prepared, and western blot/RT-qPCR performed as previously described [34]. Primers and antibodies are listed in Table S1.
Plasmids, retroviral transduction and BM transplantation (BMT)
The MSCV-MLL-FLAG-Af4 plasmid was kindly provided by Michael Thirman (University of Chicago) through MTA [32]. Nontargeting (NT) or Igf2bp3 sgRNA was cloned into an in-house MSCV-hU6-sgRNA-EFS-mCherry vector [35]. Retroviral transduction and BMT are previously described [34, 36]. 5-FU enriched BM and Lin− cells were spin-infected four times with MSCV-MLL-FLAG-Af4 virus at 30 °C for 45 min with polybrene and selected with 400 μg/ml G418 for 7 days. MLL-Af4 Cas9-GFP cells were retrovirally infected with MSCV-hU6-sgRNA-EFS-mCherry.
Mice
C57BL/6J and B6J.129(Cg)-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J (Cas9-GFP BL/6J) mice were from Jackson Laboratory. The UCI Transgenic Mouse Facility utilized CRISPR-Cas9 to insert loxP sites flanking exon 2 of Igf2bp3 to generate Igf2bp3f/f mice. To generate conditional KO, Igf2bp3f/f mice were bred with Vav1-Cre mice. Consistent with prior reports, this strategy led to “leaky” Cre expression, resulting in germline deletion [37,38,39]. To isolate floxed and deletion (del) alleles, mice were back-crossed onto C57BL/6 mice with successful germline, Mendelian transmission of del and floxed alleles in two successive generations (Table S2). Mice heterozygous for del allele were mated, leading to homozygous Igf2bp3 deletion and Igf2bp3del/del mice (I3KO) used in this study. Blinding or randomization was not applied to mice experiments.
Cell culture and flow cytometry
RS4;11, SEM, 70Z/3, and HEK293T cell lines were cultured as previously described [34]. Lin− cells were cultured in IMDM with 15% FBS supplemented with SCF, IL-6, FLT3, and TPO. CD11b+ cells were isolated from splenic tumors for positive selection by MACS (Miltenyi). Blood, BM, thymus, and spleen were collected from mice at indicated time points and staining performed as previously described [22]. Antibodies are provided in Table S1. Flow cytometry was performed on a BD FACS LSRII and analysis using FlowJo software.
Histopathology
Fixation, sectioning, and analysis were performed as previously described (DSR) [36].
Competitive repopulation assay and secondary leukemia transplantation
Competitive repopulation experiments are previously described [22]. For leukemia transplantation, BM was collected from WT/MLL-Af4 or I3KO/MLL-Af4 mice that succumbed to leukemia at 10–14 weeks post transplantation and injected into 8-week-old immunocompetent CD45.1+ female mice.
RNA-seq
Single-end, strand-specific RNA sequencing was performed on Illumina HiSeq3000 for Lin− and CD11b+ samples, 15–20 million reads/sample (UCLA Technology Center for Genomics & Bioinformatics). Analysis is previously described [22]. RNA-seq reads were mapped to the mouse genome assembly mm10 using STAR version X. Repeat sequences were masked using Bowtie 2 [40] and RepeatMasker [41]. Differentially expressed genes (DEGs) were identified using DESeq2 [42] (CD11b+) and fdrtool [43] (Lin−). Multiple testing correction used the Benjamini–Hochberg method. Significant DEGs have adjusted P value < 0.1 and log2FC > 1. Data collection and parsing were completed with bash and python2.7. Statistical analyses were performed using R version 3.5.1. Enrichment analyses were completed with Metascape [44] and gene set enrichment analysis (GSEA) using GSEAPreranked after π-value calculation [45,46,47].
Alternative splicing estimation
Mixture of Isoforms (MISO) Bayesian inference model v0.5.4 with mm10 “exon-centric annotation” quantified alternative splicing events [48]. Percent spliced in (PSI) was quantified for each event by number of read counts supporting both events and unique reads to each isoform. Delta PSI was calculated by subtracting from WT. Significant differential events had delta PSI > 0.1, Bayes factor ≥ 10, and sum of exclusion and inclusion reads ≥ 10.
Enhanced crosslinking-immunoprecipitation (eCLIP)
eCLIP was completed on a minimum of two biological replicates with two technical replicates and size matched input (smInput) samples (Eclipse BioInnovations). Overall, 5 × 105 cells were UV crosslinked (245 nm, 400 mJoules/cm2), RNAse I treated, and immunoprecipitated with anti-IGF2BP3 antibody (MBL RN009P) coupled to magnetic Protein G beads. Paired-end RNA-seq was performed on Illumina HiSeq4000 (UCSF Genomics Core Facility). Peaks were called using CLIPper [49] and filtered on smInput (FS1). HOMER [50] annotatePeaks.pl and findMotifs.pl provided peak genomic locations and motif enrichment. Background for peaks within DEGs was simulated using bedtools [51] and shuffled 1000 times.
Statistics
Data represent mean ± SD for continuous numerical data, unless otherwise noted in figure legends. One-way ANOVA followed by Bonferroni’s multiple comparisons test (>2 groups) or two-tailed Student’s t tests were performed using GraphPad Prism software.
Results
IGF2BP3 is integrated into the MLL-AF4 transcriptional program
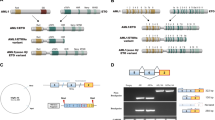
To understand the overlap of transcriptional and posttranscriptional regulation in MLL-rearranged leukemia, we compared IGF2BP3-regulated targets with a published MLL-Af4 ChIP-Seq dataset [22, 32]. Transcripts modulated by IGF2BP3 were significantly enriched for MLL-Af4-bound genes (Fig. 1a; Supplementary Fig. 1a). Interestingly, IGF2BP3 itself was a direct transcriptional target of MLL-Af4, with binding sites within the first intron and promoter region (Supplementary Fig. 1b) [32]. To confirm, we performed ChIP-PCR assays on RS4;11 and SEM, human MLL-AF4 translocated B-ALL cell lines, and determined that the first intron of IGF2BP3 is strongly bound by MLL-AF4 (Fig. 1b; Supplementary Fig. 1c). This MLL-AF4 binding was abrogated when SEM cells were treated with the DOT1L inhibitor, EPZ5676, and the bromodomain inhibitor, IBET-151 (Fig. 1c; Supplementary Fig. 1d) [52]. Furthermore, we observed an MLL-AF4-dose-dependent increase in luciferase reporter activity, using the promoter region upstream of the IGF2BP3 transcription start site (Supplementary Fig. 1e). In the murine pre-B 70Z/3 cell line and primary murine BM cells, transduction with retroviral MLL-Af4) [32] caused an ~64-fold upregulation of Igf2bp3 mRNA (Fig. 1d, e). Concordantly, IGF2BP3 protein was upregulated in MLL-Af4-transduced primary BM cells (Fig. 1f). Furthermore, enforced expression of another MLL fusion protein, MLL-AF9, and other non-MLL leukemia drivers, including AML1-ETO, MYC, and NRAS in primary HSPCs, show that the upregulation of Igf2bp3 is specific to MLL-Af4 (Supplementary Fig. 1f). These findings of Igf2bp3 specificity are in line with those that we and others have previously reported, as well as in publicly available datasets [22, 26, 53]. Interestingly, induced expression of structurally and functionally related paralogs Igf2bp1 and Igf2bp2 was noted with enforced expression of non-MLL-Af4 oncogenic drivers, again in concordance with observations in human leukemia (Supplementary Fig. 1g, h) [22, 26, 54]. Taken together, these findings demonstrate that MLL-Af4 specifically drives the expression of Igf2bp3 in vivo.

a GSEA of differentially expressed genes from IGF2BP3 depleted RS4;11 cells shows significant negative enrichment with MLL-AF4 ChIP targets (nominal P value: 0.001, FDR: 0.001, Normalized ES: −1.54)). b Schematic of MLL-AF4 binding site in intron 1 of IGF2BP3 (top). ChIP-qPCR shows fold enrichment for IGF2BP3 and CDKN1B with MLL and AF4 IP in RS4;11. Normalized to UNTR20, an untranscribed region (t test; *P < 0.05, **P < 0.01). c Fold enrichment from ChIP-qPCR of SEM cells show reduced binding of MLL-AF4 to IGF2BP3 with treatment of the DOT1L inhibitor, EPZ5676. Normalized to DMSO. (t test; **P < 0.01, ***P < 0.001, ****P < 0.0001). d Expression of MLL through RT-qPCR of 70Z/3 transduced with either control (Ctrl) or MLL-Af4 vector selected with G418 and MLL expression at the RNA level in the BM of WT recipients transplanted with Ctrl or MLL-Af4 HSPCs. e Induction of Igf2bp3 at the RNA level in selected 70Z/3 with MLL-Af4 and in the BM of WT recipients transplanted with Ctrl or MLL-Af4 HSPCs (bottom) (t test; **P < 0.01). f Induction of IGF2BP3 at the protein level in BM from mice transplanted with MLL-Af4-transduced WT donor HSPCs.
Normal hematopoiesis is maintained in Igf2bp3 KO mice
To test the in vivo requirement for IGF2BP3 in leukemogenesis, we generated an Igf2bp3 KO (I3KO) mouse. We initially generated a floxed Igf2bp3 allele (f/f; Supplementary Fig. 2a) using CRISPR-Cas9. In the course of mating these mice with Vav1-Cre mice, we serendipitously generated a germline knockout allele (del), which we isolated and characterized (Supplementary Fig. 2b). This has been previously reported in the Vav1-Cre mouse strain, which displays “leaky” Cre expression resulting in germline deletion [37,38,39]. Mendelian inheritance was confirmed for the isolated germline del allele, although distribution of genotypes was marginally skewed (Table S2). Deletion of Igf2bp3 was confirmed at the DNA, RNA, and protein level (Supplementary Fig. 2c–e). These Igf2bp3del/del (I3KO) mice were used in this study. Immunophenotyping of I3KO mice showed no significant differences in numbers of HSPCs in the BM compared to WT (Supplementary Fig. 2f). I3KO mice showed similar numbers of myeloid-lineage progenitors (CMPs, GMPs, and MEPs) (Supplementary Fig. 2g), normal B-cell development [55] (Supplementary Fig. 2h), and normal numbers of mature B lymphoid, T lymphoid, and myeloid lineages in the BM and spleen (Supplementary Fig. 2i, j). Hence, I3KO mice demonstrate preserved normal, steady-state adult hematopoiesis.
Igf2bp3 deletion increases the latency of MLL-Af4 leukemia and survival of mice
Next, we queried MLL-Af4-mediated leukemogenesis in I3KO mice, utilizing BMT (Supplementary Fig. 3a). Retroviral MLL-Af4 transduction was equivalent between WT and I3KO donor BM, based on DNA copy number (Supplementary Fig. 3b) and western blot analysis (Supplementary Fig. 3c). Following transplantation of transduced HSPCs, Igf2bp3 loss significantly increased both leukemia-free and overall survival of MLL-Af4 mice (Fig. 2a, b). The median survival of I3KO/MLL-Af4 mice was greater than 157 days, compared to 103 days for control mice. White blood cell (WBC) and myeloid cell counts in I3KO/MLL-Af4 mice were significantly reduced, compared with the control mice (Fig. 2c; Supplementary Fig. 3d). On average, I3KO/MLL-Af4 mice became overtly leukemic much later than the control mice peripheral blood (112 versus 70 days) (Fig. 2c). Concordantly, peripheral blood smears showed reduced circulating blasts in I3KO/MLL-Af4 mice (Supplementary Fig. 3e). Together, these findings indicated that Igf2bp3 is required for efficient MLL-Af4-mediated leukemogenesis.

a Leukemia-free survival of mice transplanted with control (Ctrl) or MLL-Af4-transduced HSPCs from WT or Igf2bp3 KO mice (Kaplan–Meier method with log-rank test; ****P < 0.0001). b Overall survival of mice transplanted with Ctrl or MLL-Af4-transduced HSPCs from WT or I3KO mice (n = 12 WT/Ctrl, n = 24 WT/MLL-Af4, n = 7 I3KO/Ctrl, n = 22 I3KO/MLL-Af4; Kaplan–Meier method with log-rank test; ****P < 0.0001). c Time course of WBC in the PB of mice transplanted with Ctrl or MLL-Af4-transduced HSPCs from WT or I3KO mice (data represented as means of three experiments; n = 4 Ctrl, n = 8 MLL-Af4 per experiment). d Spleen weights of mice transplanted with Ctrl or MLL-Af4-transduced HSPCs from WT or I3KO mice at 14 weeks (n = 4 Ctrl, n = 8 MLL-Af4; one-way ANOVA followed by Bonferroni’s multiple comparisons test; ****P < 0.0001). e H&E staining of liver and spleen of mice transplanted with mice transplanted with MLL-Af4-transduced HSPCs from WT or I3KO mice at 14 weeks. Scale bar: 100 μm; CV central vein; W white pulp; R red pulp; Leu leukemia; arrows showing infiltration. f Quantitation of CD11b+Ki67+ cells in the spleen at 14 weeks post transplantation (n = 4 Ctrl, n = 8 MLL-Af4; one-way ANOVA followed by Bonferroni’s multiple comparisons test; *P < 0.05). g (Left) Number of CD11b+ in the SP of recipient mice that received Ctrl or MLL-Af4-transduced HSPCs from WT or I3KO mice at 14 weeks (one-way ANOVA followed by Bonferroni’s multiple comparisons test; **P < 0.01). (Right) Corresponding representative FACS plots showing CD11b+ and B220+ cells in the SP.
Igf2bp3 modulates disease severity in MLL-Af4-driven leukemia
The MLL-Af4 model utilized here causes a highly penetrant, aggressive form of leukemia in mice. In timed experiments, I3KO/MLL-Af4 transplanted mice showed a highly significant, approximately fourfold reduction in spleen weights at 14 weeks post transplant compared to WT/MLL-Af4 mice (Fig. 2d). I3KO/MLL-Af4 mice showed reduced infiltration of the spleen and liver by leukemic cells, which obliterated normal tissue architecture in WT/MLL-Af4 mice (Fig. 2e). In line with this, I3KO/MLL-Af4 transplanted mice showed a significant reduction in CD11b+ cells, which were less proliferative (CD11b+Ki67+), both in the spleen (~30-fold) and BM (~2.5-fold) at 14 weeks (Fig. 2f, g; Supplementary Fig. 3f, g). Thus, Igf2bp3 deletion significantly reduces tumor burden and attenuates disease severity in MLL-Af4 transplanted mice.
Igf2bp3 is required for LIC function in vitro
Several studies highlight the importance of LICs in both human and mouse leukemia. In the MLL-Af4 model, LICs show expression of CD11b and c-Kit [17, 32, 56]. Given our findings of delayed initiation and decreased disease severity, we characterized these LICs (CD11b+c-Kit+) in I3KO/MLL-Af4 transplanted mice, finding a significant tenfold decrease in numbers in the spleen and fivefold decrease in the BM at 14 weeks (Fig. 3a, b). After confirming deletion of IGF2BP3 at the protein level in immortalized HSPCs (Lin−) from WT/MLL-Af4 and I3KO/MLL-Af4 mice, we turned to endpoint colony-forming unit assays (CFU) to characterize MLL-Af4 LIC dependence on IGF2BP3 (Fig. 3c). Deletion of Igf2bp3 resulted in an approximately twofold reduction in total colonies and a significant decrease in CFU-GM progenitors (Fig. 3d). To confirm this, we utilized an orthogonal method for CRISPR-Cas9-mediated Igf2bp3 deletion. Briefly, Lin− cells from Cas9-GFP mice were transduced with MSCV-MLL-Af4 virus. After selection, MLL-Af4 Cas9-GFP Lin− cells were transduced with a retroviral vector containing either a NT sgRNA or sgRNA targeting Igf2bp3 (I3sg) (Fig. 3e). Importantly, Igf2bp3 is deleted after MLL-Af4 transformation, a distinction from the prior method (Fig. 3f, g). Deletion of Igf2bp3 led to a significant reduction in total colony numbers and various colony morphologies (Fig. 3h). The differences in overall colony-forming capacity between the two systems are likely the result of utilizing different methodologies, but in both systems, Igf2bp3 deficiency led to decreased colony formation.

a Quantification of CD11b+c-Kit+ cells in the spleen of recipient mice at 14 weeks post transplantation (n = 4 Ctrl, n = 8 MLL-Af4; one-way ANOVA followed by Bonferroni’s multiple comparisons test; **P < 0.01). b Quantitation of CD11b+c-Kit+ cells in the BM 14 weeks post transplantation (n = 4 Ctrl, n = 8 MLL-Af4; one-way ANOVA followed by Bonferroni’s multiple comparisons test; **P < 0.01, ***P < 0.001). c Expression of IGF2BP3 of in WT/MLL-Af4 and I3KO/MLL-Af4 immortalized Lin− cells at the protein level. d Colony formation assay of WT/MLL-Af4 and I3KO/MLL-Af4 immortalized Lin− cells (t test; **P < 0.01). e Schematic of collection of Cas9-GFP MLL-Af4 Lin− cells and CRISPR-Cas9-mediated deletion of Igf2bp3. f Expression of Igf2bp3 in Cas9-GFP MLL-Af4 Lin− cells in nontargeting (NT) and Igf2bp3 deleted (I3sg) cells by RT-qPCR. g Expression of IGF2BP3 in NT and I3sg Cas9-GFP MLL-Af4 Lin− cells at the protein level. h Colony formation assay of NT and I3sg deleted Cas9-GFP MLL-Af4 Lin− cells (t test; *P < 0.05, **P < 0.01, ***P < 0.001).
Igf2bp3 is necessary for the function of MLL-Af4 LICs in vivo
Since Igf2bp3 deletion reduces LIC numbers and impairs LIC function, we next determined if Igf2bp3 affects LIC capability to initiate MLL-Af4 leukemia in vivo. First, to investigate baseline HSC function in I3KO mice, we completed competitive repopulation BMT by transplanting lethally irradiated CD45.1 recipient mice with 50% of WT or I3KO CD45.2 donor BM and 50% CD45.1 donor BM. We found no defect in engraftment over time in I3KO recipients (Supplementary Fig. 4a). Moreover, we determined no differences in multilineage hematopoietic reconstitution ability of I3KO donor cells, as immature lineages in the BM and mature lineages in the periphery were intact (Supplementary Fig. 4b–h). With no baseline differences in reconstitution by normal HSPCs, we investigated if Igf2bp3 impacted the number of effective LICs in secondary transplantation. Equal numbers (106, 105, and 104) of leukemic BM cells from WT and I3KO mice were transplanted into immunocompetent CD45.1 mice. At 4 weeks post transplantation, mice that received 106 I3KO/MLL-Af4 cells had significantly reduced donor CD45.2+ engraftment (Fig. 4a). With 105 and 104 cells, we no longer observed measurable leukemic burden in recipient mice (Fig. 4a), suggesting that LIC active cell frequency in I3KO/MLL-Af4 mice is lost between 106 and 105 cells (Fig. 4a) [57]. WBC and splenic weights were significantly decreased in I3KO/MLL-Af4 transplanted mice (Fig. 4b–d). Histologically, leukemic infiltration was absent in the spleen and liver of 105 I3KO/MLL-Af4 transplanted mice (Fig. 4e). Thus, Igf2bp3 deletion results in significant reduction in reconstitution of MLL-Af4 transplanted mice, suggesting that Igf2bp3 is necessary for the self-renewal capability of LICs in vivo.

a Percentage of CD45.2+ in the peripheral blood of secondary transplanted mice from leukemic WT/MLL-Af4 or I3KO/MLL-Af4 donor mice at 106, 105, and 104 BM cells at 4 weeks post transplantation (For all panels in this figure: n = 6 recipient mice per genotype for 106 cells and n = 10 recipient mice per genotype for 105 and 104 cells; t test; *P < 0.05, ***P < 0.001, ****P < 0.0001). b WBC from PB of secondary transplanted mice from WT/MLL-Af4 or I3KO/MLL-Af4 BM 3–4 weeks post transplant (t test; **P < 0.01). c Splenic weights of secondary transplanted mice at 4–5 weeks (t test; *P < 0.05, ***P < 0.001, ****P < 0.0001). d Images of splenic tumors in secondary mice transplanted with 10,000 BM cells from WT/MLL-Af4 mice (left) or I3KO/MLL-Af4 mice (right) at 5 weeks. e H&E staining of liver and spleen of secondary transplant recipients that received 105 cells at 4 weeks. Scale bar: liver, 200 μm; spleen, 100 μm; CV central vein, W white pulp, R red pulp, Leu leukemia; arrows showing infiltration.
IGF2BP3 supports oncogenic gene expression networks in LIC-enriched and bulk leukemia cells
To identify differentially expressed transcripts, we sequenced RNA from WT/MLL-Af4 and I3KO/MLL-Af4 Lin− and CD11b+ bulk leukemia cells after confirming expression of MLL and Igf2bp3 (Fig. 3c; Supplementary Fig. 5a–e). Differential expression analysis by DEseq2 revealed 208 upregulated and 418 downregulated transcripts in CD11b+ cells, and 189 upregulated and 172 downregulated transcripts in Lin− cells (Fig. 5a, b; Tables S3 and 4) [42]. We identified a significant enrichment in transcripts associated with the KEGG term transcriptional misregulation in cancer in both datasets, using Metascape for enrichment analyses [44] (Fig. 5c, d). Interestingly, discrete oncogenic signaling pathways were enriched in Lin− (PI3K/AKT) and CD11b+ cells (GTPase, MAPK pathway) (Fig. 5c, d). This was confirmed by GSEA, with significant enrichment for the Hallmark KRAS pathway in CD11b+ cells (Supplementary Fig. 5f) and GO Oxidative phosphorylation in Lin− cells (Supplementary Fig. 5g). To validate the RNA-seq data in Lin− cells, we focused on enriched differentially regulated genes including Csf2rb, Notch1, Cd69, and Hoxa cluster of transcripts, including Hoxa9, Hoxa10, and Hoxa7. We observed a significant decrease in steady-state mRNA levels for these transcripts in I3KO/MLL-Af4 Lin− cells by RT-qPCR (Fig. 5e). In I3KO/MLL-Af4 CD11b+ cells, we confirmed that transcripts encoding Ccnd1, Maf, Mafb, Itga6, Klf4, and Akt3 were decreased (Fig. 5f). Furthermore, we determined that there was a significant decrease in Ras GTPase activity in I3KO cells by ELISA assay (Fig. 5g). Together, these data demonstrate that IGF2BP3 plays a major role in amplifying the expression of many cancer-related genes in Lin− and CD11b+ cells.

a Volcano plot of differentially expressed genes determined using DESeq analysis on RNA-seq samples from WT/MLL-Af4 or I3KO/MLL-Af4 Lin− cells. Dotted lines represent onefold change in expression (vertical lines) and adjusted P < 0.1 cutoff (horizontal line). IGF2BP3 eCLIP-seq targets are highlighted in red. b Volcano plot of differentially expressed transcripts determined using DESeq analysis on RNA-seq samples from WT/MLL-Af4 or I3KO/MLL-Af4 CD11b+ cells. Dotted lines represent onefold change in expression (vertical lines) and adjusted P < 0.1 cutoff (horizontal line). IGF2BP3 eCLIP-seq targets are highlighted in red. c GO Biological Processes and KEGG pathway enrichment determined utilizing the Metascape enrichment analysis webtool on MLL-Af4 Lin− IGF2BP3 DESeq dataset with an adjusted P < 0.05 cutoff. d GO biological processes and KEGG pathway enrichment determined utilizing the Metascape enrichment analysis webtool on MLL-Af4 CD11b+ IGF2BP3 DESeq dataset with an adjusted P < 0.05 cutoff. Bar graphs are ranked by P value and overlap of terms within gene list. e Expression of leukemogenic target genes in WT/MLL-Af4 and I3KO/MLL-Af4 Lin− cells by RT-qPCR (n = 4; t test; *P < 0.05, **P < 0.01, ****P < 0.0001). f Expression of Ras signaling pathway genes in WT/MLL-Af4 and I3KO/MLL-Af4 CD11b+ cells by RT-qPCR (n = 4; t test; *P < 0.05, **P < 0.01, ***P < 0.001). g Ras GTPase activity by ELISA in WT/MLL-Af4 and I3KO/MLL-Af4 CD11b+ cells (n = 3; t test; *P < 0.05).
eCLIP analysis reveals a putative role for IGF2BP3 in precursor mRNA (pre-mRNA) splicing
To determine how IGF2BP3 modulates gene expression in MLL-Af4 leukemia, we performed eCLIP-seq (Fig. 5a, b; Tables S3 and 4; Supplementary Fig. 6a). We found that a significant fraction of the differentially expressed mRNAs are bound by IGF2BP3 (Supplementary Fig. 6b). Motif analysis confirmed an enrichment of CA-rich elements (Supplementary Fig. 6c) [58]. Although the majority of peaks were present within introns, we observed cell type-specific differences in the locations of exonic IGF2BP3 binding sites (Fig. 6a). The eCLIP data revealed numerous peaks within pre-mRNA in both Lin− and CD11b+ cells, suggesting a potential role in splicing regulation. To characterize this observation, we utilized MISO analysis to identify differentially spliced transcripts [48]. Across both cell lines, we identified hundreds of transcripts with IGF2BP3-dependent changes in alternative splicing, including 97 differential splicing events in Lin− and 261 splicing events in CD11b+ cells (Supplementary Fig. 6d). After merging all replicate eCLIP data for each cell type, we determined the position of eCLIP peaks relative to splice sites for splicing events identified by MISO (Fig. 6b). Most event types exhibited both increases and decreases in PSI, whereas intron retention (RI) events showed a consistent reduction in splicing in the I3KO/MLL-Af4 cells (Fig. 6c). A significant fraction of alternatively spliced transcripts contained IGF2BP3 binding sites in proximity of the splicing event (Supplementary Fig. 6e), strongest near the 3′ splice site (3′ss), with additional signal near the 5′ splice site. This pattern was observed for each distinct splicing event class that MISO identified, with retained introns exhibiting the strongest bias towards the 3′ss (Supplementary Fig. 6f). Notably, this positional bias in the data was noted for differentially expressed MLL-Af4 target genes, such as Hoxa9, Hoxa7, and Cd69 (Fig. 6d). To understand the impact on isoform-specific expression, RT-qPCR primers were designed to nonspecifically detect multiple isoforms or to specifically detect alternatively spliced isoforms (shorter isoforms) and full-length isoforms. As an example, reductions in both isoforms (full-length Hoxa9 and truncated Hoxa9T [59]), as well as in the total level of Hoxa9, were observed in I3KO cells (Fig. 6e). Similar reductions were observed in all isoforms for Hoxa7 and Cd69 (Fig. 6e). Furthermore, there was an alteration in the ratio of the alternative to full-length isoform for all three genes (Fig. 6f), highlighting an effect on alternative splicing. Hence, the net effect of IGF2BP3 may be multipronged—with a strong impact on steady-state mRNA levels and an additional impact on splicing—in leukemia stem and progenitor cells.

a Genomic locations of IGF2BP3 eCLIP peaks in WT/MLL-Af4 Lin− cells and CD11b+ cells. Cell type differences in location of exonic peaks were noted: in CD11b+ cells, a greater proportion of exonic peaks were found in 3′UTRs, whereas a greater proportion of peaks mapped to internal exons in Lin− cells. b Histogram showing normalized IGF2BP3 eCLIP peak counts and distance from IGF2BP3 eCLIP peak of 5′ (5ss) and 3′ (3ss) splice sites in WT/MLL-Af4 CD11b+ (top) cells and Lin− cells (bottom). c Distribution of types of alternative splicing patterns for WT/MLL-Af4 or I3KO/MLL-Af4 Lin− and CD11b+ cells using MISO analysis. Delta psi values plotted indicate difference in isoforms. The event types included are the following, with abbreviations: A3SS alternative 3′ splice sites, A5SS alternative 5’ splice sites, AFE alternative first exons, ALE alternative last exons, MXE mutually exclusive exons, RI retained introns, SE skipped exons, Bound IGF2BP3 eCLIP target. d UCSC Genome Browser snapshots of the Hoxa9, Hoxa7, and Cd69 loci. Each panel shows the exon–intron structure of the gene and unique read coverage from three eCLIP biological replicates from WT/MLL-Af4 Lin− cells. The maximum number of reads at each position is indicated to the left of each histogram. e Expression of Hoxa9, Hoxa7, and Cd69 splice variants in WT/MLL-Af4 and I3KO/MLL-Af4 Lin− cells by RT-qPCR utilizing primers which detect only its respective alternative splice isoforms (Hoxa9T, Alt), full-length isoforms (-FL), and both isoforms (n = 3–4; t test; **P < 0.01, ***P < 0.001). f Relative expression ratio of alternative splice isoform to full-length isoform (alternative/full-length) in WT/MLL-Af4 and I3KO/MLL-Af4 Lin− cells by RT-qPCR (n = 3; t test; *P < 0.05, **P < 0.01).
Discussion
Here, we have shown the central importance of the RBP IGF2BP3 in MLL-AF4-driven leukemia. MLL-AF4-driven leukemogenesis is characterized by massive transcriptional dysregulation [8]. We confirm here that Igf2bp3 is a direct transcriptional target MLL-AF4. Interestingly, we determined that IGF2BP3 itself seems to positively regulate MLL-AF4 transcriptional targets. Together, these data suggest that IGF2BP3 and MLL-AF4 form a novel posttranscriptional feed-forward loop, enhancing leukemogenic gene expression. It is not clear if IGF2BP3 may play a role in other leukemia subtypes given its relatively restricted pattern of expression in MLL-translocated leukemia. However, IGF2BP3 overexpression is noted in a wide range of cancer types—with different oncogenic transcriptional programs—and further work is needed to define whether this paradigm may be operant in other hematologic and nonhematologic cancer.
Our prior work showed that IGF2BP3 is required for B-ALL cell survival and overexpression in BM of mice leads to a pathologic expansion of HSPCs [22]. Here, we found that deletion of Igf2bp3 in MLL-Af4 leukemia caused a striking delay in leukemia development and significantly increased the survival of MLL-Af4 mice. Furthermore, Igf2bp3 deficiency greatly attenuated the aggressiveness of leukemic disease. Given that MLL-Af4 drives an AML in mice [32], our current work suggests that IGF2BP3 is a powerful modulator of the leukemic phenotype in the myeloid lineage, in addition to the previously observed effects in human B-ALL cells. The lineage of the leukemia induced by MLL-Af4 in mice may be a limitation of the study, as we cannot conclude an in vivo function for Igf2bp3 in murine B-ALL. However, Lin et al. showed that there were important pathogenetic similarities between the MLL-Af4-induced pro-B-ALL and AML in mice [32]. In this light, MLL-AF4 leukemia in humans often shows lineage infidelity and plasticity, which has led to difficulties with targeted therapy [2, 60, 61]. We propose that IGF2BP3 may prove to be a valuable therapeutic target in MLL-AF4 leukemia, given its function in the pathogenesis of this unique molecular subtype of acute leukemia.
In this study, Igf2bp3 regulated the numbers and function of LICs. Importantly, the effect of Igf2bp3 deletion was restricted to LICs and did not significantly impact normal HSC function. Deletion of Igf2bp3 led to an MLL-Af4 LIC disadvantage in vivo and in vitro. LICs have been defined as cells that can self-renew and have the capability to produce downstream bulk leukemia cells, and their persistence is thought to contribute to relapse after treatment in several different leukemia subtypes [62]. However, the details of human LICs in MLL-AF4 leukemia are less well known [28, 63]. The role of IGF2BP3 in such cells and in relapse of leukemia is of great interest and a future direction for our work.
Previously, we discovered IGF2BP3 interacts primarily with the 3′UTR of target transcripts via iCLIP-seq [22]. Unexpectedly in this study, we determined IGF2BP3 targets transcripts within intronic regions and splice sites in addition to the 3′UTR. These findings may result from utilizing the improved eCLIP technique and the implementation of the technique on primary cells instead of cell lines. Of note, a recent study showed IGF2BP3 may regulate alternative splicing of PKM in lung cancer [64]. We also found IGF2BP3-dependent dynamic splicing events, including retained introns, alternative 3′ss, and skipped exons. Intron retention has been reported to be a mechanism of transcriptome diversification in cancer and, specifically, leukemia [65, 66]. Moreover, studies have highlighted the importance of splicing to mRNA export, and that splicing factor mutations, such as those in U2AF1, result in translational misregulation in myeloid malignancy [67, 68]. Our unexpected, novel discovery, together with our prior work, shows that IGF2BP3 likely regulates specific mRNA operons and functions at multiple posttranscriptional levels, as has been described for other RBPs [69].
As an RBP, IGF2BP3 function is intimately connected to the underlying transcriptional program—IGF2BP3 can only act on specifically induced transcripts in the cell type where it is expressed. Hence, the unique gene sets that are bound and regulated by IGF2BP3 in Lin− and CD11b+ cells are not entirely unexpected, given that transcription changes as LICs differentiate into bulk leukemic cells. This is similar to miRNAs, which posttranscriptionally regulate distinct gene expression programs in distinct cell types [70]. The significant enrichment of IGF2BP3-bound mRNAs in differentially regulated and differentially spliced transcripts confirms a direct regulatory effect. However, further work is required to confirm functional relationships between the specific transcripts that are regulated and the phenotypic effects driven by IGF2BP3.
IGF2BP3 differentially regulated transcripts included MLL-AF4 target genes Hoxa9, Hoxa10, Hoxa7, and Cd69 [32]. HOXA9, HOXA10, and HOXA7 are induced by MLL-AF4 and HOXA9 is required for MLL-rearranged leukemia survival [71]. We determined significant downregulation of both alternatively spliced and full-length isoforms for Hoxa9, Hoxa7, and Cd69. The relationship between leukemogenesis and splicing regulation is complex—while Hoxa9T, the homeodomain-less splice variant, is not sufficient for transformation alone, it is required with full-length Hoxa9 for leukemogenic transformation [59, 72]. Thus, Igf2bp3 may act through alteration of splicing regulation and upregulation of MLL-Af4 target leukemogenic genes to promote leukemogenesis and impact MLL-Af4 LIC function. Importantly, Igf2bp3 is not required for steady-state hematopoiesis, in contrast to HOXA9, and may represent a more attractive therapeutic target.
In addition, we found that IGF2BP3 targets and modulates the expression of many transcripts within the Ras signaling pathway and its downstream effector pathways. RAS proteins control numerous cellular processes such as proliferation and survival and are amongst the most commonly mutated genes in cancer [73]. Interestingly, while MLL-AF4 leukemia has a paucity of additional mutations, the mutations that are present are found in the RAS signaling pathway [74]. In addition, MEK inhibitors have shown selective activity against MLL-rearranged leukemia cell lines and primary samples [75]. Hence, IGF2BP3 regulates multiple pathways known to be important in MLL-AF4 leukemia.
Here, we determined Igf2bp3 is required for the efficient initiation of MLL-Af4-driven leukemia and function of LICs. Mechanistically, IGF2BP3 binds to hundreds of transcripts and modulates their expression through posttranscriptional mechanisms including regulation of steady-state mRNA levels and pre-mRNA splicing. We describe a novel positional bias for IGF2BP3 binding in leukemic cells isolated from an in vivo model, a notable advance in the field. In summary, IGF2BP3 is an amplifier of leukemogenesis by targeting and regulating the leukemic transcriptome initiated by MLL-AF4, thereby controlling multiple critical downstream effector pathways required for disease initiation and severity. Our findings highlight IGF2BP3 as a necessary regulator of MLL-AF4 leukemia and a potential therapeutic target for this disease.
Data availability
Data have been deposited onto the NCBI Gene Expression Omnibus repository (GSE156115).
References
Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33.
Rayes A, McMasters RL, O’Brien MM. Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer. 2016;63:1113–5.
Pui C-H, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29:551–65.
Jude CD, Climer L, Xu D, Artinger E, Fisher JK, Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–37.
Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, et al. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–17.
Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–28.
McMahon KA, Hiew SYL, Hadjur S, Veiga-Fernandes H, Menzel U, Price AJ, et al. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007;1:338–45.
Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–72.
Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med. 2004;10:500–7.
Marschalek R. Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol. 2011;152:141–54.
Takahashi S, Yokoyama A. The molecular functions of common and atypical MLL fusion protein complexes. Biochim Biophys Acta. 2020;1863:194548.
Yokoyama A. Transcriptional activation by MLL fusion proteins in leukemogenesis. Exp Hematol. 2017;46:21–30.
Slany RK. The molecular biology of mixed lineage leukemia. Haematologica. 2009;94:984–93.
Krivtsov AV, Hoshii T, Armstrong SA. Mixed-lineage leukemia fusions and chromatin in leukemia. Cold Spring Harb Perspect Med. 2017;7:a026658.
Meyer C, Burmeister T, Gröger D, Tsaur G, Fechina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–84.
Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–68.
Somervaille TCP, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM. et al. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009;4:129–40.
Guenther MG, Lawton LN, Rozovskaia T, Frampton GM, Levine SS, Volkert TL, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;22:3403–8.
Wallace JA, Kagele DA, Eiring AM, Kim CN, Hu R, Runtsch MC, et al. miR-155 promotes FLT3-ITD-induced myeloproliferative disease through inhibition of the interferon response. Blood. 2017;129:3074–86.
Elcheva IA, Spiegelman VS. Targeting RNA-binding proteins in acute and chronic leukemia. Leukemia. 2021;35:360–76.
Park S-M, Gönen M, Vu L, Minuesa G, Tivnan P, Barlowe TS, et al. Musashi2 sustains the mixed-lineage leukemia–driven stem cell regulatory program. J Clin Investig. 2015;125:1286–98.
Palanichamy JK, Tran TM, Howard JM, Contreras JR, Fernando TR, Sterne-Weiler T, et al. RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J Clin Investig. 2016;126:1495–511.
Mueller F, Bommer M, Lacher U, Ruhland C, Stagge V, Adler G, et al. KOC is a novel molecular indicator of malignancy. Br J Cancer. 2003;88:699–701.
Schaeffer DF, Owen DR, Lim HJ, Buczkowski AK, Chung SW, Scudamore CH, et al. Insulin-like growth factor 2 mRNA binding protein 3 (IGF2BP3) overexpression in pancreatic ductal adenocarcinoma correlates with poor survival. BMC Cancer. 2010;10:59.
Lochhead P, Imamura Y, Morikawa T, Kuchiba A, Yamauchi M, Liao X, et al. Insulin-like growth factor 2 messenger RNA binding protein 3 (IGF2BP3) is a marker of unfavourable prognosis in colorectal cancer. Eur J Cancer. 2012;48:3405–13.
Stoskus M, Gineikiene E, Valceckiene V, Valatkaite B, Pileckyte R, Griskevicius L. Identification of characteristic IGF2BP expression patterns in distinct B-ALL entities. Blood Cells Mol Dis. 2011;46:321–6.
Chen W, Li Q, Hudson WA, Kumar A, Kirchhof N, Kersey JH. A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood. 2006;108:669–77.
Metzler M, Forster A, Pannell R, Arends MJ, Daser A, Lobato MN, et al. A conditional model of MLL-AF4 B-cell tumourigenesis using invertor technology. Oncogene. 2006;25:3093–103.
Bursen A, Schwabe K, Rüster B, Henschler R, Ruthardt M, Dingermann T, et al. The AF4·MLL fusion protein is capable of inducing ALL in mice without requirement of MLL·AF4. Blood. 2010;115:3570–9.
Tamai H, Miyake K, Takatori M, Miyake N, Yamaguchi H, Dan K, et al. Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia. 2011;25:888–91.
Montes R, Ayllon V, Gutierrez-Aranda I, Prat I, Hernandez-Lamas MC, Ponce L, et al. Enforced expression of MLL-AF4 fusion in cord blood CD34+ cells enhances the hematopoietic repopulating cell function and clonogenic potential but is not sufficient to initiate leukemia. Blood. 2011;117:4746–58.
Lin S, Luo Roger T, Ptasinska A, Kerry J, Assi Salam A, Wunderlich M, et al. Instructive role of MLL-fusion proteins revealed by a model of t(4;11) pro-B acute lymphoblastic leukemia. Cancer Cell. 2016;30:737–49.
Janardhan HP, Milstone ZJ, Shin M, Lawson ND, Keaney JF Jr., Trivedi CM. Hdac3 regulates lymphovenous and lymphatic valve formation. J Clin Investig. 2017;127:4193–206.
Fernando TR, Contreras JR, Zampini M, Rodriguez-Malave NI, Alberti MO, Anguiano J, et al. The lncRNA CASC15 regulates SOX4 expression in RUNX1-rearranged acute leukemia. Mol Cancer. 2017;16:126.
Kumar A, Hellen J, Tuong Tiffany M, Tran Tasha L, Lin D, Casero MO, et al. Focused CRISPR-Cas9 genetic screening reveals USO1 as a vulnerability in B-cell acute lymphoblastic leukemia. Scientific Reports 2021;11. https://doi.org/10.1038/s41598-021-92448-w.
O’Connell RM, Balazs AB, Rao DS, Kivork C, Yang L, Baltimore D. Lentiviral vector delivery of human interleukin-7 (hIL-7) to human immune system (HIS) mice expands T lymphocyte populations. PLoS ONE. 2010;5:e12009.
de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, et al. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–25.
Heffner CS, Herbert Pratt C, Babiuk RP, Sharma Y, Rockwood SF, Donahue LR, et al. Supporting conditional mouse mutagenesis with a comprehensive cre characterization resource. Nat Commun. 2012;3:1218.
Joseph C, Quach Julie M, Walkley Carl R, Lane Steven W, Lo Celso C, Purton, et al. Deciphering hematopoietic stem cells in their niches: a critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell. 2013;13:520–33.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinform. 2009;25:1–4.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24:1461–2.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523.
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545.
Xiao Y, Hsiao T-H, Suresh U, Chen H-IH, Wu X, Wolf SE, et al. A novel significance score for gene selection and ranking. Bioinformatics. 2014;30:801–7.
Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–15.
Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, Parra M, et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol. 2013;20:1434–42.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan W-I, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–33.
Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné M-C, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37.
Elcheva IA, Wood T, Chiarolanzio K, Chim B, Wong M, Singh V, et al. RNA-binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia. 2020;34:1354–63.
Hardy RR, Hayakawa KB. Cell development pathways. Annu Rev Immunol. 2001;19:595–621.
Somervaille TCP, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell. 2006;10:257–68.
Brien CA, Kreso A, Jamieson CHM. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16:3113.
Schneider T, Hung L-H, Aziz M, Wilmen A, Thaum S, Wagner J, et al. Combinatorial recognition of clustered RNA elements by the multidomain RNA-binding protein IMP3. Nat Commun. 2019;10:2266.
Stadler CR, Vegi N, Mulaw MA, Edmaier KE, Rawat VPS, Dolnik A, et al. The leukemogenicity of Hoxa9 depends on alternative splicing. Leukemia. 2014;28:1838–43.
Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–10.
Haddox CL, Mangaonkar AA, Chen D, Shi M, He R, Oliveira JL, et al. Blinatumomab-induced lineage switch of B-ALL with t(4:11)(q21;q23) KMT2A/AFF1 into an aggressive AML: pre- and post-switch phenotypic, cytogenetic and molecular analysis. Blood Cancer J. 2017;7:e607.
Magee Jeffrey A, Piskounova E, Morrison Sean J. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21:283–96.
Bardini M, Woll PS, Corral L, Luc S, Wittmann L, Ma Z, et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia. 2015;29:38–50.
Xueqing H, Jun Z, Yueqiang J, Xin L, Liya H, Yuanyuan F, et al. IGF2BP3 may contributes to lung tumorigenesis by regulating the alternative splicing of PKM. Front Bioeng Biotechnol. 2020;8:679.
Dvinge H, Bradley RK. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015;7:45.
Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell. 2019;35:369–84.
Akef A, Lee ES, Palazzo AF. Splicing promotes the nuclear export of β-globin mRNA by overcoming nuclear retention elements. RNA. 2015;21:1908–20.
Akef A, McGraw K, Cappell SD, Larson DR. Ribosome biogenesis is a downstream effector of the oncogenic U2AF1-S34F mutation. PLoS Biol. 2020;18:e3000920.
Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8:533–43.
Lechman Eric R, Gentner B, Ng Stanley WK, Schoof Erwin M, van Galen P, Kennedy James A, et al. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell. 2016;29:214–28.
Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, et al. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood. 2009;113:2375–85.
He M, Chen P, Arnovitz S, Li Y, Huang H, Neilly MB, et al. Two isoforms of HOXA9 function differently but work synergistically in human MLL-rearranged leukemia. Blood Cells Mol Dis. 2012;49:102–6.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22.
Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47:330.
Lavallée V-P, Baccelli I, Krosl J, Wilhelm B, Barabé F, Gendron P, et al. The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat Genet. 2015;47:1030–7.
Acknowledgements
This work was supported by the Tumor Cell Biology Training Grant NIH T32 CA009056 (TMT), Tumor Immunology Training Grant NIH T32 CA009120 (TLL), NIH/NIGMS R35 GM130361 (JRS), NIH/NCI R01 CA166450 (DSR), NIH/NCI R21 CA197441 (DSR), American Society of Hematology Bridge Grant (DSR), UCLA Jonsson Comprehensive Cancer Center Seed Grant (DSR), and STOPCancer/Barbara and Gary Luboff Mitzvah Fund Seed Grant (DSR). Flow cytometry was performed in the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research UCLA Flow Cytometry Core Resource and the UCLA JCCC/CFAR Flow Cytometry Core Facility that is supported by NIH AI-28697, P30CA016042, the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA. The authors acknowledge the support of the Chao Family Comprehensive Cancer Center Transgenic Mouse Facility (TMF) Shared Resource, supported by the National Cancer Institute of the National Institutes of Health under award number P30CA062203. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors would like to thank Jon Neumann (TMF), Michael O. Alberti, and Jorge Contreras for their expertise and helpful discussions.
Author information
Authors and Affiliations
Contributions
TMT, JSB, NN, JP, JMD, TLL, JKP, AKJ, MP, and JK performed experiments. TMT, JP, and SK analyzed results and made the figures. OS provided experimental resource. TMT and DSR designed the research and wrote the paper. TMT, JSB, NN, JP, TLL, JKP, OS, JK, JRS, and DSR reviewed and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All mouse experimental procedures were conducted with the approval of the UCLA Chancellor’s Animal Research Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tran, T.M., Philipp, J., Bassi, J.S. et al. The RNA-binding protein IGF2BP3 is critical for MLL-AF4-mediated leukemogenesis. Leukemia 36, 68–79 (2022). https://doi.org/10.1038/s41375-021-01346-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-021-01346-7
This article is cited by
-
Ppm1d truncating mutations promote the development of genotoxic stress-induced AML
Leukemia (2023)
-
RNA binding proteins in MLL-rearranged leukemia
Experimental Hematology & Oncology (2022)
-
Multi-omics analysis reveals RNA splicing alterations and their biological and clinical implications in lung adenocarcinoma
Signal Transduction and Targeted Therapy (2022)
-
RNA-binding proteins of KHDRBS and IGF2BP families control the oncogenic activity of MLL-AF4
Nature Communications (2022)
