
Abstract
The prevalence and functional impact of somatic mutations in nonleukemic T cells is not well characterized, although clonal T-cell expansions are common. In immune-mediated aplastic anemia (AA), cytotoxic T-cell expansions are shown to participate in disease pathogenesis. We investigated the mutation profiles of T cells in AA by a custom panel of 2533 genes. We sequenced CD4+ and CD8+ T cells of 24 AA patients and compared the results to 20 healthy controls and whole-exome sequencing of 37 patients with AA. Somatic variants were common both in patients and healthy controls but enriched to AA patients’ CD8+ T cells, which accumulated most mutations on JAK-STAT and MAPK pathways. Mutation burden was associated with CD8+ T-cell clonality, assessed by T-cell receptor beta sequencing. To understand the effect of mutations, we performed single-cell sequencing of AA patients carrying STAT3 or other mutations in CD8+ T cells. STAT3 mutated clone was cytotoxic, clearly distinguishable from other CD8+ T cells, and attenuated by successful immunosuppressive treatment. Our results suggest that somatic mutations in T cells are common, associate with clonality, and can alter T-cell phenotype, warranting further investigation of their role in the pathogenesis of AA.
Similar content being viewed by others


Introduction
Acquired aplastic anemia (AA) is an immune-mediated bone marrow failure syndrome in which cytotoxic T cells mediate the destruction of hematopoietic stem and progenitor cells (HSPCs) [1,2,3]. Characteristic features of immune-mediated AA are clonal alterations in HSPCs: X-chromosome skewing [4], cytogenetic abnormalities [5, 6], uniparental disomy of the 6p [7], somatic phosphatidylinositol glycan class A gene mutations leading to paroxysmal nocturnal hemoglobinuria [8], and somatic mutations in genes related to clonal hematopoiesis (CH) [9,10,11,12,13,14]. It has been suggested that some clonal changes in target cells are enriched by selective pressure from autoreactive T cells, and hence they may serve as an immune escape mechanism [1, 15].
Oligoclonal CD8+ T cells are frequently detected in AA and they are able to induce apoptosis of autologous HSPC [16, 17]. Furthermore, T cells in AA patients are known to be aberrantly active [18,19,20], but their antigenic target on HSPC remains unknown.
Previously, we and others have detected somatic mutations in T cells in patients with various immune-mediated disorders: newly diagnosed rheumatoid arthritis [21], Felty’s syndrome [22], pure red cell aplasia [23], multiple sclerosis [24, 25], chronic graft-versus-host disease [26], and T-cell large granular lymphocytic (T-LGL) leukemia [27,28,29]. In T-LGL leukemia and Felty’s syndrome activating STAT3 mutations in CD8+ T cells are the hallmark of the disease, but STAT3 mutations have also been discovered in 11% of AA patients [30].
Although the role of somatic events in lymphocytes is not known in detail in patients with autoimmune diseases, they may alter the lymphocyte phenotype and function, and in that manner contribute to the pathogenesis. Mutations may either arise de novo in mature lymphocytes during cell division or mutations of CH may drift to lymphocytes, which is a phenomenon called lympho-myeloid CH [31, 32]. In the current study, we characterized the spectrum of somatic mutations in T cells in patients with AA. Furthermore, we evaluated their origins and described their potential impact on single-cell resolution at the diagnosis, relapse, and the recovery of AA.
We show that somatic mutations in T cells are common in AA and even in healthy individuals. These findings extend our understanding of the presence of somatic mutations in nonmalignant diseases and provide basis for the further development of novel therapeutic and diagnostic approaches in lymphocyte-mediated autoimmune diseases.
Methods
Samples analyzed by immunogene panel sequencing
Peripheral blood and bone marrow were collected from patients suffering from immune-mediated AA (n = 24), see Table S1 for clinical characteristics of the cohort. Patients were recruited from the Department of Hematology in the Helsinki University Hospital and from other participating university hospitals (Karolinska Institute, Sweden, and Cleveland Clinic, Cleveland, OH, USA). This study was approved by the local Ethics Committees, and the principles of Helsinki Declaration were followed. All patients had given their written informed consent before sample collection. Age-matched healthy blood donor buffy coats were provided by Finnish Red Cross Blood Service (n = 20).
Diagnosis of AA
The diagnosis of immune-mediated AA was based on hypocellular (cellularity < 30%) bone marrow and exclusion of constitutional genetic defects and chemical and physiological damage. The severity was based on Camitta’s criteria [33]. Moderate AA was defined as decreased bone marrow cellularity and cytopenias not qualifying for severe AA and exclusion of other causes.
Sample preparation and immunogene panel sequencing
Sample preparation and immunogene panel sequencing of CD4+ and CD8+ T cells was performed as previously described [34]. A custom gene panel based on 2533 candidate genes along pathways important in innate and adaptive immunity was used. See Supplementary Methods for further information. The sequencing was performed at the Institute for Molecular Medicine Finland, HiLIFE.
Other genomic datasets
We analyzed raw whole-exome sequencing (WES) data from a previously published dataset of 37 AA patients from Yoshizato et al. [13]. In our analysis, we included CD3− and CD3+ sample from each patient. We also utilized raw WES data from skin samples of nine AA patients, sequenced as described previously [27]. WES data from skin samples were used as a part of panel of normals (PON) for filtering variants detected with WES and immunogene panel sequencing (see Supplementary Methods).
All cohorts used in somatic variant analysis are summarized in Table 1 and study design is illustrated in Fig. S1.
Identification and analysis of potentially pathogenic somatic variants
All datasets were processed and analyzed according to a previously described GATK practice [35], see Supplementary Methods for more detailed description. Following this processing, a joint recalling of newly detected variants and known hotspot variants (listed in Table S2) across all samples was done by running MuTect2 in genotyping mode. Variants detected in both CD4+ and CD8+ T cells of same patient were interpreted as lymphoid precursor (LP) variants, and the rest of the variants were categorized as CD8+ and CD4+ specific variants (list of detected variants in Table S3). Adjusted somatic mutation burden and mutational signatures were calculated and variant effects were predicted as described in Supplementary Methods.
Amplicon validation
Amplicon sequencing was performed to validate a selected set of mutations found in the immunogene panel sequencing (Table S4). Details of amplicon sequencing and analysis are provided in Supplementary Methods.
Analysis of clonal T-cell populations
Sorted CD8+ T-cell fractions of available samples were analyzed with a multiplexed PCR assay that targets the variable CDR3 region of the rearranged TCRβ locus [36]. Next-generation sequencing was performed, and data were analyzed with the ImmunoSEQ analysis tools provided by Adaptive Biotechnologies. Clonality was calculated as 1 − Shannon’s entropy for all rearrangements [37]. TCR Vβ family based flow cytometry and sorting are described in Supplementary Methods.
Single-cell gene expression and V(D)J transcript profiling and data analysis
Frozen MNC from PB or BM were sorted with BD Influx Cell sorter and the gene and V(D)J transcript profiles were studied with 10X Genomics Chromium Single Cell V(D)J and 5′ Gene Expression platform. A detailed description of sample processing and sequencing is provided in Supplementary Methods and Fig. S2.
Detailed description of bioinformatic analyses is provided in Supplementary Methods and Figs. S3–S5. Tables S5 and S6 contain lists of differentially expressed genes used in cluster annotation.
Statistical testing
Due to non-Gaussian data distributions, nonparametric tests were used, including Mann–Whitney U test in comparisons between two groups, Kruskal–Wallis in comparisons between three or more groups and Spearman’s rank test in correlation with R (3.6.0).
Results
Clonal CD8+ T-cell populations are associated with increased somatic mutation burden
We performed immunogene panel deep sequencing of CD4+ and CD8+ T cells from 24 AA patients and 20 healthy controls (age median [range], respectively: 63 [23–84] and 61 [44–66], Fig. S6). Mean sequencing depth was 299x [249x for AA and 359x for healthy samples] (Fig. S7A, B). We divided the variants to three categories: CD4+ specific or CD8+ specific variants (detected only in CD4+ or CD8+ T cells) and LP variants (detected in both CD4+ and CD8+ T cells, see “Methods”). We detected altogether 731 somatic variants in 519 genes in patients and healthy, with mean [median] of 8.3 [7] variants per sample (1.46 [1.22] variants/Mb) and mean variant allele frequency (VAF) of 4.5% (Fig. 1A). Nonsynonymous variants were enriched in AA patients compared to healthy controls (frequencies of nonsynonymous variants from all variants were 74.8% and 68.8% respectively, p = 0.058). The VAFs of CD4+ and CD8+ specific variants were not significantly different in AA patients (Fig. S7C). The complete list of variants discovered by the immunogene panel sequencing is presented in Table S3, and all nonsynonymous variants including their VAFs in individual patients are shown in Fig. S8.

A Density plot of variant allele frequencies of CD4+ and CD8+ specific variants detected with immunogene panel sequencing. B Fraction-specific somatic mutation burden in AA and healthy samples. Mann–Whitney U test was used in pairwise comparisons within each disease group and cell fraction, only statistically significant (p < 0.05) differences are marked. C Mutation burden in CD8+ T cells was associated with T-cell clonality (Spearman ρ = 0.44, p = 0.012). Each group is shown separately on the left panel (Spearman test values for AA: ρ = 0.71, p = 0.012; healthy: ρ = 0.21, p = 0.37). D Number of somatic mutations in CD8+ T cells did not have significant correlation with age at sample collection, although a positive trend was observed.
Adjusted somatic mutation burden (see Supplementary Methods) was the highest in CD8+ T cells in patients with AA (Fig. 1B). Both in AA and healthy controls, CD4+ cells had a lower mutation burden than CD8+ T cells (median for each group after outlier removal: AA CD8 1.53, AA CD4 0.87, healthy CD8 0.78, healthy CD4 0.65, Fig. 1B). In pairwise comparisons, AA CD8+ T cells had a significantly higher mutation burden compared to AA CD4+ T cells (p = 0.029). The number of variants detected in each sample did not correlate with sequencing depth (ρ = −0.15, p = 0.27, Fig. S7B).
We validated a selected set of mutations with amplicon sequencing, as shown in Table S4. Of 15 variants detected with immunopanel sequencing, 14 were confirmed with amplicon sequencing in at least one sample, if several samples from same patient were tested.
The T-cell clonality index for each CD8+ T-cell sample (see “Methods”) was defined using TCRB sequencing. The adjusted somatic CD8+ mutation burden significantly correlated with CD8+ T-cell clonality (ρ = 0.44, p = 0.012) but not with patient age (ρ = 0.22, p = 0.16) (Fig. 1C, D), despite a positive trend was observed with age and fraction-specific mutation burden in both CD8+ and CD4+ T cells (Fig. 1D, Fig. S9). When examined separately, a strong correlation was observed between the mutation load and cytotoxic T-cell clonality in AA but not in healthy controls (AA CD8 ρ = 0.71, p = 0.012, Fig. 1C).
Somatic mutations in T cells accumulate into key immune regulatory pathways
CD8+ T cells in patients with AA harbored most potentially pathogenic variants affecting the JAK-STAT signaling (Fig. 2A, B) and MAPK signaling pathways (Fig. S10), annotated by Kyoto Encyclopedia of Genes and Genomes (KEGG) [38]. Mutations in these pathways were also detected in CD4+ T cells of AA patients.

A Mean target coverage of each sample is presented in the top panel. Variants on homopolymer regions are marked with white dots. B Mutations marked on JAK-STAT signaling pathway based on KEGG database [38], complemented with closely related MAPK-Erk and PI3K-Akt-mTOR pathways. Patients with homopolymer region variants are colored with blue. Patients with pathogenic-predicted variants are marked with an asterisk (*). C Mutational signatures from pooled CD4+ and CD8+ T-cell somatic variants from each patient group.
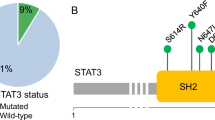
JAK-STAT pathway mutations (in CD8+ or CD4+ T cells) were detected in 75% of AA patients (18/24) and 40% of healthy controls (8/20) (Fig. 2A, B, Fig. S11). The frequency of patients with CD4+ or CD8+ specific JAK-STAT pathway mutations was higher in AA than in healthy controls (p = 0.031). In CD8+ T cells, JAK1 and STAT3 were recurrently mutated (both 8.3%, 2/24 AA patients’ CD8+ T cells). STAT3 mutations were only observed in AA patients’ CD8+ T cells. Both STAT3 mutations were p.Y640F, which is a known mutation hotspot in STAT3 exon 21 [27, 39]. JAK1 mutations were in contrast detected in both CD4+ and CD8+ T cells and across patient and healthy groups. All JAK1 mutations were located in homopolymer regions (Fig. 2A, homopolymer regions marked with white dots), known to be challenging in NGS based variant analysis.
The variant effects were predicted as described in Supplementary Methods. In the JAK-STAT pathway, genes with damaging-predicted mutations in immune-mediated AA patients included IFNG, JAK3, PTPN11, STAT3, AKT2, IL2, and PTPN2 (Fig. S11). We discovered only one damaging-predicted CD4+ or CD8+ specific JAK-STAT pathway mutation per sample in AA patients.
Potentially pathogenic CD4+ or CD8+ specific MAPK signaling pathway variants were detected in 88% of AA patients (21/24). Also, 75% of healthy controls had CD4+ or CD8+ specific variants in MAPK signaling pathway. Recurrently mutated genes in immune-mediated AA patients included ARRB2, CACNA1A, FLNB, NLK, and PPP3CC. All nonsynonymous variants in MAPK pathway genes are presented in Fig. S10.
To investigate possible mechanisms inducing somatic mutations in T cells, we assessed mutational signature analysis of the variants. We included both synonymous and nonsynonymous single-nucleotide variants (SNVs) and pooled the variants across CD4+ and CD8+ T cells per group. Age-related signature 1 and defective mismatch-repair signature 15 accounted for the majority of mutations in both groups (Fig. 2C). Signature 29, which has been linked to guanine damage, was specifically detected in AA.
For SNV types across different groups, the most frequent alteration was C > T, a characteristic transversion type of replication as well as of aging-associated mutagenesis (Fig. S12A). Most nonsilent mutations were missense (Fig. S12B) and there was no significant difference in mutation types between groups (chi-squared test p value = 0.18).
CH mutations transmit to T cells
To understand timing of mutation acquisition, an integrative analysis of our panel and previously published exome sequencing data was performed. In our data, we found 15 nonsynonymous LP variants (occurring concurrently in both CD4+ and CD8+ T cells) in 14 AA patients (Fig. 3A and list of all variants in Table S3) with a mean allele frequency of 11.4% [median 7.8%]. Older AA patients inclined to have more LP variants (Fig. S13). With the exception of NF1, these mutated genes have not been previously linked to CH. While CD4+ and CD8+ T cells harbored many mutations in other genes linked to CH (e.g., DNMT3A, BCORL1, TERT, and LAMB4), they were not concurrently detected in both T-cell fractions (Fig. 3B).

A All variants which were called from both CD4+ and CD8+ T cells in same patient and are hence interpreted to be derived from thymic precursors. MNV means two or more single-nucleotide variants in succession. B CD4+ and CD8+ specific variants in genes previously reported to be associated to CH in AA [13].
By reanalyzing WES data from Yoshizato et al. [13], we investigated mutations in CH genes in T cells (Fig. S14). Most frequent mutations in CD3− cells (T-cell depleted fraction) were in DNMT3A, BCOR/BCORL1, and ASXL1 genes (found in 7, 6, and 6 patients, respectively). In some cases, these mutations were also detected in CD3+ cells (T-cell enriched fraction) of the same patient, but in other instances they were present exclusively in CD3+ cells (BCOR and BCORL1). When detected in CD3+ cells, ASXL1 or DNMT3A mutations were always concurrently present in CD3− cells, possibly due to a clonal advantage exclusive to myeloid cells. JAK mutations (n = 2) were found in both CD3− and CD3+ fractions. One STAT3 mutation (c.G1012T, p.V338F) was detected in CD3− MNC with 3.3% VAF. This particular mutation was not found in COSMIC v90 [39].
Index patients
To understand the effect of the variants on cell phenotype, we performed paired single-cell RNA and TCRαβ sequencing (scRNA + TCRab-seq) analysis for samples from two index patients. We chose patients who carried mutations on JAK-STAT and MAPK signaling pathways and had available samples from different stages of disease.
Patient AA-4 was a 52-year-old previously healthy female who was first hospitalized due to severe H1N1-influenza infection, and afterward she gradually developed worsening pancytopenia. The first bone marrow sample was taken already at the time of H1N1 infection (because of neutropenia), but the actual diagnosis of very severe AA was made 17 months later (Fig. 4A). She was first treated with cyclosporine A (CsA) and corticosteroids (CS) without response. Two months later she was treated with equine antithymocyte globulin (ATG)-based immunosuppression which resulted in completely normalized blood counts after 4 months. Our first blood sample analyzed with scRNA + TCRab-seq was obtained immediately prior the ATG treatment and follow-up samples were received at 5 and 37 months. At the last sampling, the patient had been without any immunosuppressive treatment with normal blood counts for 25 months.

A Samples analyzed with scRNA + TCRab-seq from index patient AA-4 are marked on the clinical timeline. In addition, BM biopsies were taken at the time of influenza A infection and preceding severe AA diagnosis. Blood counts are plotted in upper four panels (B-Hb = hemoglobin (g/l), B-neut = neutrophils (E9/l), B-trom = thrombocytes (E9/l), E-retik = reticulocyte percentage of erythrocytes). Corticosteroid (prednisolone) and cyclosporine A (CyA) dosages (mg) are plotted in two panels below. Timing of ATG treatment and influenza A infection are marked with arrows. B Somatic STAT3 Y640F mutation was validated with amplicon sequencing of sorted Vbeta 5.1+ fraction, but the mutation was neither detected in Vbeta 14+ nor in Vbeta 5.1− fraction (in sample 2). Picture of BAM files opened at variant site (chr17:42322464) in Integrative Genomics Viewer. C Most expanded CTL clones of AA-4 at diagnosis (sample 1) and remission (sample 3), based on TCRb sequencing. On the x axis are different time points and y axis shows the frequency of each TCRb clone from whole CTL TCRb repertoire. At diagnosis, there were two ~20% CTL clones, which significantly diminished during immunosuppressive treatment. Vbeta 5.1 and Vbeta 14 correspond to expanded T-cell clones TCRBV05-01 and TCRBV19-01, respectively.
We found somatic STAT3 mutation p.Y640F in CD8+ cells of AA-4, which was confirmed to be restricted to one CD8+ T-cell clone (Vbeta 5.1) by flow cytometry sorting and amplicon sequencing. The variant was not detected in Vbeta 5.1− cells or in the other expanded CD8+ T-cell clone (Vbeta 14+) (Fig. 4B). The VAF in sorted Vbeta 5.1 clone was 46%, indicating heterozygosity. The STAT3 p.Y640F mutation was already detected with amplicon sequencing in the bone marrow sample taken at the time of influenza A infection (6% VAF), indicating infiltration of mutated T cells in the bone marrow at the first appearance of cytopenias. The VAF of the mutation in CD8+ T cells decreased from 16 to 1% (analyzed by the immunogene panel) after successful immunosuppression, consistent with shrinkage of the expanded T-cell clone TCRVB05-01 (corresponding to Vbeta 5.1) with TCRb sequencing (Fig. 4C).
scRNA + TCRab-seq identified 21 different transcriptomic clusters including seven CD8+ T-cell clusters and five CD4+ T clusters (Fig. 5A, B). Almost all cells from STAT3 mutated TCRBV05-01 clone (cluster 9) clustered separately. We compared the gene expression profile of this cluster to other CD8+ clusters and found cells to express aberrantly cytotoxic genes (such as PRF1, NKG7, DUSP1), genes associated with T-cell exhaustion (LAG3, TIGIT), and cytokines included in cell migration (CCL3, CCL4, CCL3L1, and CCL4L1, Fig. 5C).

A Two-dimensional UMAP projection of the transcriptomes of CD45+ lymphocyte cells pooled from three time points from peripheral blood. A total of 24,000 cells are annotated in 21 distinct clusters, 7 of which can be annotated as CD8+ T cells. *STAT3 mutated T-cell cluster. B Dot plot showing the canonical markers used to annotate the clusters. C Volcano plot showing differentially expressed genes between the TCRBV05-01 associated cluster (to the right, significant shown in red) against other CD8+ clusters (to the left, significant shown in blue). D Violin plot showing the scaled STAT3 expression in CD8+ clusters at diagnosis. E Violin plot showing the score of genes having at least one occurence of the transcription factor binding site within 4 kb of STAT3 binding site at diagnosis. F The relative abundances of CD8+ clusters during treatment. G Violin plot showing the significantly downregulated cytotoxic genes (PRF1, GZMB, GZMH, GNLY, LYZ) and stable STAT3 expression in the TCRBV05-01 associated cluster.
At diagnosis, the TCRBV05-01 associated cluster expressed the highest level of STAT3 and genes located within 4 kb of its binding site (Fig. 5D, E). After treatment, the TCRBV05-01 cluster diminished, concordant with the decreased STAT3 VAF (Fig. 5F). The therapy also affected the phenotype of the cluster, as cells lost expression of multiple cytotoxic genes, including PRF1, GZMB, and GNLY. STAT3 expression was not significantly altered between time points (Fig. 5G).
Patient AA-3 was a 58-year-old previously healthy female. She was diagnosed with severe AA after 2 years of macrocytosis and mild thrombocytopenia. Initial treatment included CsA and CS with no response, and 3 months after the diagnosis she was treated with equine ATG-based immunosuppression (Fig. 6A). No response was observed by 6 months, and rabbit ATG-based immunosuppressive therapy was administered. A year later (in summer 2014) hemoglobin was normal, and platelets and neutrophils temporarily improved. When the blood counts declined during the second year, the second remission was achieved with the reinitiation of CsA and CS; since then, she has continued on low-dose immunosuppression with moderate thrombocytopenia. The first sample was obtained before first ATG, the second sample after second ATG (before treatment response), and the third before the retreatment at the relapse.

A Samples analyzed with scRNA + TCRab-seq are marked on the clinical timeline. Blood counts are plotted in upper four panels (B-Hb = hemoglobin (g/l), B-neut = neutrophils (E9/l), B-trom = thrombocytes (E9/l), E-retik = reticulocyte percentage of erythrocytes). Corticosteroid (prednisolone) and cyclosporine A (CyA) dosages (mg) are plotted in two panels below. Timing of ATG treatments is marked with arrows. B In AA-3, TCRBV04-03 clone was expanding from sample 1 (diagnosis) to relapse (sample 3). On the x axis are different time points and y axis shows the frequency of each TCRb clone from whole CTL TCRb repertoire. C Amplicon sequencing confirmed all mutations in Vbeta 7.2+ (corresponding to TCRBV04-03) fraction of CD8+ T cells. y axis shows variant allele frequencies within Vbeta 7.2+ CD8+ T cells at different time points (shown at x axis). D Possible subclonal architecture of the mutations.
Patient AA-3 had somatic mutations in several genes, including KRAS, NFATC2, PTPN22, TNFAIP3, and JAGN1. KRAS A146P, JAGN1 T473A, and TNFAIP3 D212fs mutations were predicted to be pathogenic, and same KRAS mutation has been found in several hematologic neoplasms (genomic mutation ID in COSMIC v91: COSV55541748). Similarly to AA-4, these mutations were confirmed to be restricted to a single CD8+ T-cell clone (Vbeta 7.2), as the variants were not detected in sorted monocytes, B cells, or Vbeta 7.2− T cells. Vbeta 7.2 clone, corresponding to TCRBV04-03 in TCRb deep sequencing, increased despite immunosuppressive treatment (Fig. 6B). VAFs in sorted Vbeta 7.2 clone from sample 3 and additional sample taken a year later are shown in Fig. 6C. An additional fourth sample was only used for validation of variants with amplicon sequencing, which revealed a possible subclonal architecture, as presented in Fig. 6D.
In single-cell analysis of this patient’s CD45+ lymphocyte cells from the bone marrow samples at three time points, we identified 21 different clusters including seven CD8+ T-cell clusters but only two CD4+ T clusters (Fig. 7A, B). We identified 350 cells with matching TCRb nucleotide sequence to the TCRBV04-03. Most of the cells from the clone were CD8+ terminally differentiated effector memory cells (TEMRA), supporting the cytotoxic role of the TCRBV04-03 clone (Fig. 7C).

A Two-dimensional UMAP projection of the transcriptomes of CD45+ lymphocyte cells pooled from three time points from bone marrow. A total of 22,407 cells are annotated in 21 distinct clusters, 7 of which that can be annotated as CD8+ T cells but only 2 as CD4+ T cells. B Dot plot showing the canonical markers used to annotate the clusters. C Focused two-dimensional UMAP projection of re-embedded CD8+ lymphocyte cells. The encircled dots are the TCRBV04-03 clone colored by different time points, where turquoise is the diagnostic (sample 1), yellow is follow-up (sample 2), and red is from relapse (sample 3). Most of the TCRBV04-03 clonotype cells can be classified as 1 CD8+ TEMRA. D The proportion of TCRBV04-03 clonotype cells from the total CD8+ repertoire from the single-cell TCRab data. E The relative abundances of TCRBV04-03 clone’s phenotype during treatment. F Volcano plot showing differentially expressed genes between clonotype of interest (to the right, significant shown in red) and cells from other clonotypes from the 1 CD8+ TEMRA cluster (to the left, significant shown in blue). G Gene set enrichment analysis (GSEA) results from the differential expression analysis. Shown here are GO categories enriched (FDR qval < 0.05) to the TCRBV04-03 clone.
The TCRBV04-03 clone increased after the CsA, CS, and repeated ATG administration, and it was largest in the relapse sample (4.8% of the whole CD8+ repertoire, Fig. 7D). Similarly, the phenotype of the TCRBV04-03 clone changed from diagnosis to relapse (Fig. 7C, E).
To understand the effect of the somatic mutations on the phenotype of the TCRBV04-03 clonotype, we performed differential expression analysis between the TEMRA cells, comparing the cells from the TCRBV04-03 clonotype against the other cells in TEMRA cluster. In the relapse sample, the number of differentially expressed genes between the TCRBV04-03 clonotype in comparison to other TEMRA cells was the highest compared to other samples (56 genes up in TCRBV04-03 clonotype and 77 genes up in other TEMRA cells, genes listed in Table S7). Several genes associated with the immune response such as STAT4, NFKB1, and IFNGR1 were upregulated in the TCRBV04-03 clonotype (Fig. 7F). In gene set enrichment analysis, the upregulated pathways included type I interferon production, NF-kappaB signaling, and lymphocyte activation (Fig. 7G).
Discussion
Accumulation of somatic mutations is inherent in normal cell division and aging. Mutations can lead to abnormal cell function and uncontrolled cell proliferation, as is well established in cancer. Recently, it has also been shown that somatic mutations occur in healthy human tissues [40,41,42,43] and can play a role in the pathogenesis of other nonmalignant diseases [44]. In the current work, we addressed the key questions of somatic mutations in T cells in patients with immune-mediated AA: in which cell subset they occur, at which state of immune cell development they emerge, which genes and pathways are affected, and correlation with clinical findings.
By gene panel sequencing of T cells, we discovered that somatic variants in immune-related genes are strikingly common in T cells. The mutation burden in CD8+ T cells was higher than in CD4+ T cells. This observation is in line with previous findings [21, 22, 24] and has both technical and biological explanations. CD8+ T cells expand to large clones [45], especially following strong antigen stimuli such as EBV and CMV infections [46,47,48]. During rapid proliferation cells are more susceptible to mutations. Larger T-cell clones have higher VAFs, which facilitates mutation detection. However, in our data, the mutation burden correlated with the T-cell clonality within AA patients’ CD8+ T cells but not in healthy CD8+ T cells. This observation implies that a technical detection threshold could not alone explain higher mutation burden in CD8+ T cells of AA patients.
In accordance with our previous findings [21, 22], we were able to confirm that CD8+ specific somatic mutations were restricted to single T-cell clones, indicating post-thymic emergence. We were not able to study the clonal architecture of somatic variants in the whole cohort, as the majority of the variants had low VAFs and may have occurred in different T-cell clones. Thus, the hierarchical ordering of variants based on VAFs does not necessarily reflect the true order of mutation appearance, as other factors, such as antigen-driven expansion of T cells, may distort the hierarchy. The clonal structure of mutations have been recently studied in myeloid malignancies with single-cell DNA sequencing [49] and that approach could elucidate the role and order of different mutations in T cells as well.
We also showed that mutations in genes related to CH are common in T cells, and CH mutation transmission to T cells was frequently observed in the exome sequencing dataset of AA patients’ CD3+ and CD3− MNCs [13]. The lack of sequencing data from myeloid compartment distinctly limits our analysis. Previously, it has been shown that CH mutations are more rare in T cells compared to myeloid cells [50,51,52], but as the majority of publications on CH are based on whole blood or total MNC fraction sequencing, the mutation-specific patterns and prevalence of CH-related mutations in T cells remain to be conclusively defined.
In immune-mediated AA, we found that T-cell somatic mutations were especially frequent in the JAK-STAT and MAPK pathways. It should be noted that healthy controls also harbored mutations in both pathways, which may implicate that mutations in these pathways may be beneficial to T cells in general. Both of these pathways are important for T-cell development and function. The JAK-STAT pathway plays a central role in cytokine signaling transmitting activation and proliferation signals in T cells [53]. MAPK pathway is a major pathway induced by TCR stimulation [54] and it also regulates apoptosis [55]. However, with our panel, we were only able to analyze mutations in 2533 preselected genes, and the full spectrum of somatic mutations in T cells needs to be studied in future with exome or whole genome sequencing.
To address the effect of somatic mutations on cell phenotype, we combined single-cell RNA sequencing with TCRab sequencing. STAT3 p.Y640F mutated CD8+ T cells had a strikingly altered immunophenotype compared to other CD8+ T cells, forming their own unique phenotype cluster. The clone also showed higher expression of STAT3 target genes, which is consistent with previous studies showing STAT3 activation following p.Y640F mutation in LGL leukemia, anaplastic large cell lymphoma, inflammatory hepatocellular adenomas, and murine hematopoietic stem cells [27, 56,57,58]. STAT3 has been shown to mediate effector T-cell resistance to suppression in patients with type 1 diabetes [59].
STAT3 mutated cells had highly cytotoxic immune phenotype similarly as KRAS mutated T cells in the other AA index patient. The treatment response in these two patients was different and correlated well with findings in the clone size and phenotype. Thus, our results suggest that immunosuppressive treatment may alter not only the clone size but also the cytotoxic function of mutated CD8+ T cells, and that these changes may be linked to clinical outcomes. To generalize our results and to study effect of different mutations on clinical outcomes, a bigger cohort of AA patients would be needed with detailed clinical information available.
As the antigen target in immune-mediated AA is still unknown, we were not able to test the autoreactivity of clonal T cells. It is also undetermined whether the somatic mutations are initial drivers or secondary events following autoantigen-driven clonal expansions [60]. However, as both in our study and previous studies [21, 26] it has been shown that mutated T-cell clones persist for several years, they may maintain the aberrant immune responses. Growing evidence suggests that in myeloid cells, somatic mutations are associated with inflammatory phenotypes and increased risk for cardiovascular diseases [61,62,63]. Besides, donor CH may be related to enhanced adaptive immune response in bone marrow transplant recipients, as recipients with CH have lower relapse rate but higher rates for graft-versus-host disease [64]. Furthermore, Fraietta et al. showed that a TET2-deficient CAR-T cell clone had less T-cell exhaustion in response to stimulus and killed tumor cells more efficiently [65].
In conclusion, somatic mutations in T cells particularly in the JAK-STAT and MAPK pathways are common in patients with immune-mediated AA. Mutations are associated with T-cell clonality but not distinctly with age, differentiating them from typical CH mutations. Our findings imply that somatic mutations are one putative mechanism of aberrant T-cell activation. Treatments that target the mutated clones could have therapeutic value in immune-mediated AA.
Data availability
The scRNA + TCRab-seq raw data have been deposited at the European Genome-phenome Archive (https://ega-archive.org), which are hosted by the EBI and the CRG, under accession number EGAS00001004994. The processed scRNA + TCRab-seq data are available in ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-9969. Immunogene panel sequencing raw data are available from the corresponding author upon request.
Code availability
The source code of single-cell sequencing bioinformatic analyses is available at GitHub (https://github.com/janihuuh/aa_som_mut_manu).
References
Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–19.
Luzzatto L, Risitano AM. Advances in understanding the pathogenesis of acquired aplastic anaemia. Br J Haematol. 2018;182:758–76.
Young NS. Aplastic anemia. N Engl J Med. 2018;379:1643–56.
Mortazavi Y, Chopra R, Gordon-Smith EC, Rutherford TR. Clonal patterns of X-chromosome inactivation in female patients with aplastic anaemia studies using a novel reverse transcription polymerase chain reaction method. Eur J Haematol. 2000;64:385–95.
Afable MG, Wlodarski M, Makishima H, Shaik M, Sekeres MA, Tiu RV, et al. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011;117:6876–84.
Mikhailova N, Sessarego M, Fugazza G, Caimo A, De Filippi S, van Lint MT, et al. Cytogenetic abnormalities in patients with severe aplastic anemia. Haematologica. 1996;81:418–22.
Katagiri T, Sato-Otsubo A, Kashiwase K, Morishima S, Sato Y, Mori Y, et al. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood. 2011;118:6601–9.
Tichelli A, Gratwohl A, Würsch A, Nissen C, Speck B. Late haematological complications in severe aplastic anaemia. Br J Haematol. 1988;69:413–8.
Lane AA, Odejide O, Kopp N, Kim S, Yoda A, Erlich R, et al. Low frequency clonal mutations recoverable by deep sequencing in patients with aplastic anemia. Leukemia. 2013;27:968–71.
Kulasekararaj AG, Jiang J, Smith AE, Mohamedali AM, Mian S, Gandhi S, et al. Somatic mutations identify a subgroup of aplastic anemia patients who progress to myelodysplastic syndrome. Blood. 2014;124:2698–704.
Heuser M, Schlarmann C, Dobbernack V, Panagiota V, Wiehlmann L, Walter C, et al. Genetic characterization of acquired aplastic anemia by targeted sequencing. Haematologica. 2014;99:e165–7.
Babushok DV, Perdigones N, Perin JC, Olson TS, Ye W, Roth JJ, et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet. 2015;208:115–28.
Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. 2015;373:35–47.
Albitar A, Townsley D, Ma W, De Dios I, Funari V, Young NS, et al. Prevalence of somatic mutations in patients with aplastic anemia using peripheral blood cfDNA as compared with BM. Leukemia. 2018;32:227–9.
Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016;128:337–47.
Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004;364:355–64.
Giudice V, Feng X, Lin Z, Hu W, Zhang F, Qiao W, et al. Deep sequencing and flow cytometric characterization of expanded effector memory CD8+CD57+ T cells frequently reveals T-cell receptor Vβ oligoclonality and CDR3 homology in acquired aplastic anemia. Haematologica. 2018;103:759–69.
Li B, Guo L, Zhang Y, Xiao Y, Wu M, Zhou L, et al. Molecular alterations in the TCR signaling pathway in patients with aplastic anemia. J Hematol Oncol. 2016;9:32–9.
Li B, Liu S, Niu Y, Fang S, Wu X, Yu Z, et al. Altered expression of the TCR signaling related genes CD3 and FcεRIγ in patients with aplastic anemia. J Hematol Oncol. 2012;5:6–7.
Sheng W, Liu C, Fu R, Wang H, Qu W, Ruan E, et al. Abnormalities of quantities and functions of linker for activations of T cells in severe aplastic anemia. Eur J Haematol. 2014;93:214–23.
Savola P, Kelkka T, Rajala HL, Kuuliala A, Kuuliala K, Eldfors S, et al. Somatic mutations in clonally expanded cytotoxic T lymphocytes in patients with newly diagnosed rheumatoid arthritis. Nat Commun. 2017;8:15869.
Savola P, Brück O, Olson T, Kelkka T, Kauppi MJ, Kovanen PE, et al. Somatic STAT3 mutations in Felty syndrome: an implication for a common pathogenesis with large granular lymphocyte leukemia. Haematologica. 2018;103:304–12.
Kawakami T, Sekiguchi N, Kobayashi J, Imi T, Matsuda K, Yamane T, et al. Frequent STAT3 mutations in CD8+ T cells from patients with pure red cell aplasia. Blood Adv. 2018;2:2704–12.
Valori M, Jansson L, Kiviharju A, Ellonen P, Rajala H, Awad SA, et al. A novel class of somatic mutations in blood detected preferentially in CD8+ cells. Clin Immunol. 2017;175:75–81.
Van Horebeek L, Hilven K, Mallants K, Van Nieuwenhuijze A, Kelkka T, Savola P, et al. A robust pipeline with high replication rate for detection of somatic variants in the adaptive immune system as a source of common genetic variation in autoimmune disease. Hum Mol Genet. 2019;28:1369–80.
Kim D, Park G, Huuhtanen J, Lundgren S, Khajuria RK, Hurtado AM, et al. Somatic mTOR mutation in clonally expanded T lymphocytes associated with chronic graft versus host disease. Nat Commun. 2020;11:2246–17.
Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–13.
Jerez A, Clemente MJ, Makishima H, Koskela H, LeBlanc F, Peng Ng K, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120:3048–57.
Andersson EI, Tanahashi T, Sekiguchi N, Gasparini VR, Bortoluzzi S, Kawakami T, et al. High incidence of activating STAT5B mutations in CD4-positive T-cell large granular lymphocyte leukemia. Blood. 2016;128:2465–8.
Jerez A, Clemente MJ, Makishima H, Rajala H, Gómez-Seguí I, Olson T, et al. STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood. 2013;122:2453–9.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33.
Thol F, Klesse S, Köhler L, Gabdoulline R, Kloos A, Liebich A, et al. Acute myeloid leukemia derived from lympho-myeloid clonal hematopoiesis. Leukemia. 2017;31:1286–95.
Camitta BM, Nathan DG, Forman EN, Parkman R, Rappeport JM, Orellana TD. Posthepatitic severe aplastic anemia–an indication for early bone marrow transplantation. Blood. 1974;43:473–83.
Savola P, Martelius T, Kankainen M, Huuhtanen J, Lundgren S, Koski Y, et al. Somatic mutations and T-cell clonality in patients with immunodeficiency. Haematologica. 2020;105:2757–68
Dufva O, Kankainen M, Kelkka T, Sekiguchi N, Awad SA, Eldfors S, et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat Commun. 2018;9:1567.
Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–107.
Harden JL, Hamm D, Gulati N, Lowes MA, Krueger JG. Deep sequencing of the T-cell receptor repertoire demonstrates polyclonal T-cell infiltrates in psoriasis. F1000Res. 2015;4:460.
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–7.
Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–9.
Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–7.
Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–4.
Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–7.
Nikolaev SI, Vetiska S, Bonilla X, Boudreau E, Jauhiainen S, Rezai Jahromi B, et al. Somatic activating KRAS mutations in arteriovenous malformations of the brain. N Engl J Med. 2018;378:250–61.
Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee J-Y, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci USA. 2014;111:13139–44.
Callan MF, Steven N, Krausa P, Wilson JD, Moss PA, Gillespie GM, et al. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat Med. 1996;2:906–11.
Maini MK, Gudgeon N, Wedderburn LR, Rickinson AB, Beverley PC. Clonal expansions in acute EBV infection are detectable in the CD8 and not the CD4 subset and persist with a variable CD45 phenotype. J Immunol. 2000;165:5729–37.
Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, et al. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J Immunol. 2002;169:1984–92.
Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587:477–82.
Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484.
Arends CM, Galan-Sousa J, Hoyer K, Chan W, Jäger M, Yoshida K, et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia. 2018;32:1908–19.
Buscarlet M, Provost S, Zada YF, Bourgoin V, Mollica L, Dubé M-P, et al. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood. 2018;132:277–80.
O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. 2015;66:311–28.
Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619.
Holmström TH, Schmitz I, Söderström TS, Poukkula M, Johnson VL, Chow SC, et al. MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. EMBO J. 2000;19:5418–28.
Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27:516–32.
Pilati C, Amessou M, Bihl MP, Balabaud C, Nhieu JTV, Paradis V, et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J Exp Med. 2011;208:1359–66.
Couronné L, Scourzic L, Pilati C, Valle Della V, Duffourd Y, Solary E, et al. STAT3 mutations identified in human hematologic neoplasms induce myeloid malignancies in a mouse bone marrow transplantation model. Haematologica. 2013;98:1748–52.
Ihantola E-L, Viisanen T, Gazali AM, Näntö-Salonen K, Juutilainen A, Moilanen L, et al. Effector T cell resistance to suppression and STAT3 signaling during the development of human type 1 diabetes. J Immunol. 2018;201:1144–53.
Kerr CM, Clemente MJ, Chomczynski PW, Przychodzen B, Nagata Y, Adema V, et al. Subclonal STAT3 mutations solidify clonal dominance. Blood Adv. 2019;3:917–21.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–21.
Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–7.
Frick M, Chan W, Arends CM, Hablesreiter R, Halik A, Heuser M, et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. 2019;37:375–85
Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–12.
Acknowledgements
The authors would like to thank Hanna Lähteenmäki, Tiina Kasanen, and Jay Klievink from the Hematology Research Unit Helsinki for valuable technical assistance. The authors would also like to thank Noora Aarnio from the Biomedicum Flow Cytometry for her expertise and assistance with cell sorting. Jenni Lahtela and Emma Saarinen from Technology Centre of Institute of Molecular Medicine Finland (FIMM) are acknowledged for their excellent work on single-cell sequencing. Single-cell sequencing, immunogene panel sequencing, and skin sample WES were performed at FIMM Single-cell Analytics and Sequencing units, which are supported by HiLIFE and Biocenter Finland. The authors acknowledge Finnish Red Cross Blood Service for providing healthy control samples. The authors acknowledge Helsinki Biobank for providing bone marrow biopsies and the Finnish Hematology Registry and Clinical Biobank (FHRB) for providing MNC samples of AA patients. The authors thank all the patients for their generous participation. The FHRB Biobank is supported by the Finnish Association of Hematology, Finnish Red Cross Blood Service, Institute for Molecular Medicine Finland, and participating hospitals in Finland. The authors acknowledge IT Center for Science Ltd for data storage and computational resources and the computational resources provided by the Aalto Science-IT project. The research was funded with research grants from the European Research Council (M-IMM and STRATIFY projects), Academy of Finland, Sigrid Juselius Foundation, Cancer Foundation Finland, Blood Disease Research Foundation, and Instrumentarium Science Foundation.
Funding
Open access funding provided by University of Helsinki including Helsinki University Central Hospital.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
SM has received honoraria and research funding from Novartis, Pfizer, and Bristol Myers Squibb (not related to this study). The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lundgren, S., Keränen, M.A.I., Kankainen, M. et al. Somatic mutations in lymphocytes in patients with immune-mediated aplastic anemia. Leukemia 35, 1365–1379 (2021). https://doi.org/10.1038/s41375-021-01231-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-021-01231-3
This article is cited by
-
The constitutive activation of STAT3 gene and its mutations are at the crossroad between LGL leukemia and autoimmune disorders
Blood Cancer Journal (2024)
-
Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer
Signal Transduction and Targeted Therapy (2023)
-
Lymphoid clonal hematopoiesis: implications for malignancy, immunity, and treatment
Blood Cancer Journal (2023)
-
TGF-β-p-STAT1-LAIR2 axis has a “self-rescue” role for exhausted CD8+ T cells in hepatocellular carcinoma
Cellular Oncology (2023)
-
STAT1 overexpression triggers aplastic anemia: a pilot study unravelling novel pathogenetic insights in bone marrow failure
Clinical and Experimental Medicine (2023)
