
Abstract
Ikaros encodes a transcription factor that functions as a tumor suppressor in T-cell acute lymphoblastic leukemia (T-ALL). The mechanisms through which Ikaros regulates gene expression and cellular proliferation in T-ALL are unknown. Re-introduction of Ikaros into Ikaros-null T-ALL cells resulted in cessation of cellular proliferation and induction of T-cell differentiation. We performed dynamic, global, epigenomic, and gene expression analyses to determine the mechanisms of Ikaros tumor suppressor activity. Our results identified novel Ikaros functions in the epigenetic regulation of gene expression: Ikaros directly regulates de novo formation and depletion of enhancers, de novo formation of active enhancers and activation of poised enhancers; Ikaros directly induces the formation of super-enhancers; and Ikaros demonstrates pioneering activity by directly regulating chromatin accessibility. Dynamic analyses demonstrate the long-lasting effects of Ikaros DNA binding on enhancer activation, de novo formation of enhancers and super-enhancers, and chromatin accessibility. Our results establish that Ikaros’ tumor suppressor function occurs via global regulation of the enhancer and super-enhancer landscape and through pioneering activity. Expression analysis identified a large number of novel signaling pathways that are directly regulated by Ikaros and Ikaros-induced enhancers, and that are responsible for the cessation of proliferation and induction of T-cell differentiation in T-ALL cells.
Similar content being viewed by others


Introduction
The IKZF1 gene encodes the Ikaros protein, which acts as a tumor suppressor and master regulator of hematopoiesis [1,2,3]. Impaired Ikaros function is associated with the development of high-risk B-ALL, primary immunodeficiency [4,5,6,7,8,9], as well as 5–8% of T-ALL and 11% of early T-cell precursor (ETP) leukemia [10]. The Ikaros protein is a transcription factor that regulates expression of its target genes via chromatin remodeling [11,12,13,14,15,16,17,18,19,20].
The role of Ikaros in the epigenetic regulation of gene expression has been extensively studied in normal hematopoiesis and B-ALL, but much less so in T-ALL [21,22,23,24,25,26]. Ikaros haplo-knockout mice develop T-ALL with 100% penetrance with T-cell differentiation arrested at the CD4−CD8− double-negative 3 (DN3) stage of development [27]. During the process of malignant transformation to T-ALL, Ikaros haploinsufficient thymocytes lose their remaining wild-type Ikaros allele; thus, T-ALL cells in these mice have an Ikaros-null genotype [27]. Re-introduction of Ikaros into Ikaros-null T-ALL cells from these mice results in the cessation of cellular proliferation, and induction of T-cell differentiation [28]. These data demonstrate that Ikaros is a critical tumor suppressor in T-ALL. Re-introduction of Ikaros into Ikaros-null T-ALL captures the role of Ikaros in transition from the malignant state (Ikaros-null T-ALL) to the non-malignant state (following Ikaros re-introduction). Thus, this is an optimal system for studying Ikaros tumor suppressor function and its role in the cessation of cell growth and the induction of differentiation.
In this report, we use Ikaros-null T-ALL cells to study the role of Ikaros as an epigenetic regulator of gene expression, cellular proliferation, and T-cell differentiation. To understand Ikaros activity over time, we performed extensive analyses at several time points following the re-introduction of Ikaros in Ikaros-null, T-ALL cells. Results revealed previously unknown Ikaros roles in regulating chromatin accessibility, as well as enhancer and super-enhancer activation. These data led to a new model of Ikaros function as a regulator of gene expression and the epigenetic landscape in leukemia.
Materials and methods
Cell culture and viral transduction
The JE131 cell line (referred to here as DN3) is an early T-ALL cell line derived from Ikaros knockout mice that spontaneously develop T-ALL [28]. DN3 cells were transduced using an MSCV-based bicistronic retroviral vector that expresses Ikaros and GFP. Wild-type cells as well as retrovirally transduced cells were collected at 1-, 2-, and 3-day time points for experiments.
Microarray
Total RNA was extracted using QIAGEN RNeasy Mini Kit (Qiagen, 74104, Hilden Germany). cDNA was hybridized to the mouse 430 2.0 array.
ChIP-seq
ChIP-seq assays for Ikaros and histone modifications were performed using antibodies and methods in Supplementary data and as previously described [29, 30].
ATAC-seq
ATAC-seq was performed as previously described [31] with minor changes, described in Supplementary Data. Samples were sequenced using the Illumina HiSeq 2500 at the sequencing core facility at Pennsylvania State University College of Medicine.
Results
Ikaros regulates diverse cellular pathways by binding to upstream regulatory sequences
The tumor suppressor function of Ikaros was studied using an optimal system: Ikaros re-introduction into Ikaros-null mouse T-ALL cells (DN3 cells) [28]. DN3 cells were transduced with retrovirus that contains HA-tagged wild-type murine Ikaros or empty vector (as a negative control). Gene expression, chromatin accessibility, Ikaros occupancy, and global epigenomic signature was compared prior to, and during the 3 days following, Ikaros re-introduction (Figs. S1–S3). The level of Ikaros protein in DN3 cells following retroviral transduction was similar to its physiological levels in wild-type thymocytes (Fig. S4). This assures that the observed effects of Ikaros are not the result of excessive overexpression. Re-introduction of Ikaros into DN3 cells resulted in cessation of cellular proliferation (Fig. S5a) and induction of T-cell differentiation (Fig. S5b). These results support previously reported effects following Ikaros re-introduction into Ikaros-null T-ALL [28].
Analysis of genome-wide occupancy revealed dynamic, Ikaros-mediated changes in the regulation of gene expression. To better characterize the interactions between Ikaros and DNA elements, we divided the genome into four categories: Gene Body (GENCODE annotated gene body regions), Promoter (≤ 3 kb up and down stream of TSS regions for each annotated gene), Enhancer (> 3 kb away from TSS with overlapping H3K4me1 peak signal regions), and Gene Desert (the remainder of the genome). Ikaros binds genome-wide to all four categories of DNA elements and, despite similar Ikaros expression at days 1, 2, and 3, the number of binding sites in each class is significantly larger at day 1 as compared with day 2 and day 3 (Fig. 1a) Ikaros primarily binds to promoter/enhancer regions at day 1 and day 2, but predominantly binds to gene desert regions at day 3 (Fig. 1b). Interestingly, Ikaros occupancy at the promoters of its target genes is quite diverse during the 3 days following its re-introduction (Fig. 1c). This suggests that Ikaros transiently and sequentially regulates diverse sets of its target genes. Motif analysis of ChIP-seq peak regions shows enrichment for Ikaros consensus binding sequences (–GGAA– and –GGGA–) (Fig. 1d, Fig. S6). Ikaros binding to the promoters of its target genes on day 1 can result in either transcriptional repression or activation (Fig. 1e).

Dynamic regulation of gene expression by Ikaros following re-introduction into Ikaros-null T-ALL cells. a, b Dynamic changes in Ikaros binding to different genome regions. Number of Ikaros binding peaks within Promoters, Enhancers, Gene Body and Gene Desert regions following Ikaros re-introduction. c Ikaros binds the regulatory elements of different target genes during each of the days following its re-introduction into DN3 cells. The number of Ikaros target genes during each day is shown. d Motif analysis of Ikaros binding sites at day 1. e Differentially expressed gene analysis at day 1 vs. day 0
Gene ontology (GO) term (Fig. S7) and pathway enrichment analysis (Fig. S8) as well as GSEA (gene set enrichment analysis) (Figs. S9-S10) showed Ikaros regulating stem cell pluripotency, multiple cancer pathways, and T-cell differentiation. Together, these data demonstrate that Ikaros has the strongest effect immediately after re-introduction into leukemia cells. This suggests that alterations in gene expression and the epigenetic landscape (described below) following Ikaros re-introduction are a result of its tumor suppressor activity. Results show that Ikaros can regulate different sets of genes depending on the stage of differentiation.
Ikaros has pioneering activity
A “typical” transcription factor (TF) binds to open chromatin resulting in altered expression of its target genes [32, 33]. However, a few TFs are able to bind to condensed chromatin, and induce chromatin de-condensation and increased accessibility [34, 35]. This is often associated with the induction of chromatin modifications that lead to the early activation process, or “gene priming”, which is characterized by de novo formation of the H3K4me1 mark [32, 36, 37]. Such TFs are called Pioneer TFs and usually function as master regulators of tissue differentiation (e.g., Gata and FoxA factors) [32, 38,39,40]. Because Ikaros functions as a master regulator of hematopoiesis, we tested the hypothesis that Ikaros functions as a Pioneer TF to produce de novo open chromatin. Chromatin accessibility was measured using ATAC-seq [31, 41]. To determine if a transcription factor has pioneer activity, it is essential to determine the chromatin state before and after the factor is expressed in a cell [32]. Binding of a transcription factor to closed chromatin that results in the formation of open chromatin is the critical test for pioneer activity [34,35,36]. Our data show that Ikaros binding at day 1 following its re-introduction in Ikaros-null T-ALL cells induces de novo open chromatin (positive ATAC-seq signal) in over 3400 regions with previously closed chromatin (negative ATAC-seq signal) at day 0 (prior to Ikaros re-introduction) (Fig. 2a). Induction of open chromatin via Ikaros binding was associated with de novo formation of H3K4me1, H3K4me3, and, to a lesser extent, H3K27ac marks (Fig. 2a, b). Motif analysis showed enrichment of Ikaros’ core-binding motif in the de novo ATAC-seq peak regions (Fig. 2c). Most of the Ikaros-induced de novo open chromatin regions are located within promoters or enhancers (Fig. 2d). Ikaros-induced de novo open chromatin at distal regulatory regions regulates genes involved in cellular differentiation and signaling pathways that control cellular proliferation (Fig. 2e, f).

Ikaros functions as a pioneer transcription factor. a Heatmaps of Ikaros ChIP-seq, ATAC-seq, H3K4me1 and H3K27Ac ChIP-seq signals at open chromatin regions gained at day 1 vs. day 0. b Examples of de novo open chromatin regions and associated genes. Ikaros-induced epigenetic changes are shaded in gray. c Motif analysis for de novo open chromatin regions. d De novo open chromatin regions classified by function of the DNA element. e, f Gene ontology and pathway enrichment analysis of genes associated with de novo open chromatin regions
We analyzed whether Ikaros binding that produces de novo open chromatin has a lasting effect. Results showed that Ikaros binding at day 1 is transient, and that over 90% of Ikaros-induced de novo open chromatin sites are not occupied by Ikaros at day 2 and day 3 (Fig. S11). However, despite a lack of Ikaros occupancy, open chromatin induced by Ikaros binding at day 1 continued to persist at over 600 sites at day 2, and at over 200 sites at day 3 (Fig. S11). Overall, the data in Fig. 2 demonstrate that Ikaros binds to closed chromatin which results in de novo chromatin accessibility at promoters and distal regulatory regions and the priming of these elements by induction of H3K4me1. The transient Ikaros binding at day 1 has a long-lasting effect on chromatin accessibility during induction of differentiation and cessation of cellular proliferation. These data support Ikaros’ role as a Pioneer TF. The long-lasting effect of Ikaros pioneering activity suggests that Ikaros sets the stage for the continuation of T-cell differentiation.
Ikaros regulates de novo formation and depletion of enhancers
Ikaros re-introduction into Ikaros-null T-ALL, results in the formation of a large number of de novo enhancers as evidenced by de novo H3K4me1 enriched sites, as compared with wild-type DN3 cells (Fig. 3a). The de novo-formed enhancers showed enrichment of Ikaros’ core-binding motif (Fig. 3b). Analysis revealed over 6000 de novo enhancers with Ikaros occupancy and H3K4me1 enrichment (Fig. 3c, d). De novo enhancer regions are significantly enriched with Ikaros peaks, as compared with random genomic regions (Fig. S12). These data strongly suggest that Ikaros DNA binding at day 1 directly induces formation of de novo enhancers, and directly determines the enhancer landscape during induction of T-cell differentiation.

Ikaros binding induces de novo formation and depletion of enhancers. a Heatmaps of H3K4me1 ChIP-seq signals at genome-wide de novo enhancer regions at day 1, after Ikaros introduction, vs. day 0. Signals are centered on H3K4me1 peaks at day 1. b Motif enrichment analysis for de novo enhancer regions. c Heatmaps of Ikaros and H3K4me1 ChIP-seq signals at de novo-formed enhancers with Ikaros direct binding regions at day 0 vs. day 1. Signals are centered on Ikaros peaks at day 1. d Examples of de novo-formed enhancers that are induced by Ikaros binding. Ikaros-induced epigenetic changes are shaded in gray. e Heatmaps of H3K4me1 ChIP-seq signals at genome-wide depleted enhancer regions at day 1, after Ikaros introduction, vs. day 0. Signals are centered on H3K4me1 peaks at day 0. f Motif enrichment analysis for depleted enhancer regions. g Heatmaps of Ikaros and H3K4me1 ChIP-seq signals at depleted enhancers with Ikaros direct binding regions on day 1 vs. day 0. Signals are centered on Ikaros peaks at day 1
Further analyses showed that re-introduction of Ikaros into DN3 cells can produce the opposite effect—a depletion of a large number of existing enhancers, as evidenced by the loss of H3K4me1 enrichment (Fig. 3e). Depleted enhancers showed enrichment of the Ikaros’ consensus DNA-binding motif (Fig. 3f). Over 800 depleted enhancers showed a loss of H3K4me1 enrichment concomitant with direct Ikaros binding (Fig. 3g, S13). A permutation assay confirmed significantly higher enrichment for Ikaros’ occupancy at depleted enhancers as compared with random genomic regions (Fig. S14). Together, these results indicate a direct role for Ikaros in enhancer depletion.
A dynamic analysis of the epigenetic signature and Ikaros occupancy of de novo and depleted enhancers showed that Ikaros binding to these regions is transient. Most of the de novo and depleted enhancers showed no Ikaros binding at day 2 (Figures S15-S16). However, the epigenetic changes induced by Ikaros binding at day 1 were preserved at day 2 in 37% and 40% of de novo and depleted enhancers, respectively. At day 3—2 days after the loss of Ikaros binding—over 20% of Ikaros-induced de novo enhancers and 36% of Ikaros-depleted enhancers maintained the epigenetic signature that was induced by Ikaros binding to these regions at day 1. These data demonstrate the long-lasting effect of Ikaros binding on the de novo formation and depletion of enhancers.
Ikaros binding induces de novo formation of active enhancers
Enhancers can be in a “poised” state (enriched for H3K4me1 but not H3K27ac) or an “active” state (enriched for both H3K4me1 and H3K27ac) [33, 42]. Poised enhancers do not affect gene expression, while active enhancers positively regulate transcription of target genes. We analyzed the effect of Ikaros on the de novo formation of active (de novo-activated) enhancers and on the activation of previously poised (poised-activated) enhancers. Re-introduction of Ikaros into DN3 cells results in the formation of over 13,000 de novo-activated enhancers (Fig. 4a). De novo-activated enhancers have a positive effect on expression of genes near their location (Fig. 4b) and have enrichment for the Ikaros core binding motif (Fig. 4c). Over 3000 de novo-activated enhancers had de novo enrichment of both H3K4me1 and H3K27ac concomitant with Ikaros occupancy (Fig. 4d, e). This represents 22% of all de novo-activated enhancers and is consistent with significantly higher enrichment for Ikaros occupancy at these regions (Fig. S17). This shows that Ikaros binding directly induced the de novo formation of a very large number of active enhancers as a part of its tumor suppressor/differentiation function.

Ikaros binding induces de novo formation of active enhancers. a Heatmaps of H3K4me1 and H3K27Ac signals at genome-wide de novo formed activated enhancer regions at day 1, after Ikaros introduction, vs. day 0. Signals are centered on H3K4me1 peak at day 1. b Expression values for genes associated with activated enhancer regions at day 1 (red) vs. day 0 (black). c Motif analysis for activated enhancer regions. d Heatmaps of Ikaros, H3K4me1, and H3K27Ac ChIP-seq signals at de novo formed activated enhancers with Ikaros direct binding regions at day 1 vs. day 0. Signals are centered on Ikaros peaks at day 1. e Examples of de novo-formed active enhancers that are induced by Ikaros binding and associated genes
Ikaros induces activation of “poised” enhancers
The re-introduction of Ikaros resulted in the activation of 5541 enhancers that were previously in a “poised” state (H3K4me1+/H3K27ac–) (Fig. 5a). Activation of poised enhancers positively regulates expression of the genes near these enhancers (Fig. 5b). A motif analysis revealed a strong enrichment for the core Ikaros binding motif (Fig. 5c). Ikaros binding directly resulted in the activation of 942 enhancers that were previously in a “poised” state (Fig. 5d, e, S18). This represents 18% of all activated enhancers and indicates that Ikaros plays a critical role in the direct activation of poised enhancers.

Ikaros binding induces activation of “poised” enhancers. a Heatmaps of H3K4me1 and H3K27Ac ChIP-seq signals at genome-wide activated enhancer regions (from previously “poised state”) at day 1, after Ikaros introduction, vs. day 0. Signals are centered on H3K4me1 peaks at day 1. b Increased expression of genes associated with activated enhancer regions at day 1 (red) vs. day 0 (black). c Motif analysis for activated enhancer regions. d Heatmaps of Ikaros, H3K4me1, and H3K27Ac ChIP-seq signals at activated enhancer regions (from the previously “poised” state at day 0) with Ikaros direct binding at day 0 vs. day 1 after induction of Ikaros. Signals are centered on Ikaros peaks at day 1. e Examples of activated enhancers (from the previously “poised” state at day 0) that are activated by Ikaros binding and associated genes
We analyzed the dynamic effect of Ikaros binding on de novo-activated and poised-activated enhancers. Ikaros binding at day 1, which results in enhancer activation, is transient, because most Ikaros-induced active enhancers at day 1 showed no Ikaros binding at day 2 (Figs. S19-S20). However, the epigenetic changes induced by Ikaros binding at day 1 were strongly preserved at day 2 in 42% and 74.5% of de novo-activated and poised-activated enhancers, respectively. At day 3, 2 days after the loss of Ikaros binding, over 10% of Ikaros-induced de novo-activated and 39.5% of Ikaros-induced poised-to-activated enhancers maintained the epigenetic signature that was induced by Ikaros binding. These data demonstrate the long-lasting effect of Ikaros binding on enhancer activation, which is particularly pronounced on poised-to-activated enhancers (Fig. S20). Because enhancer activation has a strong positive effect on target gene expression, these results reveal a novel mechanism by which Ikaros binding regulates the expression of a very large number of genes as part of its tumor suppressor function in T-ALL.
Ikaros binding induces de novo formation of super-enhancers
Super-enhancers are potent DNA regulatory regions that contain clusters of enhancers that regulate expression of large numbers of cell-specific genes [43,44,45]. Ikaros re-introduction in DN3 cells results in de novo formation of a very large number (609) of super-enhancers at day 1, as compared with only 24 super-enhancers present in Ikaros-null DN3 leukemia cells (Fig. 6a). Over 90% of the newly formed super-enhancers showed enrichment for Ikaros occupancy (Fig. 6b, c). Gene expression analysis demonstrated higher expression levels of the genes in close proximity to super-enhancers as compared with genes near all enhancers (super-enhancers and regular enhancers combined) (Fig. 6d), which suggests that Ikaros-induced formation of super-enhancers has a profound, positive effect on expression of genes that are regulated by super-enhancers. Ikaros-induced super-enhancers regulate genes involved in both T-cell differentiation as well as chromatin organization and regulation (Fig. 6e, f).

Ikaros induces de novo formation of super-enhancers. a Super-enhancers at day 0 (black) and day 1 (red). Enhancer regions are plotted in increasing order based on their input-normalized H3K4me1 ChIP-Seq signal. Super-enhancers are defined as the population of enhancers above the inflection point of the curve. b The number of super-enhancers at day 0 (black) and day 1 (red). The number of super-enhancers at day 1, with Ikaros binding (orange) or not (blue). c Examples of newly formed, Ikaros-bound, super-enhancers. d Higher expression of the genes in close proximity to super-enhancers vs. the genes near total enhancers—including both super-enhancers and regular enhancers. e, f Gene ontology and pathway enrichment analysis of genes associated with super-enhancers
Analysis of the dynamic effects of Ikaros on the formation of super-enhancers showed that Ikaros binding that results in the de novo formation of super-enhancers is longer-lasting than Ikaros binding that results in the activation of “classic” enhancers. Over 90% of Ikaros-induced super-enhancers show Ikaros occupancy at day 2 and have preserved super-enhancer activity. This is in striking contrast to Ikaros occupancy of “regular” enhancers, which is mostly present at day 1 (Fig. S21 vs. Figs. S19-S20). At day 3, Ikaros binding to the super-enhancer regions is lost, although 55% of the regions continue to maintain an epigenetic signature characteristic of super-enhancers (Fig. S21). These data demonstrate that Ikaros binding produces a strong, long-lasting effect on the formation and maintenance of super-enhancers and suggest that super-enhancers have a critical role in tumor suppression in T-ALL.
Comparative analysis of the enhancer and super-enhancer landscape showed remarkable preservation between mouse and human T-ALL (Fig. S22). There is also a high similarity in the enhancer/super-enhancer landscape between DN3 cells, following Ikaros re-introduction, and mouse thymocytes, indicating Ikaros’ role in thymocyte differentiation.
Discussion
Re-introduction of Ikaros into Ikaros-null mouse T-ALL (DN3) cells provided an opportunity to analyze dynamic changes in Ikaros tumor suppressor activity in T-ALL. This approach identified previously unknown Ikaros functions in epigenetic regulation of gene expression:
(a) Pioneer activity: Pioneer TFs initiate the coordination of regulatory mechanisms by opening previously closed chromatin and by inducing histone modifications that allow the formation of putative enhancers (H3K4me1) or promoters (H3K4me3) [32]. Ikaros binding at day 1 resulted in the opening of over 3400 previously inaccessible chromatin sites. This is accompanied by de novo enrichment of H3K4me1 and H3K4me3 modifications, and formation of de novo enhancers and promoters. These data strongly support the conclusion that Ikaros has pioneer activity, and triggers coordinated regulation of gene expression resulting in tumor suppression and induction of T-cell differentiation. Ikaros thus joins PU.1, C/EBPa, and EBF1 as a pioneer factor in hematopoiesis [32, 46,47,48,49,50,51,52].
(b) Activation of enhancers: Putative enhancers are characterized by the presence of H3K4me1 histone modifications. In the absence of H3K27ac, these enhancers are termed “poised” or “primed” enhancers [36, 53]. The presence of H3K27ac modifications defines “active” enhancers that positively regulate transcription of their target genes [36, 53, 54]. Our data showed that Ikaros binding to DNA can either induce de novo formation of active enhancers (transforming H3K4me1–/H3K27ac– DNA sites into H3K4me1+/H3K27ac+ sites) or activate “poised” enhancers (H3K4me1+/H3K27ac–). This is a novel mechanism by which Ikaros regulates gene expression. Ikaros binding to active enhancers has been observed in the past [55, 56], but our results demonstrate for the first time that Ikaros DNA-binding directly induces the formation of active enhancers.
(c) Formation of super-enhancers: Super-enhancers are clusters of enhancers with very high levels of H3K27ac [57,58,59]. The role of super-enhancers in T-ALL and the targeting of super-enhancers as a treatment for T-ALL have been proposed [44, 45]. Our data demonstrated that Ikaros induces the formation of super-enhancers as a part of its tumor suppressor function and induction of T-cell differentiation. Because super-enhancers regulate target genes more strongly than “regular” enhancers (Fig. 6d) [26], the role of Ikaros in the formation of super-enhancers is an important, novel aspect of its function.
Our data also demonstrate a role for Ikaros in de novo enhancer formation and depletion, but this was less pronounced than the above-outlined Ikaros activities.
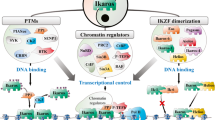
In conclusion, our results identify novel Ikaros functions in the epigenetic regulation of gene expression (Fig. 7). These include Ikaros’ ability to induce formation of activated enhancers and super-enhancers, and thus act as a positive regulator of gene expression when associated with enhancer and super-enhancer regions. It is likely that Ikaros regulates the enhancer/super-enhancer landscape by recruiting chromatin remodeling complexes. One possible candidate is the SWI/SNF complex, which is reported to associate with Ikaros. Further experiments will be directed toward identification of such complex(es). The dynamic approach used to study Ikaros’ effect on epigenetic signature, chromatin accessibility, and gene expression over several time points was crucial in uncovering these previously unknown Ikaros functions. This suggests that the optimal method for determining the function of tumor suppressors and/or oncogenes is to perform dynamic analyses of their effect in gain-of-function and/or loss-of-function experiments. Future analyses in different hematopoietic malignancies will help to provide a more complete picture of Ikaros’ role in tumor suppression.

Novel Ikaros functions in the epigenetic regulation of gene expression. Results presented here reveal novel functions of Ikaros: de novo formation or depletion of enhancers; de novo formation of active enhancers; activation of previously “poised” enhancers; and formation of super-enhancers. Ikaros shows pioneering factor activity while binding to promoter and/or enhancer regions
References
Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science. 1992;258:808–12.
Lo K, Landau NR, Smale ST. LyF-1, a transcriptional regulator that interactswith a novel class of promoters for lymphocyte-specific genes. Mollecular Cell Biol. 1991;11:5229–43.
Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–56.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–80.
Goldman FD, Gurel Z, Al-Zubeidi D, Fried AJ, Icardi M, Song C, et al. Congenital pancytopenia and absence of B lymphocytes in a neonate with a mutation in the Ikaros gene. Pedia Blood Cancer. 2012;58:591–7.
Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21:1258–66.
Kuiper RP, Waanders E, van der Velden VH, van Reijmersdal SV, Venkatachalam R, Scheijen B, et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia. 2010;24:1258–64.
van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russell LJ, et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood. 2013;122:2622–9.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63.
Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, et al. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity. 1999;10:345–55.
Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell. 1997;91:845–54.
Ge Z, Song EJ, Kawasawa YI, Li J, Dovat S, Song C. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget. 2016;7:37740–54.
Ge Z, Zhou X, Gu Y, Han Q, Li J, Chen B, et al. Ikaros regulation of the BCL6/BACH2 axis and its clinical relevance in acute lymphoblastic leukemia. Oncotarget. 2017;8:8022–34.
Ge Z, Gu Y, Xiao L, Han Q, Li J, Chen B, et al. Co-existence of IL7R high and SH2B3 low expression distinguishes a novel high-risk acute lymphoblastic leukemia with Ikaros dysfunction. Oncotarget. 2016;7:46014–27.
Ge Z, Gu Y, Zhao G, Li J, Chen B, Han Q, et al. High CRLF2 expression associates with IKZF1 dysfunction in adult acute lymphoblastic leukemia without CRLF2 rearrangement. Oncotarget. 2016;7:49722–32.
Ge Z, Guo X, Li J, Hartman M, Kawasawa YI, Dovat S, et al. Clinical significance of high c-MYC and low MYCBP2 expression and their association with Ikaros dysfunction in adult acute lymphoblastic leukemia. Oncotarget. 2015;6:42300–11.
Ge Z, Gu Y, Han Q, Zhao G, Li M, Li J, et al. Targeting high dynamin-2 (DNM2) expression by restoring ikaros function in acute lymphoblastic leukemia. Sci Rep. 2016;6:38004.
Koipally J, Renold A, Kim J, Georgopoulos K. Repression by Ikaros and Aiolos is mediated through histone deacetylase complexes. EMBO J. 1999;18:3090–100.
Song C, Pan X, Ge Z, Gowda C, Ding Y, Li H, et al. Epigenetic regulation of gene expression by Ikaros, HDAC1 and Casein Kinase II in leukemia. Leukemia. 2016;30:1436–40.
Brown KE, Baxter J, Graf D, Merkenschlager M, Fisher AG. Dynamic repositioning of genes in the nucleus of lymphocytes preparing for cell division. Mol Cell. 1999;3:207–17.
Ferreiros-Vidal I, Carroll T, Taylor B, Terry A, Liang Z, Bruno L, et al. Genome-wide identification of Ikaros targets elucidates its contribution to mouse B-cell lineage specification and pre-B-cell differentiation. Blood. 2013;121:1769–82.
Su RC, Brown KE, Saaber S, Fisher AG, Merkenschlager M, Smale ST. Dynamic assembly of silent chromatin during thymocyte maturation. Nat Genet. 2004;36:502–6.
Zhang J, Jackson AF, Naito T, Dose M, Seavitt J, Liu F, et al. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat Immunol. 2012;13:86–94.
Song C, Gowda C, Pan X, Ding Y, Tong Y, Tan BH, et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood. 2015;126:1813–22.
Hu Y, Zhang Z, Kashiwagi M, Yoshida T, Joshi I, Jena N, et al. Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev. 2016;30:1971–90.
Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell. 1995;83:289–99.
Kathrein KL, Lorenz R, Innes AM, Griffiths E, Winandy S. Ikaros induces quiescence and T-cell differentiation in a leukemia cell line. Mol Cell Biol. 2005;25:1645–54.
Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903.
Fujiwara T, O’Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, et al. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol Cell. 2009;36:667–81.
Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21 9 1–9.
Mayran A, Drouin J. Pioneer transcription factors shape the epigenetic landscape. J Biol Chem. 2018;293:13795–804.
Choukrallah MA, Matthias P. The interplay between chromatin and transcription factor networks during B cell development: who pulls the trigger first? Front Immunol. 2014;5:156.
Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014;28:2679–92.
Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–41.
Mayran A, Khetchoumian K, Hariri F, Pastinen T, Gauthier Y, Balsalobre A, et al. Pioneer factor Pax7 deploys a stable enhancer repertoire for specification of cell fate. Nat Genet. 2018;50:259–69.
Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell. 2011;8:96–105.
Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279–89.
Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, et al. Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 1998;17:244–54.
Cirillo LA, Zaret KS. An early developmental transcription factor complex that is more stable on nucleosome core particles than on free DNA. Mol Cell. 1999;4:961–9.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8.
Huang J, Liu X, Li D, Shao Z, Cao H, Zhang Y, et al. Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev Cell. 2016;36:9–23.
Shin HY. Targeting super-enhancers for disease treatment and diagnosis. Mol Cells. 2018;41:506–14.
Wong RWJ, Ishida T, Sanda T. Targeting general transcriptional machinery as a therapeutic strategy for adult T-cell leukemia. Molecules. 2018;23:5.
Wong RWJ, Ngoc PCT, Leong WZ, Yam AWY, Zhang T, Asamitsu K, et al. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood. 2017;130:2326–38.
Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, et al. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell. 2014;54:844–57.
Carotta S, Wu L, Nutt SL. Surprising new roles for PU.1 in the adaptive immune response. Immunol Rev. 2010;238:63–75.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89.
van Oevelen C, Collombet S, Vicent G, Hoogenkamp M, Lepoivre C, Badeaux A, et al. C/EBPalpha activates pre-existing and de novo macrophage enhancers during induced pre-B cell transdifferentiation and myelopoiesis. Stem Cell Rep. 2015;5:232–47.
Gyory I, Boller S, Nechanitzky R, Mandel E, Pott S, Liu E, et al. Transcription factor Ebf1 regulates differentiation stage-specific signaling, proliferation, and survival of B cells. Genes Dev. 2012;26:668–82.
Treiber T, Mandel EM, Pott S, Gyory I, Firner S, Liu ET, et al. Early B cell factor 1 regulates B cell gene networks by activation, repression, and transcription-independent poising of chromatin. Immunity. 2010;32:714–25.
Boller S, Ramamoorthy S, Akbas D, Nechanitzky R, Burger L, Murr R, et al. Pioneering activity of the C-terminal domain of EBF1 shapes the chromatin landscape for B cell programming. Immunity. 2016;44:527–41.
Cico A, Andrieu-Soler C, Soler E. Enhancers and their dynamics during hematopoietic differentiation and emerging strategies for therapeutic action. FEBS Lett. 2016;590:4084–104.
Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–54.
Katerndahl CDS, Heltemes-Harris LM, Willette MJL, Henzler CM, Frietze S, Yang R, et al. Antagonism of B cell enhancer networks by STAT5 drives leukemia and poor patient survival. Nat Immunol. 2017;18:694–704.
Unnikrishnan A, Guan YF, Huang Y, Beck D, Thoms JA, Peirs S, et al. A quantitative proteomics approach identifies ETV6 and IKZF1 as new regulators of an ERG-driven transcriptional network. Nucleic Acids Res. 2016;44:10644–61.
Vahedi G, Kanno Y, Furumoto Y, Jiang K, Parker SC, Erdos MR, et al. Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature. 2015;520:558–62.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–19.
Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34.
Acknowledgements
This work was supported by R01CA209829 (KJP and SD); R01CA213912 (SD and CS) F30CA221109 (JLP); R01DK110108 and R01CA204044 (SH); R35GM124820 and R01HG009906 (FY); the PhRMA foundation (BZ); a Hyundai Hope on Wheels Scholar Grant, and the Four Diamonds Fund of the Pennsylvania State University College of Medicine (to SD and CS); Bear Necessities Pediatric Cancer Foundation, Alex’s Lemonade Stand Foundation, and the John Wawrynovic Leukemia Research Scholar Endowment (to SD); and the St. Baldrick’s Foundation and Bear Necessities (CG).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Kimberly J. Payne is CEO and owns stock and Sinisa Dovat is on the board of directors of Elf Zone, Inc., a startup company developing therapies for B-cell acute lymphoblastic leukemia. Elf Zone, Inc. did not provide support for this work. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ding, Y., Zhang, B., Payne, J.L. et al. Ikaros tumor suppressor function includes induction of active enhancers and super-enhancers along with pioneering activity. Leukemia 33, 2720–2731 (2019). https://doi.org/10.1038/s41375-019-0474-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-019-0474-0
This article is cited by
-
IKAROS: from chromatin organization to transcriptional elongation control
Cell Death & Differentiation (2023)
-
Protein kinase CK2 – diverse roles in cancer cell biology and therapeutic promise
Molecular and Cellular Biochemistry (2023)
-
IKAROS and MENIN coordinate therapeutically actionable leukemogenic gene expression in MLL-r acute myeloid leukemia
Nature Cancer (2022)
-
Increased Incidence of IKZF1 deletions and IGH-CRLF2 translocations in B-ALL of Hispanic/Latino children—a novel health disparity
Leukemia (2021)
-
Dual targeting of MTOR as a novel therapeutic approach for high-risk B-cell acute lymphoblastic leukemia
Leukemia (2021)
