
Abstract
Current cytogenetic risk stratification in primary myelofibrosis (PMF) is two-tiered: ‘favorable’ and ‘unfavorable’. Recent studies have suggested prognostic heterogeneity within the unfavorable risk category. In 1002 consecutive patients, we performed stepwise analysis of impact on survival from individual and prognostically ordered cytogenetic abnormalities, leading to a revised three-tiered risk model: ‘very high risk (VHR)’—single/multiple abnormalities of −7, i(17q), inv(3)/3q21, 12p−/12p11.2, 11q−/11q23, or other autosomal trisomies not including + 8/ + 9 (e.g., +21, +19); ‘favorable’—normal karyotype or sole abnormalities of 13q−, +9, 20q−, chromosome 1 translocation/duplication or sex chromosome abnormality including -Y; ‘unfavorable’—all other abnormalities. Median survivals for VHR (n = 75), unfavorable (n = 190) and favorable (n = 737) risk categories were 1.2 (HR 3.8, 95% CI 2.9–4.9), 2.9 (HR 1.7, 95% CI 1.4–2.0) and 4.4 years and survival impact was independent of clinically derived prognostic systems, driver and ASXL1/SRSF2 mutations. The revised model was also effective in predicting leukemic transformation: HRs (95% CI) were 4.4 (2.0–9.4) for VHR and 2.0 (1.2–3.4) for unfavorable. The impact of driver mutations on survival was confined to favorable and that of ASXL1/SRSF2 mutations to favorable/unfavorable cytogenetic risk categories. The current study clarifies the prognostic hierarchy of genetic risk factors in PMF and provides a more refined three-tiered cytogenetic risk model.
Similar content being viewed by others

Introduction
Current prognostication in primary myelofibrosis (PMF) relies on information from clinical variables, karyotype and mutations. The prototype international prognostic scoring system (IPSS) [1] was first described in 2009 and was designed to predict survival in newly diagnosed patients. In 2010, IPSS was modified into the dynamic IPSS (DIPSS) [2], in order to allow prognostication of patients seen at any time during their clinical course. Both IPSS and DIPSS employed five clinical risk factors, including older age, anemia, leukocytosis, constitutional symptoms and circulating blasts, in order to construct a four-tiered risk categorization system. In 2011, DIPSS-plus [3] was introduced in order to account for IPSS/DIPSS-independent risk factors, including thrombocytopenia, red cell transfusion need and karyotype. The inclusion of cytogenetic information to clinically derived risk models for PMF has boosted their performance, especially in predicting leukemia-free survival [3,4,5].
Observations regarding the prognostic contribution of karyotype in PMF span over 30 years. In 1985 [6], a small study of 28 patients was unable to demonstrate a survival difference between normal and abnormal karyotype. In 1988 [7], Demory et al. first noticed the adverse impact of abnormal karyotype on survival in PMF; this was subsequently confirmed by a series of reports in 1994 [8], 1996 [9] and 1997 [10]. In 2001 [11], +8 and 12p−, and in 2005 [12], −7/7q− were specifically associated with poor survival in PMF. In 2005 [13] and 2009 [14], we underlined the prognostic distinction between ‘abnormal’ and ‘unfavorable’ karyotype. In 2010 [15], we confirmed the favorable (sole 20q−, 13q−, +9 abnormalities) and unfavorable (+8 and complex karyotype) impact of specific lesions. We validated these findings in a 2011 study [16], using 433 informative cases of PMF, and also identified sole chromosome 1 translocation/duplication as favorable and −7/7q− as unfavorable abnormalities; the particular study was used to promote the currently employed two-tiered cytogenetic risk stratification in PMF: [3] unfavorable karyotype is assigned to complex karyotype or sole or two abnormalities of +8, −7/7q−, i(17q), inv(3), −5/5q−, 12p− or 11q23 rearrangement while all other scenarios, including normal karyotype, were considered ‘favorable’ [15, 16]. Unfavorable karyotype in PMF occurs in ~14% of patients and is associated with median survival of 2 years, compared to 5.2 years in its absence; [16] unfavorable karyotype was also associated with higher risk of leukemic transformation with reported 5-year risk of 46% versus 7% in patients with ‘favorable’ karyotype [16].
The prognostic relevance of karyotype in PMF was further highlighted by the demonstration of a dismal prognosis associated with monosomal karyotype and abnormalities of inv(3) and i(17q) [17]. The particular observation suggested significant prognostic heterogeneity within the conventional unfavorable karyotype category. At the same time, studies in other myeloid malignancies have implicated specific abnormalities as being responsible for the prognostic relevance of broad categories such as complex or monosomal karyotype [18, 19]. Another recent development concerns the discovery of prognostically relevant mutations in PMF and the prospect of their interaction with karyotype [20]. PMF is characterized by three ‘driver’ mutations, which are often mutually exclusive and are currently thought to be pathogenetically and prognostically relevant: [21] JAK2, CALR and MPL. The respective mutational frequencies of these mutations are estimated at 65, 20, and 5%, whereas 10% of patients may not express any one of these three mutations and are accordingly referred to as ‘triple-negative’ [21]. Other mutations of prognostic relevance in PMF include ASXL1 and SRSF2 [20, 22]. Given the breadth of cytogenetic abnormalities in PMF [23], and the prospect of genetically oriented prognostic models [20], it is important to periodically revisit with cytogenetic risk stratification in PMF, pending access to higher number of informative cases.
Methods
After approval from the Mayo Clinic institutional review board, clinical and laboratory data, including cytogenetic information, were collected from patients at the time of diagnosis or referral to the Mayo Clinic; the study date spanned from 4 November 1977 through 16 April 2015. The median time between time of cytogenetic studies and death or last follow-up was 3.1 years with maximum follow-up as long as 31 years; ~60% of patients were evaluated within one year of initial diagnosis. Diagnoses of PMF and leukemic transformation were according to World Health Organization (WHO) criteria [24]. Cytogenetic analysis and reporting was done according to the International System for Human Cytogenetic Nomenclature (ISCN) criteria [25]. Cytogenetic analysis in all instances was performed on fresh bone marrow aspirates, placed in hypotonic trypsin-colcemid solution and processed according to standard techniques for chromosome analysis using GTL banding with trypsin and Leishman stain; we utilized two 24-hour and 48-hour cultures without stimulation [26]. Thrombolytic agents were added to clotted bone marrow specimens in order to improve success rates [27]. From approximately the year 2000, the clinical laboratory has transitioned from manual cutting of chromosomes from Kodachrome prints to digital computer imaging.
Chromosomal abnormalities were considered clonal if the same structural abnormality or extra chromosome appears in at least 2 and monosomy in at least three metaphases. For the purposes of the current study, a minimum of 10 metaphases was analyzed before assigning a normal karyotype status. A complex karyotype was defined as the presence of three or more distinct structural or numeric abnormalities. Monosomal karyotype was defined as two or more distinct autosomal monosomies or single autosomal monosomy associated with at least one structural abnormality [28]. Driver mutational status was classified into favorable and unfavorable category based on the presence or absence of type 1/like CALR mutations; [29] high molecular risk (HMR) mutations studied in the current report included ASXL1 and SRSF2 [20]. Driver and other mutations were detected by Illumina platform-supported targeted amplicon next generation or direct sequencing, as previously described [20, 30,31,32].
We followed a stepwise approach (Supplemental Fig. 1), in order to prognostically assign specific cytogenetic abnormalities into distinct risk categories. The process first considered sole abnormalities occurring in at least 10 incident cases and normal karyotype as the reference for survival data comparisons. Other notable but less frequent abnormalities were then considered in the context of both single and multiple abnormalities, in order to overcome statistical limitations from inadequate sample size. Historical relevance was also considered in establishing operational cytogenetic groups. Overall and leukemia-free survivals were calculated from time of referral to the Mayo Clinic, commensurate with time of cytogenetic analysis. Standard statistical methods were used to determine significance of differences among groups in the distribution of continuous or nominal variables. Overall and leukemia-free survival data were prepared by the Kaplan–Meier method and compared by the long –rank test. Cox proportional hazard regression model was applied for multivariable analysis. p−value < 0.05 was considered significant. The Stat View (SAS Institute, Cary, NC, USA) statistical package was used for all calculations.
Results
Patients
A total of 1002 patients with PMF (median age 65 years; 62% males) and available cytogenetic information were considered. The study population was selected from an initial group of 1067 patients with available cytogenetic reports; 65 patients were excluded because they did not meet the 10 metaphase minimum criteria for designation of normal karyotype (n = 63) or detection of a minor t(9;22)(q34;q11) clone (n = 2). The presenting clinical and laboratory features of the study population, stratified by the presence or absence of abnormal karyotype and by the most frequent sole abnormalities are outlined in Table 1. DIPSS risk distribution was 11% high, 43% intermediate-2, 33% intermediate-1 and 13% low [2]. Driver mutational information was available in 637 patients and included 66% JAK2, 15% CALR type 1/like, 4% CALR type 2/like, 5% MPL and 10% triple-negative. On informative cases, ASXL1 was mutated in 38% and SRSF2 in 14% (Table 1). Median follow-up was 3.1 years, during which time 748 (73%) deaths, 75 (7.5%) leukemic events and 52 (5.2%) AlloSCT were recorded. Treatment was consistent with what was considered standard of care at the time.
Cytogenetic findings and phenotypic correlates of the most frequent sole abnormalities
Abnormal karyotype was reported in 449 (45%) patients. Compared to normal karyotype (n = 553; 55%), abnormal karyotype was associated with older age (p = 0.02), lower hemoglobin level (p = 0.001), higher red cell transfusion requirement (p = 0.03), lower leukocyte count (p = 0.007), lower platelet count (p < 0.001), higher circulating blast count (p = 0.001), higher risk DIPSS (p = 0.002), and lower incidence of ASXL1 mutations (p = 0.01; Table 1). Among the 449 (45%) cases with abnormal karyotype, 320 (32%) harbored sole, 68 (7%) two and 61 (6%) three or more abnormalities; by definition, therefore, 61 (6%) patients had complex karyotype and of these 25 (2.3%) were classified as monosomal karyotype. The most frequent sole abnormalities were 20q− (n = 74; ~7%), 13q− (n = 56; ~6%), + 8 (n = 26; ~3%) and +9 (n = 14; 1.4%); less frequent sole abnormalities included 7q− (n = 12), -Y (n = 9) and a sex chromosome abnormality other than –Y (n = 10). Phenotypic correlative studies involving the most frequent sole abnormalities showed significant associations between older age and 20q−, +9 and +8 (p = 0.03); lower hemoglobin level and 20q− and +8 (p = 0.04); higher leukocyte count and +9 and 13q− (p = 0.001); higher platelet count and 13q− (p < 0.001); and ASXL1 mutations and +9 and 20q− (p = 0.004) (Table 1).
Infrequent sole abnormalities included monosomy 7 (n = 7), 5q− (n = 6) + 21 (n = 5), 12p−/12p11.2 (n = 5), 11q−/11q23 (n = 4), i(17q) (n = 4) and inv(3)/3q21 (n = 3) abnormalities; the number of informative cases for these infrequent abnormalities with historical relevance to prognosis was higher when both single and multiple abnormalities were considered: monosomy 7 (n = 18), 5q− (n = 11), + 21 (n = 9), 12p−/12p11.2 (n = 11), 11q−/11q23 (n = 13), i(17q) (n = 6), and inv(3)/3q21 (n = 5). Other sole abnormalities of broader category included sole translocations/duplications of chromosome 1 (n = 21), sole translocations not involving chromosome 1 (n = 25) and ‘other’ sole abnormalities that were otherwise not classified (n = 31; Table 2).
Prognostic classification of cytogenetic groups: step 1
A stepwise approach was undertaken (Supplemental Fig. 1), in order to prognostically classify specific cytogenetic abnormalities. Single abnormalities with at least 10 occurrences were initially considered; 20q− (n = 74), 13q− (n = 56), +8 (n = 26), +9 (n = 14) and 7q− (n = 12) (Table 2). Amongst these, 20q−, 13q− and +9 were associated with survival data that was not significantly different from that of normal karyotype (n = 553) (Table 2; supplemental Fig. 2); the results remained unchanged when analysis was adjusted for age. In contrast, survival of patients with either +8 or 7q− was significantly worse than that seen with normal karyotype (Table 2; supplemental Fig. 2). Age-adjusted survival data were similar for 20q− vs 13q− vs +9 and for +8 vs 7q− (supplemental Fig. 2). Furthermore, each one of 20q−, 13q− and +9, when compared to the combined +8/7q− group, was associated with significantly longer survival (Table 2). Accordingly, sole abnormalities of +8 and 7q− were grouped together, to serve as the initial template for ‘unfavorable’ risk category, and sole abnormalities of 20q−, 13q−, and +9 were classified with normal karyotype to form the initial template for the prognostically superior ‘favorable’ risk category (age-adjusted p = 0.007; HR 0.6, 95% CI 0.4–0.8).
Prognostic classification of cytogenetic groups: step 2
We next focused on specific abnormalities that have historically been marked as being particularly detrimental for survival, including monosomy 7, inv(3)/3q21, i(17q), 11q−/11q23, and 12p−/12p11.2 (Supplemental Fig. 1) [16, 17, 33]. Considering the relative rarity of sole occurrences involving these abnormalities, they were considered in the setting of both single and multiple abnormalities, in order to assemble eighteen cases of monosomy 7, 11 of inv(3)/3q21 (n = 5) or i(17q) (n = 6), eleven of 12p−/12p11.2 and 13 of 11q−/11q23 (Table 2). Monosomy 7 was chosen over monosomal karyotype after preliminary analysis confirmed its primary prognostic contribution to ‘monosomal’ karyotype (HR 3.4, 95% CI 1.3–8.8, compared to monosomal karyotype without monosomy 7).
As expected, each one of the above-listed high risk cytogenetic categories was associated with significantly worse survival, compared to normal karyotype, with more than fivefold hazard ratio for inv(3)/3q21/i(17q) (HR 6.6, 95% CI 3.6–12.2), monosomy 7 (HR 6.3, 95% CI 3.8–10.4) and 12p−/12p11.2 (HR 5.6, 95% CI 2.9–10.5; Table 2). These latter three, but not 11q−/11q23 abnormalities, were also associated with significantly shorter survival, compared to the +8/7q− unfavorable risk category template (Table 2); significance in all instances was retained when analysis was adjusted for age. Accordingly, single/multiple abnormalities of inv(3)/3q21, i(17q), monosomy 7 and 12p−/12p11.2 were grouped together and assigned to an operational very high risk (VHR) category, and displayed similar survival data when compared to each other (p = 0.8). Therefore, at the completion of this second step process, we had established three risk category templates: favorable (normal or sole abnormalities of 20q−, 13q−, or +9; n = 697), unfavorable (sole abnormalities of +8 or 7q−; n = 38; HR 2.2, 95% CI 1.5–3.2) and VHR (single or multiple abnormalities of −7, inv(3)/3q21, i(17q) and 12p−/12p11.2; n = 40; HR 6.1, 95% CI 4.3–8.6) (supplemental Fig. 3).
Prognostic classification of cytogenetic groups: step 3
For the third and final stage analysis, we organized the remaining cytogenetic abnormalities, based on number and type of abnormalities, historical mention and reported relevance in other myeloid malignancies (Table 2). Sole abnormalities of chromosome Y loss were grouped with other sole abnormalities of a sex chromosome, in order to offset statistical limitations from small sample size (Table 2); of note, results of statistical calculations were mostly unchanged when –Y was analyzed separately (data not shown). Autosomal sole trisomies other than +9 or + 8 (e.g., +21, +19) were considered separately from two abnormalities and complex karyotype, including monosomal karyotype. Furthermore, in order to avoid confounding from prognostic contribution of a VHR abnormality (i.e., −7, inv(3)/3q21, i(17q), 12p−/12p11.2), complex and monosomal karyotype groups, as well as the ‘two abnormalities’ group, were required not to include cases with VHR abnormalities (Table 2).
Each one of the above-listed operational cytogenetic groups was subsequently compared to normal, unfavorable and VHR cytogenetic risk templates, in order to determine its best-fit category. Based on results obtained and outlined in Table 2, risk allocations were straightforward for sole chromosome 1 translocations/duplications (favorable risk), sole sex chromosome abnormalities including –Y (favorable risk), sole translocations not involving chromosome 1 (unfavorable), other sole abnormalities not otherwise classified (unfavorable) and non-monosomal and complex karyotypes without VHR abnormality (unfavorable) (Table 2). Also, careful inspection of hazard ratios (HR) and 95% confidence limits (CI) favored placement of monosomal karyotype without VHR abnormality into the unfavorable risk category (Table 2).
Risk assignment for the remaining cytogenetic groups was more complicated and required further analysis; initially, based on HR and 95% CI inspections (Table 2), we grouped sole autosomal trisomies (e.g., +21, +19) and single/multiple 11q−/11q23 abnormalities together in a ‘provisional VHR’ category; similarly, ‘two abnormalities’ without VHR abnormality and single/multiple abnormalities of 5q− were grouped together in a ‘provisional unfavorable’ category (Fig. 1). Survival re-analysis that accounted for these provisional risk groups revealed significantly shorter survival when ‘provisional unfavorable’ group was compared to favorable risk group (HR 1.4, 95% CI 1.0–1.8; p = 0.035) and similar survival data when compared to unfavorable risk group (HR 1.2, 95% CI 0.9–1.8; p = 0.12). Similarly, provisional VHR group displayed significantly worse survival, when compared to unfavorable risk group (HR 2.0, 95% CI 1.3–3.1; p = 0.0008) and similar survival data, when compared to VHR group (p = 0.08; HR 1.6, 95% CI 0.9–2.7). Accordingly, it was decided to list sole autosomal trisomies other than +8 and +9 and single/multiple 11q−/11q23 abnormalities in the VHR group, and ‘two abnormalities’ without VHR abnormality and single/multiple abnormalities of 5q−, in the unfavorable risk group.

Overall survival of 1002 patients with primary myelofibrosis stratified by more definitive and provisional cytogenetic risk categories
Final risk assignment in the revised three-tiered cytogenetic risk model
Based on the above elaborated survival analysis, the following cytogenetic abnormalities were classified into the revised ‘favorable’ cytogenetic risk category: normal karyotype and sole abnormalities of 20q−, 13q−, +9, chromosome 1 translocation/duplication and sex chromosome abnormality including –Y (n = 737); the following abnormalities were assigned to the revised ‘VHR’ cytogenetic risk category: single/multiple abnormalities of −7, inv(3)/3q21, i(17q), 12p−/12p11.2, 11q−/11q23 and autosomal trisomies (e.g., +21, +19) other than +8 or +9 (n = 75); all other abnormalities were assigned to the ‘unfavorable’ risk category (n = 190; Table 3; Fig. 2). By comparison, according to the conventional two-tiered cytogenetic risk model [3], 269 cases belonged to the unfavorable risk category; among them, 75 (28%) were reassigned to the revised VHR and 4 (1.5%) to favorable risk category.

Overall survival of 1002 patients with primary myelofibrosis stratified by the revised three-tiered cytogenetic risk model
Overall and leukemia-free survival analysis
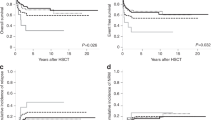
Survival data stratified by the new revised cytogenetic risk stratification are depicted in Fig. 2 and demonstrate the adverse impact of VHR, compared to both unfavorable (HR 2.2, 95% CI 1.6–3.0) and favorable (HR 3.8, 95% CI, 2.9–4.9; p < 0.0001) risk categories, and that of unfavorable, compared to favorable (HR 1.7, 95% CI 1.4–2.0; p < 0.0001) risk category. Multivariable analysis confirmed the independent prognostic contribution of the revised cytogenetic risk model in the context of DIPSS (HR 2.9; 95% CI 2.2–3.7 for VHR and 1.6, 1.3–1.9 for unfavorable), driver mutational status (HR 4.5, 95% CI 3.2–6.4 for VHR and 1.6, 1.3–2.1 for unfavorable), and HMR (i.e., ASXL1/SRSF2) mutations (HR 4.3, 95% CI 2.8–6.7 for VHR and 2.3, 1.7–3.0 for unfavorable); an all-inclusive multivariable analysis found the revised cytogenetic risk model, DIPSS, driver mutational status and HMR mutations to predict shortened survival, independent of each other: HRs (95% CI) were 4.1 (2.6–6.5) for VHR, 2.3 (1.7–3.1) for unfavorable, 2.6 (1.9–3.6) for absence of type 1/like CALR, 1.9 (1.5–2.4) for HMR mutations, 7.7 (4.3–13.9) for DIPSS high, 5.8 (3.5–9.7) for DIPSS intermediate-2 and 3.1 (1.9–5.2) for DIPSS intermediate-1.
The adverse impact of driver mutational status (i.e., type 1 CALR mutation absent vs present) was most evident in patients with favorable karyotype (Fig. 3a; HR 3.2, 95% CI 2.1–4.7; p < 0.0001) and not apparent in those with VHR (HR 1.1; 95% CI 0.5–2.5; p = 0.77) or unfavorable risk category (HR 1.5, 95% CI 0.8–2.8; p = 0.22). Similarly, the adverse impact of HMR mutations was evident in patients with favorable (HR 2.4, 95% CI 1.8–3.1; p < 0.0001) or unfavorable (HR 2.4, 95% CI 1.4–4.1; p = 0.002) karyotype but not in those with VHR (HR 1.4, 95% CI 0.6–3.2; p = 0.47) karyotype (Fig. 3b).

a Overall survival of 637 patients with primary myelofibrosis stratified by the revised cytogenetic risk model and driver mutational status; b Overall survival of 423 patients with primary myelofibrosis stratified by the revised cytogenetic risk model and high risk mutations (ASXL1 or SRSF2)
The revised cytogenetic risk model was also effective in predicting leukemic transformation (HR 4.4, 95% CI 2.0–9.4 for VHR and 2.0, 1.2–3.4 for unfavorable; Supplemental figure 4), independent of other recognized risk factors for leukemia-free survival, including presence of HMR mutations, thrombocytopenia and DIPSS; during an all-inclusive multivariable analysis that included all of these risk factors, DIPSS lost its significance whereas VHR (HR 3.0, 95% CI 1.01–9.0), unfavorable risk (HR 2.0, 95% CI 1.01–4.0), HMR mutations (HR 2.6; 95% CI 1.5–4.7) and platelet <100 × 10(9)/l (HR 2.1, 95% CI 1.2-3.9) retained their significance.
Discussion
The prognostic value of karyotype in myeloid malignancies is well recognized and integrated into formal prognostic models [34,35,36]. Prognostically relevant cytogenetic abnormalities in other myeloid malignancies do not always signify similar risk in PMF. For example, 11q− and 12p− abnormalities have been associated with good or very good risk disease in myelodysplastic syndromes (MDS) [35], while they were flagged as VHR abnormalities in PMF. Similarly, other VHR abnormalities in PMF, including i(17)(q10), +21 and +19, were classified as being intermediate risk in MDS [35]. On the other hand, monosomy 7 and inv(3)(q21.3q26.2)/t(3;3)(q21.3;q26.2) were associated with VHR/poor risk and +8 with unfavorable/intermediate risk disease, in both PMF and MDS, as well as in acute myeloid leukemia (AML) [35, 36]. The current study suggests that broad categories such as ‘complex’ or ‘monosomal’ karyotype do not necessarily imply dismal outcome if one were to account for more specific abnormalities associated with very high risk disease, which in the case of PMF included monosomy 7, i(17)(q10), inv(3)/3q21 and 12p−/12p11.2. The particular concept was recently examined in patients with AML where the prognostic relevance of monosomal karyotype was ascertained in a less complex setting and where monosomy 17 and other specific monosomies might have accounted for the adverse prognosis attached to monosomal karyotype [18, 37].
The current study reveals that more than a quarter of PMF patients currently belonging to ‘unfavorable’ cytogenetic risk category harbor VHR abnormalities, which downgrades their prognosis significantly. In this regard, it is important to note that most of the specific abnormalities currently included in the unfavorable risk category constituted VHR lesions, including single/multiple abnormalities of i(17q), inv(3), 12p− and 11q23; [3] on the other hand, +8, 7q−, and 5q− abnormalities remained in the unfavorable category in the revised risk model, along with complex karyotype without VHR abnormality. Single/multiple abnormalities of monosomy 7 were shown to exert a significantly worse prognosis, compared to otherwise monosomal karyotype without VHR abnormality; the former was accordingly included in the VHR and the latter in the unfavorable risk categories. The current study identified autosomal trisomies, including +21 and +19, to be particularly detrimental to survival, and worthy of inclusion in the VHR category. The distinction between VHR and unfavorable karyotype is practically relevant in terms of both personalized disease prognostication and treatment decision-making; in the latter regard, the presence of VHR karyotype alerts the patient and their physician regarding the urgency to pursue more aggressive treatment approaches, such as AlloSCT, whereas the same might not be true for unfavorable karyotype that is not accompanied by other prognostically adverse mutations, such as ASXL1 and SRSF2. The revised cytogenetic risk model refines the favorable risk category by limiting it to specific abnormalities with survival impact that is not significantly different than that seen with normal karyotype: single abnormalities of 20q−, 13q−, +9, chromosome 1 translocation/duplication and sex chromosome abnormality including –Y.
The current study is unique in regards to its sample size (n = 1002), maturity of survival data (73% of patients were followed till time of death) and availability of other genetic information, which allowed assessment of prognostic interaction between karyotype and mutations. In this regard, it was particularly noteworthy to find the survival impact of driver mutations being confined to patients with favorable karyotype and that of HMR mutations to patients with either favorable or unfavorable karyotype. In other words, the presence of a VHR cytogenetic abnormality superseded the prognostic contribution of both driver and HMR mutations while patients with favorable karyotype were the most susceptible in this regard. Taken together, the current study clarifies the hierarchy of prognostic contribution from genetic markers and confirms the inter-independent prognostic contribution of a newly revised three-tiered cytogenetic risk stratification, driver mutational status, HMR mutations and clinically derived prognostic scoring systems. These observations support the development of comprehensive prognostic tools that integrate these variables and therefore facilitate the process of risk-appropriate treatment selections. Finally, it is important to recognize the limitations in the current study design that include prior assumptions of adverse cytogenetic and mutation abnormalities and the fact that information on relevant mutations was neither comprehensive nor complete.
References
Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895–901.
Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115:1703–8.
Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29:392–7.
Tefferi A, Lasho TL, Jimma T, Finke CM, Gangat N, Vaidya R, et al. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc. 2012;87:25–33.
Samuelson Bannow BT, Salit RB, Storer BE, Stevens EA, Wu D, Yeung C, et al. Hematopoietic cell transplantation for myelofibrosis: dipss plus risk predicts post-transplant outcomes. Biol Blood Marrow Transplant. 2018;24(2):386–392.
Miller JB, Testa JR, Lindgren V, Rowley JD. The pattern and clinical significance of karyotypic abnormalities in patients with idiopathic and postpolycythemic myelofibrosis. Cancer. 1985;55:582–91.
Demory JL, Dupriez B, Fenaux P, Lai JL, Beuscart R, Jouet JP, et al. Cytogenetic studies and their prognostic significance in agnogenic myeloid metaplasia: a report on 47 cases. Blood. 1988;72:855–9.
Reilly JT, Wilson G, Barnett D, Watmore A, Potter A. Karyotypic and ras gene mutational analysis in idiopathic myelofibrosis. Brit J Haematol. 1994;88:575–81.
Dupriez B, Morel P, Demory JL, Lai JL, Simon M, Plantier I, et al. Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood. 1996;88:1013–8.
Reilly JT, Snowden JA, Spearing RL, Fitzgerald PM, Jones N, Watmore A, et al. Cytogenetic abnormalities and their prognostic significance in idiopathic myelofibrosis: a study of 106 cases. Br J Haematol. 1997;98:96–102.
Tefferi A, Mesa RA, Schroeder G, Hanson CA, Li CY, Dewald GW. Cytogenetic findings and their clinical relevance in myelofibrosis with myeloid metaplasia. Br J Haematol. 2001;113:763–71.
Strasser-Weippl K, Steurer M, Kees M, Augustin F, Tzankov A, Dirnhofer S, et al. Chromosome 7 deletions are associated with unfavorable prognosis in myelofibrosis with myeloid metaplasia. Blood. 2005;105:4146.
Tefferi A, Dingli D, Li CY, Dewald GW. Prognostic diversity among cytogenetic abnormalities in myelofibrosis with myeloid metaplasia. Cancer. 2005;104:1656–60.
Hussein K, Huang J, Lasho T, Pardanani A, Mesa RA, Williamson CM, et al. Karyotype complements the International Prognostic Scoring System for primary myelofibrosis. Eur J Haematol. 2009;82:255–9.
Hussein K, Pardanani AD, Van Dyke DL, Hanson CA, Tefferi A. International prognostic scoring system-independent cytogenetic risk categorization in primary myelofibrosis. Blood. 2010;115:496–9.
Caramazza D, Begna KH, Gangat N, Vaidya R, Siragusa S, Van Dyke DL, et al. Refined cytogenetic-risk categorization for overall and leukemia-free survival in primary myelofibrosis: a single center study of 433 patients. Leukemia. 2011;25:82–8.
Tefferi A, Jimma T, Gangat N, Vaidya R, Begna KH, Hanson CA, et al. Predictors of greater than 80% 2-year mortality in primary myelofibrosis: a Mayo Clinic study of 884 karyotypically annotated patients. Blood. 2011;118:4595–8.
Strickland SA, Sun Z, Ketterling RP, Cherry AM, Cripe LD, Dewald G, et al. Independent Prognostic Significance of Monosomy 17 and Impact of Karyotype Complexity in Monosomal Karyotype/Complex Karyotype Acute Myeloid Leukemia: Results from Four ECOG-ACRIN Prospective Therapeutic Trials. Leuk Res. 2017;59:55–64.
Schanz J. Helpful tool or oversimplification? concept of the monosomal karyotype from the clinical and cytogenetic point of view. Biol Blood Marrow Transplant. 2016;22:191–2.
Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27:1861–9.
Tefferi A, Nicolosi M, Mudireddy M, Szuber N, Finke CM, Lasho TL, et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1095 patients. Am J Hematol. (in press).
Tefferi A, Lasho TL, Finke CM, Elala Y, Hanson CA, Ketterling RP, et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;1:105–11.
Wassie E, Finke C, Gangat N, Lasho TL, Pardanani A, Hanson CA, et al. A compendium of cytogenetic abnormalities in myelofibrosis: molecular and phenotypic correlates in 826 patients. Br J Haematol. 2015;169:71–6.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–51.
McGowan-Jordan J, Simons A, Schmid M ISCN 2016:An International System for Human Cytogenomic Nomenclature (2016) Reprint of: Cytogenetic and Genome Research 2016, Vol. 149, No. 1-2: KARGER, 2016.
Dewald GW, Broderick DJ, Tom WW, Hagstrom JE, Pierre RV. The efficacy of direct, 24-hour culture, and mitotic synchronization methods for cytogenetic analysis of bone marrow in neoplastic hematologic disorders. Cancer Genet Cytogen. 1985;18:1–10.
St Antoine A, Ketterling MN, Sukov WR, Lowman J, Knudson RA, Sinnwell JP, et al. Application of thrombolytic drugs on clotted blood and bone marrow specimens to generate usable cells for cytogenetic analyses. Arch Pathol & Lab Med. 2011;135:915–9.
Breems DA, Van Putten WL, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KB, Mellink CH, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol: Off J Am Soc Clin Oncol. 2008;26:4791–7.
Tefferi A, Lasho TL, Tischer A, Wassie EA, Finke CM, Belachew AA, et al. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood. 2014;124:2465–6.
Tefferi A, Lasho TL, Finke C, Belachew AA, Wassie EA, Ketterling RP, et al. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia. 2014;28:1568–70.
Guglielmelli P, Rotunno G, Fanelli T, Pacilli A, Brogi G, Calabresi L, et al. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015;5:e360.
Tefferi A, Lasho T, Guglielmelli P, Finke C, Rotunno G, Elala Y, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Bloood Adv. 2016;1:21–30.
Tam CS, Abruzzo LV, Lin KI, Cortes J, Lynn A, Keating MJ, et al. The role of cytogenetic abnormalities as a prognostic marker in primary myelofibrosis: applicability at the time of diagnosis and later during disease course. Blood. 2009;113:4171–8.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC: Lyon, France, 2017.
Schanz J, Tuchler H, Sole F, Mallo M, Luno E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30:820–9.
Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–83.
Jang JE, Min YH, Yoon J, Kim I, Lee JH, Jung CW, et al. Single monosomy as a relatively better survival factor in acute myeloid leukemia patients with monosomal karyotype. Blood Cancer J. 2015;5:e358.
Acknowledgements
Funding provided by the Henry J. Predolin foundation grant (Madison, WI, USA).
Author contributions
All authors reviewed and approved the manuscript. CAH reviewed pathology data. RPK reviewed cytogenetic data. TLL analyzed and interpreted molecular data. MN and MM assisted in data extraction, statistical analysis and preparation of tables. AT, NG, KHB and AP contributed patients and participated in study design and data extraction. AT performed statistical analysis and wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tefferi, A., Nicolosi, M., Mudireddy, M. et al. Revised cytogenetic risk stratification in primary myelofibrosis: analysis based on 1002 informative patients. Leukemia 32, 1189–1199 (2018). https://doi.org/10.1038/s41375-018-0018-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0018-z
This article is cited by
-
Calreticulin and JAK2V617F driver mutations induce distinct mitotic defects in myeloproliferative neoplasms
Scientific Reports (2024)
-
Blast phase myeloproliferative neoplasm: contemporary review and 2024 treatment algorithm
Blood Cancer Journal (2023)
-
Allogeneic haematopoietic stem cell transplantation for myelofibrosis: prognostic indicators and the role of JAK2V617F measurable-residual disease monitoring by droplet-digital polymerase chain reaction
Annals of Hematology (2023)
-
International Consensus Classification of myeloid and lymphoid neoplasms: myeloproliferative neoplasms
Virchows Archiv (2023)
-
Cytogenetic abnormalities in essential thrombocythemia: Clinical and molecular correlates and prognostic relevance in 809 informative cases
Blood Cancer Journal (2022)
