
Abstract
Background
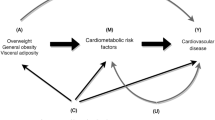
Higher body mass index (BMI) and waist-to-hip ratio (WHR) increase the risk of cardiovascular disease, but the extent to which this is mediated by blood pressure, diabetes, lipid traits, and smoking is not fully understood.
Methods
Using consortia and UK Biobank genetic association summary data from 140,595 to 898,130 participants predominantly of European ancestry, Mendelian randomization mediation analysis was performed to investigate the degree to which systolic blood pressure (SBP), diabetes, lipid traits, and smoking mediated an effect of BMI and WHR on the risk of coronary artery disease (CAD), peripheral artery disease (PAD) and stroke.
Results
The odds ratio of CAD per 1-standard deviation increase in genetically predicted BMI was 1.49 (95% CI 1.39 to 1.60). This attenuated to 1.34 (95% CI 1.24 to 1.45) after adjusting for genetically predicted SBP (proportion mediated 27%, 95% CI 3% to 50%), to 1.27 (95% CI 1.17 to 1.37) after adjusting for genetically predicted diabetes (41% mediated, 95% CI 18% to 63%), to 1.47 (95% CI 1.36 to 1.59) after adjusting for genetically predicted lipids (3% mediated, 95% −23% to 29%), and to 1.46 (95% CI 1.34 to 1.58) after adjusting for genetically predicted smoking (6% mediated, 95% CI −20% to 32%). Adjusting for all the mediators together, the estimate attenuated to 1.14 (95% CI 1.04 to 1.26; 66% mediated, 95% CI 42% to 91%). A similar pattern was observed when considering genetically predicted WHR as the exposure, and PAD or stroke as the outcome.
Conclusions
Measures to reduce obesity will lower the risk of cardiovascular disease primarily by impacting downstream metabolic risk factors, particularly diabetes and hypertension. Reduction of obesity prevalence alongside control and management of its mediators is likely to be most effective for minimizing the burden of obesity.
Similar content being viewed by others

Background
Cardiovascular disease (CVD) is the leading cause of death and disability worldwide [1]. Obesity can contribute towards CVD risk through effects on hyperglycaemia, hypertension, dyslipidaemia, and smoking behaviour [2,3,4,5]. The global prevalence of obesity has more than tripled in the last 40 years, with an even greater rise in incidence amongst children [6]. It is estimated that by 2030, approximately half of the US population will be obese [7]. While obesity prevention remains the priority, there are also treatments available to effectively manage the downstream mediators through which obesity causes CVD [8,9,10,11]. Understanding such pathways is therefore paramount to reducing cardiovascular risk.
Obesity can be measured by various means. It is defined by the World Health Organisation as a body mass index (BMI) greater than or equal to 30 kg/m2 [12], although this cut-off threshold can vary between different populations. However, BMI is not a direct measure of adiposity and is also correlated with fat-free mass [12]. Assessment of obesity using the waist-to-hip ratio (WHR) is less subject to influence from height and muscle mass and is positively associated with cardiovascular risk in individuals with a normal BMI [13, 14]. Thus, BMI and WHR represent distinct measures of body fat that may differentially affect the risk of CVD outcomes. Conventional observational studies have shown that the relationship between obesity measures such as BMI and WHR with CVD is attenuated when adjustment is made for cardiometabolic risk factors such as blood pressure, lipid traits or measures of glycaemia [15]. This has allowed for estimation of the proportion of the effect of obesity that is mediated through these intermediates [15]. However, such observational analysis is vulnerable to bias from environmental confounding factors and measurement error, both of which can result in underestimation of the proportion of effect mediated [16, 17]. The Mendelian randomization (MR) approach uses genetic variants as instruments for studying the effect of modifying an exposure on an outcome and has now been extended to perform mediation analyses [16, 18]. Such use of genetic variants whose allocation is not affected by environmental confounding factors means that MR estimates are less vulnerable to confounding from environmental factors. Furthermore, the use of genetic variants that are associated with the exposure (BMI or WHR) in large populations including individuals of different ages means that their association estimates are typically less vulnerable to measurement error or variation related to the timing of measurement [16].
The increasing availability of large-scale genome-wide association study (GWAS) data has greatly facilitated MR analyses considering cardiovascular risk factors and outcomes. In this study, we aimed to use such data within the MR framework to investigate the role of blood pressure, diabetes, fasting glucose, lipid traits, and smoking in mediating the effect of BMI and WHR on coronary artery disease (CAD), peripheral arterial disease (PAD) and stroke risk.
Methods
Ethical approval, data availability, code availability and reporting
The data used in this work are publicly available and the studies from which they were obtained are cited. All these studies obtained relevant participant consent and ethical approval. The results from the analyses performed in this work are presented in the main manuscript or its supplementary files. All code used for this work is available upon reasonable request to the corresponding author. This paper has been reported based on recommendations by the STROBE-MR Guidelines (Research Checklist) [19]. The study protocol and details were not pre-registered.
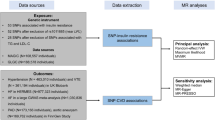
Data sources
Genetic association estimates for BMI and WHR were obtained from the GIANT Consortium GWAS meta-analysis of 806,834 and 697,734 European-ancestry individuals, respectively [20]. Genetic association estimates for systolic blood pressure (SBP) were obtained from a GWAS of 318,417 White British individuals in the UK Biobank, with the correction made for any self-reported anti-hypertensive medication use by adding 10 mmHg to the mean SBP measured from two automated recordings that were taken 2 min apart at baseline assessment [21]. Previous methodological work has supported that the addition of a constant value to the observed blood pressure in individuals taking antihypertensive medication as a strategy that optimises statistical power while minimising bias [22]. Genetic association estimates for lifetime smoking (referred to hereon as smoking) were obtained from a GWAS of 462,690 European-ancestry individuals in the UK Biobank [23]. A lifetime measure of smoking was created based on self-reported age at initiation, age at cessation and cigarettes smoked per day [23]. Genetic association estimates for liability to diabetes came from the DIAGRAM Consortium GWAS meta-analysis of 74,124 cases and 824,006 controls, all of the European ancestry [24]. Genetic association estimates for plasma fasting glucose were obtained by using PLINK software to carry out a meta-analysis of MAGIC Consortium GWAS summary data from separate analyses of 67,506 men and 73,089 women who were not diabetic [25, 26]. Genetic association estimates for fasting serum low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C) and triglycerides were obtained from the Global Lipids Genetics Consortium GWAS of 188,577 European-ancestry individuals [27]. Genetic association estimates for CAD were obtained from the CARDIoGRAMplusC4D Consortium 1000G multi-ethnic GWAS (77% European-ancestry) of 60,801 cases and 123,504 controls [28]. Genetic association estimates for PAD were obtained from the Million Veterans Programme multi-ethnic (72% European-ancestry) GWAS of 31,307 cases and 211,753 controls [29]. Genetic association estimates for stroke were obtained from the MEGASTROKE multi-ethnic (86% European-ancestry) GWAS of 67,162 cases (of any stroke) and 454,450 controls [30]. Population characteristics and specific trait definitions relating to all these summary genetic association estimates are available in their original publications. For the analyses performed in this current work, genetic variants from different studies were aligned by their effect alleles and no exclusions were made for palindromic variants. Only variants for which genetic association estimates were available for all the traits being investigated in any given analysis were considered. In order to maintain consistency in the variants employed as instruments across different analyses, proxies were not used.
Instrument selection
To estimate the total effect of BMI and WHR, respectively on the considered cardiovascular outcomes, instruments were selected as single-nucleotide polymorphisms (SNPs) that associated with BMI or WHR at genome-wide significance (P < 5 × 10−8) and were in pair-wise linkage disequilibrium (LD) r2 < 0.001. The percentage variance in BMI and WHR explained by the variants selected as their respective instruments was estimated as previously described [31]. To select instruments for mediation analysis, all SNPs related to the considered exposure (BMI or WHR) or mediators at genome-wide significance were pooled and clumped to pairwise LD r2 < 0.001 based on the lowest P-value for association with any trait. All clumping was performed using the TwoSampleMR package in R [32].
Total effects
Random-effects inverse-variance weighted (IVW) MR was used as the main analysis for estimating the total effects of genetically predicted BMI and genetically predicted WHR respectively on each of the considered CVD outcomes [33]. The contamination-mixture method, weighted median and MR-Egger were used in sensitivity analyses to explore the robustness of the findings to potential pleiotropic effects of the variants [34,35,36]. The contamination-mixture model makes the assumption that MR estimates from valid instruments follow a normal distribution that centres on the true causal effect estimate, while those calculated from invalid instrument variants follow a normal distribution centred on the null [35]. This allows for a likelihood function to be specified and maximized when allocating each variant to one of the two mixture distributions [35]. The weighted median approach orders the MR estimates from individual variants by their magnitude weighted for their precision and selects the median as the overall MR estimate, calculating standard error by bootstrapping [34]. MR-Egger regresses the variant-outcome association estimates against the variant-exposure association estimates, weighted for the precision of the variant-outcome estimates [36]. It gives a valid MR estimate and test for the presence of directional pleiotropy in scenarios where any direct effect of the variants on the outcome is not correlated to their association with the exposure [36]. The MendelianRandomization package (version 0.4.2) in R (version 3.6.3) was used for performing the IVW, contamination-mixture, weighted median MR and MR-Egger analyses [37].
Mediation analysis
To estimate the direct effect of genetically predicted BMI and genetically predicted WHR on each of the three considered CVD outcomes that were not being mediated by the investigated intermediary risk factors, summary data multivariable MR was performed [38,39,40]. Specifically, the orientations of all genetic association estimates were harmonized and the variant-outcome genetic association estimates were regressed on the variant-exposure and variant-mediator estimates, weighted for the precision of the variant-outcome association, with the intercept fixed to zero [40]. Using this approach, adjustment was made for genetically predicted SBP, diabetes, smoking and lipid traits (LDL-C, HDL-C and triglycerides together) in turn, and finally including all mediators together in a joint model. In a sensitivity analysis, genetically predicted diabetes was excluded from this joint model to remove any bias that might be introduced because of its binary nature [41]. For analyses considering genetically predicted fasting glucose in non-diabetics instead of genetically predicted diabetes, the corresponding genetic association data were substituted. Diabetes and fasting glucose were not included together in the same model.
Multivariable MR mediation analysis was performed to estimate the proportion of the effect of BMI and WHR respectively on CAD, PAD and stroke that was mediated through each of the considered risk factors, and also all of them together [16]. Specifically, the direct effect of genetically predicted BMI and genetically predicted WHR respectively were divided by their total effect and subtracted from 1, with standard errors estimated using the propagation of error method [16, 18].
Independent effects of genetically predicted BMI and WHR
The direct effects of genetically predicted BMI and genetically predicted WHR on the considered CVD outcomes that are not mediated through each other were measured by including only these two traits together as exposures in the summary data multivariable MR model described above.
Results
Total effects
The variants selected as instruments for BMI and WHR explain 5.7% and 3.6% of their variance respectively. Considering total effects, there was consistent evidence across the IVW, contamination-mixture, weighted median and MR-Egger methods that both higher genetically predicted BMI and higher genetically predicted WHR increased CAD, PAD and stroke risk (Supplementary Fig. 1). The confidence intervals of the MR-Egger estimates were wider than for the other methods, consistent with its lower statistical power [42]. The MR-Egger intercept did not provide evidence to suggest directional pleiotropy in any analysis (P > 0.05 in all analyses). In the main IVW MR analysis, the odds ratio per 1-standard deviation (SD) increase in genetically predicted BMI (4.81 kg/m2) for CAD risk was 1.49 (95% confidence interval [CI] 1.39 to 1.60), for PAD risk was 1.70 (95% CI 1.58 to 1.82), and for stroke risk was 1.22 (95% CI 1.15 to 1.29). For a 1-SD increase in genetically predicted WHR (0.09), this was 1.54 (95% CI 1.38 to 1.71) for CAD risk, 1.55 (95% CI 1.40 to 1.71) for PAD risk, and 1.30 (95% CI 1.21 to 1.40) for stroke risk.
Mediation analysis
There was attenuation in the associations of genetically predicted BMI and genetically predicted WHR with the three CVD outcomes after adjusting for genetically predicted SBP, diabetes, lipid traits (LDL-C, HDL-C and triglycerides together) and smoking, either separately or in the same joint model (Fig. 1). The 49% (95% CI 39% to 60%) increased risk of CAD conferred per 1-SD increase in genetically predicted BMI attenuated to 34% (95% CI 24% to 45%) after adjusting for genetically predicted SBP, to 27% (95% CI 17% to 37%) after adjusting for genetically predicted diabetes, to 47% (95% CI 36% to 59%) after adjusting for genetically predicted lipids, and to 46% (95% CI 34% to 58%) after adjusting for genetically predicted smoking. Adjusting for all the mediators together in the same model, the association attenuated to 14% (95% CI 4% to 26%).

The y-axis details the genetically predicted mediator(s) for which adjustments were made. Blood pressure refers to systolic blood pressure. Lipids refer to serum low-density lipoprotein cholesterol, high-density lipoprotein cholesterol and triglycerides considered together in one model. CI confidence interval, OR odds ratio, SD standard deviation.
The percentage attenuation in the total effects of genetically predicted BMI and WHR respectively on the three CVD outcomes after adjusting for the mediators is depicted in Fig. 2. For the effect of genetically predicted BMI on CAD risk, 27% (95% CI 3% to 50%) was mediated by genetically predicted SBP, 41% (95% 18% to 63%) was mediated by genetically predicted diabetes, 3% (−23% to 29%) was mediated by genetically predicted lipids, and 6% (95% CI −20% to 32%) was mediated by genetically predicted smoking. All the mediators together accounted for 66% (95% CI 42% to 91%) of the total effect of genetically predicted BMI on CAD risk.

The y-axis details the genetically predicted mediator(s) for which adjustment was made. Blood pressure refers to systolic blood pressure. Lipids refer to serum low-density lipoprotein cholesterol, high-density lipoprotein cholesterol and triglycerides considered together in one model. CI confidence interval.
A joint model including all considered mediators except genetically predicted diabetes was also constructed (Supplementary Fig. 2). Adjusting together for all the mediators except genetically predicted diabetes, the association of genetically predicted BMI with CAD risk attenuated from odds ratio 1.49 (95% CI 1.39 to 1.60) to 1.27 (95% CI 1.16 to 1.40).
There was little change in the association of either genetically predicted BMI or genetically predicted WHR with risk of the three CVD outcomes after adjusting for genetically predicted fasting glucose in non-diabetic individuals (Fig. 3).

CI confidence interval, OR odds ratio, SD standard deviation.
Independent effects of genetically predicted BMI and WHR
Both genetically predicted BMI and genetically predicted WHR had direct effects on CAD, PAD and stroke after mutual adjustment (Fig. 4). The increased CAD risk attributed to a 1-SD higher genetically predicted BMI attenuated from 49% (95% CI 39% to 60%) to 32% (95% CI 20% to 45%) after adjusting for genetically predicted WHR, and the increased CAD risk attributed to a 1-SD higher genetically predicted WHR attenuated from 54% (95% CI 38% to 71%) to 33% (95% CI 18% to 50%) after adjusting for genetically predicted BMI.

CI confidence interval, OR odds ratio, SD standard deviation.
Discussion
This study uses large-scale genetic association data within the MR paradigm to investigate the role of SBP, diabetes, lipid traits and smoking in mediating the effect of BMI and WHR on CAD, PAD and stroke risk. The results support that the majority of the effects of obesity on CVD are mediated through these risk factors, with diabetes and blood pressure being the most notable and accounting for approximately one-third and one-quarter of the effect respectively. In contrast, the analysis of genetically predicted fasting glucose in non-diabetic individuals did not provide any evidence to support its role in mediating the effect of obesity on CVD risk. Previous work has supported an effect of diabetes liability, fasting glucose and glycated haemoglobin on CVD risk [43, 44]. Taken together with our current findings, this suggests that obesity may be affecting CVD risk by increasing diabetes liability and non-fasting (postprandial) glucose levels. Similarly, while lipid traits are known to affect CVD risk [45], our current study suggests that obesity is conferring only a small proportion of its effect on CVD risk through this pathway. Consistent with this, previous work has supported an effect of BMI on HDL-C and triglyceride levels, but not LDL-C [44].
In our analyses, the sum of the estimated mediating effects of the various risk factors considered individually was comparable to their total mediating effect estimated when considering them all together in the same model, consistent with them acting through distinct mechanisms. Including genetically predicted BMI and genetically predicted WHR in the same model produced evidence consistent with these traits having direct effects on CVD risk independently of each other. It follows that rather than analysing BMI or WHR alone, they should be considered together as they capture different aspects of adiposity.
Our findings have important clinical and public health implications. Behavioural interventions to reduce obesity can have inadequate long term effects [46], pharmacological treatments may be limited by unfavourable adverse effect profiles [47], and surgical procedures are often reserved for only severe cases [48]. While preventing obesity remains the priority, this work supports that the majority of its cardiovascular consequences may also be managed by effectively controlling its downstream mediators, most notably diabetes and raised blood pressure, for which effective pharmacological interventions are available. This has relevance for the more than 640 million individuals worldwide currently living with obesity [49], and the many more forecasted to become obese in coming years [50]. Such holistic consideration of obesity together with its mediators could contribute to a shift from the single-disease focus of health systems towards prioritizing multi-morbidity and promoting individual and societal wellness [51].
Our analyses were also suggestive of some possible residual effect of BMI on CVD risk even after adjusting for all the considered mediating risk factors, consistent with metabolically healthy obesity still conferring increased CVD risk [52]. In contrast, the investigation of WHR was consistent with an absence of any direct effect on CVD risk after accounting for all mediating risk factors together, suggesting that WHR may be entirely influencing CVD through downstream metabolic traits. Taken together, these results suggest that unless the growing obesity epidemic is effectively tackled, we risk undoing the large reductions in CVD mortality achieved over past decades [1]. Population-based approaches that decrease obesity by addressing key upstream drivers such as poor diet and physical inactivity have substantial potential for impact and are also effective for reducing health inequalities [53, 54].
The results of our current study can be contrasted to those from a large-scale observational analysis of 1.8 million people across 97 studies [15, 55]. This previous work estimated that 46% (95% CI 42% to 50%) of the excess risk conferred by raised BMI on CAD and 76% (95% CI 65% to 91%) on stroke were mediated by effects on blood pressure, glucose levels and lipid traits, with blood pressure being the most important and mediation for stroke being greatest [15]. However, the approach and data used in our current study offer a number of possible improvements. Our work includes a greater repertoire of risk factors and CVD outcomes than have been considered together previously [15, 44], in particular, drawing on recently available GWAS summary data to study smoking and PAD [23, 29]. MR analysis uses randomly allocated genetic variants that represent a lifelong cumulative liability to the traits for which they serve as instruments and can therefore help overcome the environmental confounding that may bias conventional observational studies [16]. Consistent with this, our MR results indicate that these risk factors mediate a greater proportion of the effect of obesity than suggested by previous conventional observational analyses [15]. Furthermore, our MR estimates are comparable to those obtained in previous MR studies considering BMI and WHR as exposures and different types of CVD as the outcome [44, 56, 57].
Also of relevance here, we considered a genetic liability to diabetes and genetically predicted fasting glucose in non-diabetic individuals as separate risk factors. Our findings support the concept that obesity traits confer an increased risk of CVD specifically through liability to diabetes, rather than variation in fasting glucose levels within the normal physiological range. This is important because fasting glucose may have a non-linear association with CVD risk [58], only having detrimental effects beyond a certain point [59].
Our current study also has limitations. The aim of the current work was to investigate the degree to which cardiometabolic traits mediate the effects of BMI and WHR on CVD outcomes, and our study did not extend to investigate any possible role of BMI or WHR in mediating the effects of the considered cardiometabolic traits on CVD risk. The genetic association data used in this work are drawn from predominantly European populations, and should therefore be interpreted with caution when extrapolating to other ethnic groups. Diabetes is a binary outcome, and as such our consideration of genetically predicted diabetes could introduce bias into the mediation analysis because not all individuals possessing such genetic liability to develop diabetes-related traits [41]. SBP was used as a proxy for studying the effects of blood pressure more generally. Given the high degree of phenotypic and genetic correlation between blood pressure traits [60], this would seem unlikely to affect the conclusions drawn. A theoretical weakness of the MR approach relates to bias from pleiotropic effects of the genetic variants incorporated as instruments for the traits under study, whereby they may directly affect the outcome through pathways independent of the exposure or mediators being considered. Although such bias cannot be entirely excluded, it is reassuring that we obtained similar MR estimates for the total effect of BMI and WHR respectively on the three CVD outcomes when performing the IVW, contamination-mixture, weighted median and MR-Egger methods that each make different assumptions concerning the presence of pleiotropic variants [42]. Finally, there is currently no available method for assessing instrument strength within the two-sample multivariable MR setting, and we could therefore not assess potential vulnerability to weak instrument bias [38].
In conclusion, this work using the MR framework suggests that the majority of the effects of obesity on CVD risk are mediated through metabolic risk factors, most notably diabetes and blood pressure. Comprehensive public health measures that target the reduction of obesity prevalence alongside control and management of its downstream mediators are likely to be most effective for minimizing the burden of obesity on individuals and health systems alike.
References
GBD. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1151–210.
Singh GM, Danaei G, Farzadfar F, Stevens GA, Woodward M, Wormser D, et al. The age-specific quantitative effects of metabolic risk factors on cardiovascular diseases and diabetes: a pooled analysis. PLoS ONE. 2013;8:e65174.
Van Gaal LF, Mertens IL, De, Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–80.
Carreras-Torres R, Johansson M, Haycock PC, Relton CL, Davey Smith G, Brennan P, et al. Role of obesity in smoking behaviour: Mendelian randomisation study in UK Biobank. BMJ. 2018;361:k1767.
Taylor AE, Richmond RC, Palviainen T, Loukola A, Wootton RE, Kaprio J, et al. The effect of body mass index on smoking behaviour and nicotine metabolism: a Mendelian randomization study. Hum Mol Genet. 2019;28:1322–30.
Jaacks LM, Vandevijvere S, Pan A, McGowan CJ, Wallace C, Imamura F, et al. The obesity transition: stages of the global epidemic. Lancet Diabetes Endocrinol. 2019;7:231–40.
Ward ZJ, Bleich SN, Cradock AL, Barrett JL, Giles CM, Flax C, et al. Projected U.S. state-level prevalence of adult obesity and severe obesity. N Engl J Med. 2019;381:2440–50.
Wright JM, Musini VM, Gill R. First-line drugs for hypertension. Cochrane Database Syst Rev. 2018;4:CD001841.
Michos ED, McEvoy JW, Blumenthal RS. Lipid management for the prevention of atherosclerotic cardiovascular disease. N Engl J Med. 2019;381:1557–67.
Rigotti NA, Clair C. Managing tobacco use: the neglected cardiovascular disease risk factor. Eur Heart J. 2013;34:3259–67.
Davies MJ, D’Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, et al. Management of hyperglycemia in Type 2 diabetes, 2018. A consensus report by the American diabetes association (ADA) and the European association for the study of diabetes (EASD). Diabetes Care. 2018;41:2669–701.
Neeland IJ, Ross R, Despres JP, Matsuzawa Y, Yamashita S, Shai I, et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: a position statement. Lancet Diabetes Endocrinol. 2019;7:715–25.
Chen GC, Arthur R, Iyengar NM, Kamensky V, Xue XN, Wassertheil-Smoller S, et al. Association between regional body fat and cardiovascular disease risk among postmenopausal women with normal body mass index. Eur Heart J. 2019;40:2849-+.
Sahakyan KR, Somers VK, Rodriguez-Escudero JP, Hodge DO, Carter RE, Sochor O, et al. Normal-weight central obesity: implications for total and cardiovascular mortality. Ann Intern Med. 2015;163:827–35.
Lu Y, Hajifathalian K, Ezzati M, Woodward M, Rimm EB, Danaei G, et al. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet. 2014;383:970–83.
Carter AR, Sanderson E, Hammerton G, Richmond RC, Smith GD, Heron J, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. bioRxiv. 2019:835819.
Relton CL, Davey Smith G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol. 2012;41:161–76.
Burgess S, Thompson DJ, Rees JMB, Day FR, Perry JR, Ong KK. Dissecting causal pathways using Mendelian randomization with summarized genetic data: application to age at menarche and risk of breast cancer. Genetics. 2017;207:481–7.
Davey Smith G, Davies NM, Dimou N, Egger M, Gallo V, Golub R, et al. STROBE-MR: guidelines for strengthening the reporting of Mendelian randomization studies. PeerJ Preprints. 2019;7:e27857v1 https://doi.org/10.7287/peerj.preprints.27857v1.
Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet. 2019;28:166–74.
Carter AR, Gill D, Davies NM, Taylor AE, Tillmann T, Vaucher J, et al. Understanding the consequences of education inequality on cardiovascular disease: Mendelian randomisation study. BMJ. 2019;365:l1855.
Tobin MD, Sheehan NA, Scurrah KJ, Burton PR. Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med. 2005;24:2911–35.
Wootton RE, Richmond RC, Stuijfzand BG, Lawn RB, Sallis HM, Taylor GMJ, et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med. 2020;50:2435–2443.
Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50:1505–13.
Lagou V, Mägi R, Hottenga JJ, Grallert H, Perry JRB, Bouatia-Naji N, et al. Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC). Sex-dimorphic genetic effects and novel loci for fasting glucose and insulin variability. Nat Commun. 2021;12:24.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30.
Klarin D, Lynch J, Aragam K, Chaffin M, Assimes TL, Huang J, et al. Genome-wide association study of peripheral artery disease in the million veteran program. Nat Med. 2019;25:1274–9.
Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–37.
Gill D, Sheehan NA, Wielscher M, Shrine N, Amaral AFS, Thompson JR, et al. Age at menarche and lung function: a Mendelian randomization study. Eur J Epidemiol. 2017;32:701–10.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife. 2018;7:e34408.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–65.
Bowden J, Davey, Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14.
Burgess S, Foley CN, Allara E, Staley JR, Howson JMM. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun. 2020;11:376.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25.
Yavorska OO, Burgess S. Mendelian randomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46:1734–9.
Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int J Epidemiol. 2018;48:713–27.
Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181:251–60.
Burgess S, Dudbridge F, Thompson SG. Re: “Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects”. Am J Epidemiol. 2015;181:290–1.
Burgess S, Labrecque JA. Mendelian randomization with a binary exposure variable: interpretation and presentation of causal estimates. Eur J Epidemiol. 2018;33:947–52.
Slob EAW, Burgess S. A comparison of robust Mendelian randomization methods using summary data. Genet Epidemiol. 2020;44:313–29.
Ahmad OS, Morris JA, Mujammami M, Forgetta V, Leong A, Li R, et al. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nat Commun. 2015;6:7060.
Xu L, Borges MC, Hemani G, Lawlor DA. The role of glycaemic and lipid risk factors in mediating the effect of BMI on coronary heart disease: a two-step, two-sample Mendelian randomisation study. Diabetologia. 2017;60:2210–20.
Allara E, Morani G, Carter P, Gkatzionis A, Zuber V, Foley CN, et al. Genetic determinants of lipids and cardiovascular disease outcomes: a wide-angled Mendelian randomization investigation. Circ Genom Precis Med. 2019;12:e002711.
Douketis JD, Macie C, Thabane L, Williamson DF. Systematic review of long-term weight loss studies in obese adults: clinical significance and applicability to clinical practice. Int J Obes. 2005;29:1153–67.
Rucker D, Padwal R, Li SK, Curioni C, Lau DC. Long term pharmacotherapy for obesity and overweight: updated meta-analysis. BMJ. 2007;335:1194–9.
Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K, et al. Bariatric surgery: a systematic review and meta-analysis. JAMA. 2004;292:1724–37.
NCD Risk Factor Collaboration. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet. 2016;387:1377–96.
Foreman KJ, Marquez N, Dolgert A, Fukutaki K, Fullman N, McGaughey M, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet. 2018;392:2052–90.
Pearson-Stuttard J, Ezzati M, Gregg EW. Multimorbidity—a defining challenge for health systems. Lancet Public Health. 2019;4:e599–e600.
Caleyachetty R, Thomas GN, Toulis KA, Mohammed N, Gokhale KM, Balachandran K, et al. Metabolically healthy obese and incident cardiovascular disease events among 3.5 million men and women. J Am Coll Cardiol. 2017;70:1429–37.
Backholer K, Beauchamp A, Ball K, Turrell G, Martin J, Woods J, et al. A framework for evaluating the impact of obesity prevention strategies on socioeconomic inequalities in weight. Am J Public Health. 2014;104:e43–50.
Adams J, Mytton O, White M, Monsivais P. Why are some population interventions for diet and obesity more equitable and effective than others? The role of individual agency. PLoS Med. 2016;13:e1001990.
Lawlor DA, Tilling K, Davey Smith G. Triangulation in aetiological epidemiology. Int J Epidemiol. 2016;45:1866–86.
Censin JC, Peters SAE, Bovijn J, Ferreira T, Pulit SL, Magi R, et al. Causal relationships between obesity and the leading causes of death in women and men. PLOS Genet. 2019;15:e1008405.
Marini S, Merino J, Montgomery BE, Malik R, Sudlow CL, Dichgans M, et al. Mendelian randomization study of obesity and cerebrovascular disease. Ann Neurol. 2020;87:516–24.
Park C, Guallar E, Linton JA, Lee DC, Jang Y, Son DK, et al. Fasting glucose level and the risk of incident atherosclerotic cardiovascular diseases. Diabetes Care. 2013;36:1988–93.
Fuller JH, Shipley MJ, Rose G, Jarrett RJ, Keen H. Coronary-heart-disease risk and impaired glucose tolerance. The Whitehall study. Lancet. 1980;1:1373–6.
Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50:1412–25.
Acknowledgements
The authors acknowledge the contributors of the data used in this work: CARDIoGRAMplusC4D, DIAGRAM, GIANT, Global Lipids Genetics Consortium, MAGIC, MEGASTROKE, Million Veterans Programme and UK Biobank.
VA Million Veteran Program
MVP Executive Committee
J. Michael Gaziano27, Sumitra Muralidhar28, Rachel Ramoni28, Jean Beckham29, Kyong-Mi Chang30, Christopher J. O’ Donnell27, Philip S. Tsao31, James Breeling28, Grant Huang28, Juan P. Casas27
MVP Program Office
Sumitra Muralidhar28, Jennifer Moser28
MVP Recruitment/Enrollment
Stacey B. Whitbourne27, Jessica V. Brewer27, Mihaela Aslan32, Todd Connor33, Dean P. Argyres33, Philip S. Tsao31,8, J. Michael Gaziano35, Brady Stephens36, Mary T. Brophy37, Donald E. Humphries37, Luis E. Selva37, Nhan Do38, Shahpoor (Alex) Shayan38, Kelly Cho27, Lori Churby31
MVP Science
Christopher J. O’Donnell39,40, Saiju Pyarajan40,41, Scott L. DuVall42, Elizabeth Hauser43, Philip S. Tsao31, Yan Sun44, Hongyu Zhao45
Current MVP Local Site Investigators
Peter Wilson46, Rachel McArdle47, Louis Dellitalia48, Kristin Mattocks49, John Harley50, Jeffrey Whittle51, Frank Jacono52, Jean Beckham53, John Wells54, Salvador Gutierrez55, Gretchen Gibson56, Kimberly Hammer57, Laurence Kaminsky58, Gerardo Villareal34, Scott Kinlay59, Junzhe Xu27, Mark Hamner60, Roy Mathew61, Sujata Bhushan62, Pran Iruvanti63, Michael Godschalk64, Zuhair Ballas65, Douglas Ivins66, Stephen Mastorides67, Jonathan Moorman68, Saib Gappy69, Jon Klein70, Nora Ratcliffe71, Hermes Florez72, Olaoluwa Okusaga73, Maureen Murdoch74, Peruvemba Sriram75, Shing Shing Yeh76, Neeraj Tandon77, Darshana Jhala78, Samuel Aguayo79, David Cohen80, Satish Sharma81, Suthat Liangpunsakul82, Kris Ann Oursler83, Mary Whooley84, Sunil Ahuja85, Joseph Constans86, Paul Meyer87, Jennifer Greco88, Michael Rauchman89, Richard Servatius90, Melinda Gaddy91, Agnes Wallbom92, Timothy Morgan93, Todd Stapley94, Scott Sherman95, George Ross96, Philip Tsao97, Patrick Strollo98, Edward Boyko99, Laurence Meyer44, Samir Gupta44,100, Mostaqul Huq101, Joseph Fayad102, Adriana Hung103, Jack Lichy104, Robin Hurley105, Brooks Robey106, Robert Striker107
27VA Boston Healthcare System, Boston, MA, USA; 28US Department of Veterans Affairs, Washington, DC, USA; 29Durham VA Medical Center, Durham, NC, USA; 30Philadelphia VA Medical Center, Philadelphia, PA, USA; 31VA Palo Alto Health Care System, Palo Alto, CA, USA; 32Clinical Epidemiology Research Center (CERC), West Haven, West Haven VA Medical Center, West Haven, CT, USA; 33Cooperative Studies Program Clinical Research Pharmacy Coordinating Center, Albuquerque, New Mexico VA Health Care System, Albuquerque, NM, USA; 34Genomics Coordinating Center, Palo Alto, CA, USA; 35MVP Boston Coordinating Center, Boston, MA, USA; 36MVP Information Center, Canandaigua, Canandaigua VA Medical Center, Canandaigua, NY, USA; 37VA Central Biorepository, Boston, MA, USA; 38MVP Informatics, Boston, VA Boston Healthcare System, Boston, MA, USA; 39Science Operations, VA Boston Healthcare System, Boston, MA, USA; 40Genomics Core, VA Boston Healthcare System, Boston, MA, USA; 41Data and Computational Sciences, VA Boston Healthcare System, Boston, MA, USA; 42VA Informatics and Computing Infrastructure (VINCI), VA Salt Lake City Health Care System, Salt Lake City, UT, USA; 43Statistical Genetics, Durham VA Medical Center, Durham, NC, USA; 44Atlanta VA Medical Center, Decatur, GA, USA; 45West Haven VA Medical Center, West Haven, CT, USA; 46Atlanta VA Medical Center, Decatur, GA, USA; 47Bay Pines VA Healthcare System, Bay Pines, FL, USA; 48Birmingham VA Medical Center, Birmingham, AL, USA; 49Central Western Massachusetts Healthcare System, Leeds, MA, USA; 50Cincinnati VA Medical Center, Cincinnati, OH, USA; 51Clement J. Zablocki VA Medical Center, Milwaukee, WI, USA; 52VA Northeast Ohio Healthcare System, Cleveland, OH, USA; 53Durham VA Medical Center, Durham, NC, USA; 54Edith Nourse Rogers Memorial Veterans Hospital, Bedford, MA, USA; 55Edward Hines, Jr. VA Medical Center, Hines, IL, USA; 56Veterans Health Care System of the Ozarks, Fayetteville, AR, USA; 57Fargo VA Health Care System, Fargo, ND, USA; 58VA Health Care Upstate New York, Albany, NY, USA; 59VA Western New York Healthcare System, Buffalo, NY, USA; 60Ralph H. Johnson VA Medical Center, Mental Health Research, Charleston, SC, USA; 61Columbia VA Health Care System, Columbia, SC, USA; 62VA North Texas Health Care System, Dallas, TX, USA; 63Hampton VA Medical Center, Hampton, VA, USA; 64Richmond VA Medical Center, Richmond, VA, USA; 65Iowa City VA Health Care System, Iowa City, IA, USA; 66Eastern Oklahoma VA Health Care System, Muskogee, OK, USA; 67James A. Haley Veterans’ Hospital, Tampa, FL, USA; 68James H. Quillen VA Medical Center, Corner of Lamont and Veterans Way, Mountain Home, TN, USA; 69John D. Dingell VA Medical Center, Detroit, MI, USA; 70Louisville VA Medical Center, Louisville, KY, USA; 71Manchester VA Medical Center, Manchester, NH, USA; 72Miami VA Health Care System, Miami, FL, USA; 73Michael E. DeBakey VA Medical Center, Houston, TX, USA; 74Minneapolis VA Health Care System, Minneapolis, MN, USA; 75N. FL/S. GA Veterans Health System, Gainesville, FL, USA; 76Northport VA Medical Center, Northport, NY, USA; 77Overton Brooks VA Medical Center, Shreveport, LA, USA; 78Philadelphia VA Medical Center, Philadelphia, PA, USA; 79Phoenix VA Health Care System, Phoenix, AZ, USA; 80Portland VA Medical Center, Portland, OR, USA; 81Providence VA Medical Center, Providence, RI, USA; 82Richard Roudebush VA Medical Center, Indianapolis, IN, USA; 83Salem VA Medical Center, Salem, VA, USA; 84San Francisco VA Health Care System, San Francisco, CA, USA; 85South Texas Veterans Health Care System, San Antonio, TX, USA; 86Southeast Louisiana Veterans Health Care System, New Orleans, LA, USA; 87Southern Arizona VA Health Care System, Tucson, AZ, USA; 88Sioux Falls VA Health Care System, Sioux Falls, SD, USA; 89St. Louis VA Health Care System, St. Louis, MO, USA; 90Syracuse VA Medical Center, Syracuse, NY, USA; 91VA Eastern Kansas Health Care System, Leavenworth, KS, USA; 92VA Greater Los Angeles Health Care System, Los Angeles, CA, USA; 93VA Long Beach Healthcare System, Long Beach, CA, USA; 94VA Maine Healthcare System, 1 VA Center, Augusta, ME, USA; 95VA New York Harbor Healthcare System, New York, NY, USA; 96VA Pacific Islands Health Care System, Honolulu, HI, USA; 97VA Palo Alto Health Care System, Palo Alto, CA, USA; 98VA Pittsburgh Health Care System, University Drive, Pittsburgh, PA, USA; 99VA Puget Sound Health Care System, Seattle, WA, USA; 100VA San Diego Healthcare System, San Diego, CA, USA; 101VA Sierra Nevada Health Care System, Reno, NV, USA; 102VA Southern Nevada Healthcare System, North Las Vegas, NV, USA; 103VA Tennessee Valley Healthcare System, South Nashville, TN, USA; 104Washington DC VA Medical Center, Washington, DC, USA; 105W.G. (Bill) Hefner VA Medical Center, Salisbury, NC, USA; 106White River Junction VA Medical Center, Hartford, VT, USA; 107William S. Middleton Memorial Veterans Hospital, Madison, WI, USA
Funding
This work was supported by funding from the US Department of Veterans Affairs Office of Research and Development, Million Veteran Programme Grant MVP003 (I01-BX003362). This publication does not represent the views of the Department of Veterans Affairs of the US Government. The MEGASTROKE project received funding from sources specified at http://www.megastroke.org/acknowledgments.html. Details of all MEGASTROKE authors are available at http://www.megastroke.org/authors.html. This work was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre at the University Hospitals Bristol National Health Service (NHS) Foundation Trust and the University of Bristol. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. DG and JP-S are funded by the Wellcome 4i Clinical Ph.D. Programme at Imperial College London (203928/Z/16/Z). DG is supported by the British Heart Foundation Centre of Research Excellence at Imperial College London (RE/18/4/34215) and by a National Institute for Health Research Clinical Lectureship at St. George’s, University of London (CL-2020-16-001). ARC and ES are funded by and work in a unit that receives core funding from the Medical Research Council (MRC) and the University of Bristol (MC_UU00011/1). VK is funded by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant (721567). REW is a member of the MRC Integrative Epidemiology Unit at the University of Bristol funded by the MRC (MC_UU_00011/7). SMD was supported by the Department of Veterans Affairs Office of Research and Development (IK2-CX001780). SB is supported by Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (204623/Z/16/Z). PE acknowledges support from the MRC (MR/S019669/1), the NIHR Imperial Biomedical Research Centre, Imperial College London (RDF03), the UK Dementia Research Institute (DRI) at Imperial College London funded by UK DRI Ltd (funded by MRC, Alzheimer’s Society, Alzheimer’s Research UK), and Health Data Research (HDR) UK London funded by HDR UK Ltd (funded by a consortium led by the MRC 1004231). The funding sources for this work were not involved in study design, data analysis, interpretation of results or writing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
DG is employed part-time by Novo Nordisk. JP-S reports personal fees from Novo Nordisk related to consultancy outside of the submitted work. SMD has received grants from the U.S. Department of Veterans Affairs, Calico Labs, and Renalytix AI plc outside the submitted work. All other authors have no conflicts of interest to declare.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the VA Million Veteran Program are listed below Acknowledgements.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gill, D., Zuber, V., Dawson, J. et al. Risk factors mediating the effect of body mass index and waist-to-hip ratio on cardiovascular outcomes: Mendelian randomization analysis. Int J Obes 45, 1428–1438 (2021). https://doi.org/10.1038/s41366-021-00807-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-021-00807-4
This article is cited by
-
Mendelian randomization studies on coronary artery disease: a systematic review and meta-analysis
Systematic Reviews (2024)
-
Revealing brain cell-stratified causality through dissecting causal variants according to their cell-type-specific effects on gene expression
Nature Communications (2024)
-
Emerging Medical Therapies for the Treatment of Obesity in Women with Cardiovascular Diseases
Current Cardiology Reports (2023)
-
Separating the direct effects of traits on atherosclerotic cardiovascular disease from those mediated by type 2 diabetes
Diabetologia (2022)
-
Cardiovascular Disease Risk Reduction and Body Mass Index
Current Hypertension Reports (2022)
