
Abstract
The glycerol 3-phosphate shuttle (GPS) is composed of two different enzymes: cytosolic NAD+-linked glycerol 3-phosphate dehydrogenase 1 (GPD1) and mitochondrial FAD-linked glycerol 3-phosphate dehydrogenase 2 (GPD2). These two enzymes work together to act as an NADH shuttle for mitochondrial bioenergetics and function as an important bridge between glucose and lipid metabolism. Since these genes were discovered in the 1960s, their abnormal expression has been described in various metabolic diseases and tumors. Nevertheless, it took a long time until scientists could investigate the causal relationship of these enzymes in those pathophysiological conditions. To date, numerous studies have explored the involvement and mechanisms of GPD1 and GPD2 in cancer and other diseases, encompassing reports of controversial and non-conventional mechanisms. In this review, we summarize and update current knowledge regarding the functions and effects of GPS to provide an overview of how the enzymes influence disease conditions. The potential and challenges of developing therapeutic strategies targeting these enzymes are also discussed.
Similar content being viewed by others


Introduction
The first observation of glycerol 3-phosphate (G3P) oxidation was reported in 19191. With subsequent studies, the presence of two G3P dehydrogenases (GPDs) and their reactions, constituting the G3P shuttle (GPS), were discovered in the flight muscles by the 1960s2,3. Alongside the malate-aspartate shuttle (MAS), the GPS is one of the two mitochondrial NADH shuttles that transports reducing equivalents from cytosolic NADH across the inner mitochondrial membrane (IMM) to the mitochondrial electron transport chain (ETC)4. The two GPDs of GPS are NADH-dependent, cytosolic G3P dehydrogenase (GPD1) and FAD-dependent, mitochondrial G3P dehydrogenase (GPD2). GPD1 catalyzes the conversion of the glycolytic intermediate dihydroxyacetone phosphate (DHAP) to G3P using NADH. On the other hand, GPD2, which is located on the outer surface of the IMM, catalyzes the reverse reaction: it converts G3P back to DHAP with the reduction of FAD to FADH2, consequently transferring electrons to coenzyme Q (CoQ) in the ETC (Fig. 1)2,5.

GPD1, located in the cytosol, reduces the glycolytic intermediate dihydroxyacetone phosphate (DHAP) to glycerol 3-phosphate (G3P) using two electrons from NADH, thus generating NAD+. On the outer surface of the IMM, GPD2 oxidizes G3P back to DHAP by reducing FAD to FADH2 (dehydrogenase activity). It also mediates electron transfer from FADH2 to ubiquinone (Q) (oxidoreductase activity). The blue dashed arrows denote the flow of electrons (depicted as a green ball with “e”).
Because the activity of GPD1 is lower than that of lactate dehydrogenase in mammalian tissues6 and because GPS is less efficient than MAS in terms of ATP generation (1.5 ATP per NADH for GPS and 2.5 for MAS), the functional roles of GPS were not highlighted until the 1970s, when it was discovered that mammalian brown adipose tissue (BAT) exhibits high activities of GPD1 and GPD27,8. Prominent studies by Albert Lehninger suggested that the MAS accounts for all the NADH transferred to mitochondria generated from glucose to pyruvate catabolism (glycolysis) in tumor cells, might also have led to less interest in the GPS9,10. Currently, more than four decades of research have shown that GPS and its component enzymes work as central hubs in relation to glucose metabolism, lipid metabolism, and mitochondrial respiration. The overall importance of these enzymes was further evidenced by neonatal lethality and substantial metabolic alterations in mice lacking both GPD1 and GPD211. In this review, we combined several critical studies on the roles of the GPS proteins GPD1 and GPD2 to summarize the current understanding of the metabolic and functional impacts of GPS, particularly focusing on their involvement in diseases such as cancer. A list of the involvements of GPDs in various non-cancerous diseases is provided in Table 1. Due to page limitations, we might not have cited all the relevant studies, and due credit should be given to those studies. For readers seeking a detailed historical review of GPD2 up to 2013, we recommend an excellent review by Tomas Mráček and coworkers12.
Glycerol 3-phosphate dehydrogenases: individual enzymes and involvement in metabolic diseases
Glycerol 3-phosphate dehydrogenase 1 (GPD1)
Gene and protein structure
The GPD1 gene is 7306 bp in length and is located on human chr12q13.12. It includes eight exons and seven introns and encodes a protein consisting of 349 amino acids. It does not seem to be an essential gene for life, as orthologs have not been identified in many prokaryotes or even in some eukaryotes. The three-dimensional structure of human GPD1 was solved with X-ray crystallography13, revealing two distinct regions: the C-terminal domain, which has several helical structures where the substrate DHAP binds; and the N-terminal domain, which contains a β-sheet core for NADH binding. This study also revealed the substrate binding and catalytic mechanism, indicating that Arg269 and Lys120 contribute to the binding of the substrate DHAP, and Lys204 polarizes the carboxyl group of DHAP for hydride attack from NADH.
Regulation
The regulation of GPD1 activity seems to occur mostly at the transcriptional level. GPD1 has been reported to be a direct target of peroxisome proliferator-activated receptor alpha (PPARα) in the liver and PPARγ in adipose tissues14. The upregulation of GPD1 by PPARγ and the expected increase in G3P were proposed to be related to the enhanced lipid storage and insulin-stimulated fatty acid uptake in adipose tissues induced by a thiazolidinedione, a PPARγ agonist used for treating type 2 diabetes15. Additionally, androgen-mediated regulation of GPD1 for hepatic gluconeogenesis has been reported16. Another example of transcriptional regulation involves mutual positive regulation between hypoxia-inducible factor 1-alpha (HIF1α) and GPD1 in clear cell renal cell carcinoma (ccRCC)17.
Non-transcriptional regulation was also reported, whereby GPD1 expression was markedly elevated during adipogenesis in mice due to histone H3 lysine K4 (H3K4) methylation18,19. In glycerol-producing yeast, Gpd1 and Gpd2, both of which are homologous to mammalian GPD1, were shown to be inhibited by phosphorylation under different metabolic conditions by AMP-activated protein kinase (AMPK) and target of rapamycin complex 220. However, mammalian cells do not typically operate glycerol fermentation, and similar phosphorylation in higher animals has not been demonstrated. Therefore, more studies are needed to determine the existence and functional relevance of posttranslational modifications.
Functions and implications in non-cancer diseases
As GPD1 generates G3P, which connects carbohydrate and lipid metabolism and is involved in NADH/NAD+ recycling, abnormal activity of GPD1 is expected to cause metabolic diseases. There is evidence showing that GPD1 has pro-obesity effects. Enhanced GPD1 activity has been observed in morbidly obese patients, and correlations between GPD1 expression and obesity, body mass index (BMI), and fat mass were found21,22. As fatty acid synthase (FAS) and ATP citrate lyase (ACLY) activities were lower in obese patients’ adipose tissues, GPD1-generated G3P was suggested to be the driving force for triglyceride (TG) accumulation. In line with this, GPD1 expression in the skeletal muscle of obese individuals was high, and it could be reversed after weight loss surgery that improved BMI and insulin resistance, and was associated with TG accumulation23. On the other hand, other studies have shown that GPD1 plays a role in protection against hyperlipidemia or liver steatosis. Basel-Vanagaite et al. demonstrated that a truncation mutation of GPD1 is associated with increased secretion of TGs, leading to hypertriglyceridemia, which was supported by cell-based experiments24. Subsequently, several clinical mutations in GPD1 have been reported with consistent phenotypes, such as hepatomegaly, steatosis, or hypertriglyceridemia, often in infants25,26,27,28. For example, a compound heterozygous mutation in GPD1 leading to the absence of the protein in the liver of a female infant caused hepatomegaly, steatosis, and hypertriglyceridemia25. This study also suggested that GPD1 deficiency leads to lipid accumulation via two mechanisms: DHAP accumulation or decreased fatty acid oxidation. Given the pro-obesity or anti-lipid accumulation activity of GPD1, as stated above, it is difficult to propose a unified idea on the role of GPD1 in human obesity.
In comparison, mice lacking GPD1 exhibited the most extensive metabolite changes in skeletal muscle, with many minor changes in the liver and kidney (limited to those in which GAPDH is upregulated during glycolysis)29,30. Notably, these mice are not laboratory-generated GPD1 knockout (KO) mice but are a subline of BALB/c (BALB/cHeA) mice with a natural GPD1 mutation leading to the loss of GPD131,32. Despite the metabolite alterations (in G3P, DHAP, and the lactate/pyruvate ratio), the exercise tolerance and pancreatic islet function of the mutant mice were normal. On the other hand, later studies revealed that mice without GPD1 had higher fatty acid oxidation in skeletal muscle, leading to higher exercise endurance and less weight gain33,34, possibly regulated by EP300 interacting inhibitor of differentiation 1 (EID1)35. Although other roles of GPD1, such as protection from cell death upon treatment with oxidants, have been reported with a cell line36,37, confirmation is necessary in in vivo systems. Therefore, the exact roles of GPD1 in obesity, muscle function, and protection against oxidants have not been well established. In the opposite context of the GPD1 level, the overexpression of GPD1 partially rescued survival, the alpha-hydroxybutyrate level (representing the NADH/NAD+ ratio), motor function, and neuroinflammation in the Ndufs4-/- mouse modeling human neuropathies38. As the study focused on the roles of GPD1 in complex I (CI)-compromised conditions, it remains unclear whether those functions are applicable to normal conditions.
It may be worthwhile to comment on a related protein called GPD1L, whose gene was first discovered in 200239. This gene is located on human chr3p22.3, very close to SCN5A, whose mutation is associated with Brugada syndrome, an autosomal-dominant form of idiopathic arrhythmia potentially leading to sudden death40. The protein encoded by GPD1L has 84% amino acid homology to GPD1 and was proposed to be associated with the plasma membrane41. It is possible that, through its association with SCN5A, GPD1L is also associated with Brugada syndrome and sudden cardiac death42. A GPD1L mutation appears to reduce its association with SCN5A, a sodium channel, consequently lowering the inward sodium current in the heart42,43. Additionally, GPD1L expression has been shown to be downregulated in certain types of cancer, such as head and neck squamous cell carcinoma and renal cell carcinoma39,44. Given that GPD1 mutation is associated with transient infantile hypertriglyceridemia, the disease association and intracellular localization seem different from those of GPD1L.
Overall, there seem to be at least three variables to consider in understanding GPD1 functions: species, tissues, and compensation. As human GPD1 mutations were identified more recently than mutations in mice, future studies should devote more attention to the differences between the species. In mouse experiments, GPD1-deficient mice are not specifically generated for GPD1 deletion; therefore, they might have other unknown abnormalities. For tissues, mice deficient in GPD1 exhibited very different metabolite changes according to tissues. This might be related to tissue-specific compensation, e.g., less compensation by glycerol kinase in muscle than in liver or kidney. Compensation for GPD1 activity by GPD1L might also be possible, but the GPD1 gene is responsible for almost all glycerophosphate dehydrogenase activity in all adult tissues, with the liver and possibly the kidney depending entirely on GPD132. Therefore, GPD1L compensation may need to be further tested experimentally.
Glycerol 3-phosphate dehydrogenase 2 (GPD2)
Gene and protein structure
The GPD2 gene on human chr2q24.1 spans a 150,025-bp region, including 17 exons and 16 introns, and its coding sequence encodes 727 amino acid residues. Unlike GPD1, GPD2 is a conserved protein with an ortholog even in E. coli. The bacterial version, GlpD, is associated with the bacterial cell membrane, corresponding to the mitochondrial membrane in eukaryotes, and is devoid of ~100 residues homologous to calmodulin at the C-terminus45. GlpO is another ortholog of GPD2 in bacteria. GlpO is a soluble, cytosolic, and FAD-linked glycerol phosphate oxidase that can reduce O2 to H2O2 in some heme-deficient bacteria, such as Enterococcus casseliflavus46,47. To date, the three-dimensional structure of human GPD2 has not been determined, and much structural and mechanistic information has been obtained using the crystal structure of detergent-solubilized E. coli GlpD48, which has ~45% sequence similarity (30% identity) with the mammalian enzyme. Bacterial GlpD has two distinct regions with opposite electrostatic potentials. The C-terminal domain, which has a negative electrostatic surface, assists in the recognition of the phosphate group. The N-terminal domain consists of an FAD- and substrate-binding domain and has overall positive potential, enabling its interaction with negatively charged phospholipids of the membrane. Notably, the base region of the FAD-binding domain was suggested to be embedded into the lipid bilayer by ~12–15 Å, based on its observed interaction with detergent molecules. Interestingly, this region also contains a putative ubiquinone docking site comprising a hydrophobic plateau, providing insights into how electrons are transferred to CoQ. Although the bacterial enzyme structure provides valuable information on substrate binding, lipid-enzyme interactions and a possible electron transfer mechanism have yet to be confirmed in the context of mammalian enzymes. The dimeric form of bacterial GlpD in the crystal structure is consistent with a suggested functional dimer for GPD249, but this could still be due to crystal packing. A recent study of the purification of active GPD2 suggested a monomeric status50; therefore, the true oligomeric status may require future studies.
Regulation
Compared to GPD1, which is expressed in almost all tissues29, the expression of GPD2 is quite selective and is thus regulated by more factors. However, transcriptional regulation is the primary mechanism, and GPD2 is regulated by at least three different promoters, namely, promoters A, B, and C51,52,53. These promoters are associated with different 1st exons in tissue-specific manners (e.g., promoter A in the brain, promoter B in all tissues, and promoter C in the testis), which might partially explain the highly differential expression of GPD2 across tissues. Interestingly, ubiquitous promoter B, which is the only promoter regulated by thyroid hormone in the liver53, does not respond to thyroid hormone in tissues with high GPD2 expression (e.g., brown adipose tissue, brain, and testis), suggesting that some tissue-dependent factors, such as Sp1, are involved in regulation by promoter B54,55. Additional complexity in promoter B regulation was reported wherein the promoter B is sensitive to thyroid hormone in rats54 but not in humans56, thus suggesting species-differential regulation. Although it is not exactly a transcriptional regulation, miR-1 and miR-206 have been shown to regulate the 3′-UTR of GPD2 as a downstream mediator of NRF2 signaling57.
Other mechanisms, including cofactors or posttranslational modifications, can regulate GPD2. Ca2+ is an important cofactor, possibly acting through the C-terminal putative calmodulin-like domain that is absent in the E. coli GlpD structure48. As substrate binding occurs in a different region, it may be an allosteric regulator that enhances GPD2 activity by lowering the Km for the substrate58,59,60. A recent study revealed the functional relevance of the Ca2+-mediated regulation of the GPS in terms of ATP generation for the electrical activity of hippocampal neurons61. This mitochondrial Ca2+-regulated GPS activity, which involved GPD2, was suggested to act as a backup system for more prominent Ca2+-dependent ATP generation systems, such as the MAS62 and mitochondrial calcium uniporter-driven activation of Ca2+-sensitive TCA enzymes63. Other important allosteric regulators of GPD2 are free fatty acids (FFAs) and their acyl-CoA esters. Even in the presence of Ca2+, palmitoyl-CoA inhibited the activity of GPD2 at a very low concentration by decreasing G3P availability in a competitive manner64. Inhibition by FFAs is more complex in that oleate inhibited oxidoreductase activity, which transfers electrons from GPD2 to CoQ, but not dehydrogenase activity, which oxidizes G3P in a non-competitive and BSA-relievable way65. Other studies have shown that the inhibitory effects of FFAs might occur by altering membrane microviscosity or composition66,67. It might be suggested that a high concentration of FFAs may shift metabolism toward glycerolipid synthesis through a dual mechanism: 1) serving as a substrate for esterification or 2) increasing G3P by inhibiting GPD2. Not only small molecules but also protein factors have been reported to regulate GPD2. GCN5L1, known as a mitochondrial acetyltransferase, regulates GPD2 activity by protein interactions to support gluconeogenesis in the liver, as demonstrated by respiration measurements and coimmunoprecipitation68.
Post-translational or covalent modification of GPD2 also controls its activity. Phosphorylation at T10 by protein kinase delta enhances its substrate affinity in glioma cells69. Although some studies have proposed that GPD2 has catalytically important SH- groups70,71, it is only modestly inhibited by thiol-targeting chemicals. Instead, its activity is more profoundly inhibited by chemicals that modify tyrosyl, lysyl, or histidyl side chains71. Not only modification but also removal of residues affects GPD2 activity. GPD2 in prostate cancer cells forms functional dimers only after the removal of the N-terminal 42 residues49. This processing seems to be mediated by the inner membrane protease IMMP2L, which, unlike GPD2, does not exhibit a biased distribution across tissues. Nevertheless, it is unclear whether the N-terminal sequence is required for the enzyme activity per se or whether it is just for the mitochondrial targeting and inner membrane localization of GPD2. Additionally, the tissue specificity of this process needs to be further studied, especially considering the phosphorylation of T10 and the subsequent activation of GPD2 in glioma cells, as stated above69.
Functions and implications in non-cancer diseases
The high activity of GPD2 in BAT51, along with the fact that GPD2 was originally discovered in the flight muscles of Drosophila2, suggest that GPD2 plays a significant role in energy production and utilization as an important factor for thermogenesis. In one study, GPD2 KO mice did not exhibit hypothermia or defective gross thermogenicity, and the temperature increased normally in response to thyroid hormone treatment72. However, a later study employing another breed of GPD2 KO mice revealed that there was a small but noticeable decrease in energy expenditure, despite the increase in the serum thyroid hormone level73. The authors suggested that GPD2 is important in thyroid hormone-mediated thermogenesis and further showed that the absence of GPD2 caused a state of sustained cold stress that incurred compensatory heat generation through BAT and skeletal muscle uncoupling protein 3 expression. The apparent discrepancy between the two studies might be reconciled considering the supraphysiological thyroid hormone used in the first study, which involves multiple thermogenic mechanisms not dependent upon GPD2 and normal gross thermogenesis upon the deletion of possible thermogenic genes74,75. The KO mice in the second study were further characterized as having a “thrifty” phenotype by the same group76.
A common phenotype between the above two breeds is a small but significant weight loss (~20% in the first and ~5% in the second). However, another GPD2 KO breed did not experience weight loss, but defects in muscle regeneration after injury were detected77. Although muscle development and myofibril size remained normal with no gross histological defects, GPD2 KO mice exhibited impaired muscle regeneration and myoblast differentiation, which may have implications for decreasing muscle mass in obese and diabetic patients. Mechanistically, GPD2 increased the NAD+/NADH ratio accompanied by activation of the AMPK/PGC1α axis, ultimately resulting in mitochondrial biogenesis. An earlier GPD2 KO breed exhibited higher G3P and lactate/pyruvate ratios in muscle76, suggesting increased glycolysis; future studies should perform a comparison with the GPD2 regulation of muscle glycolysis. Additionally, despite the decrease in white adipose tissue (WAT) in an earlier KO breed72, the reported inconsistency in weight loss makes it difficult to determine the effects of GPD2 on WAT adiposity, weight changes, or obesity at this point. Interestingly, liver-specific GPD2 loss caused ER stress-induced liver steatosis through increased release of mitochondrial calcium via the permeability transition pore (PTP)78. A mechanistic investigation revealed that GPD2 induced the ubiquitin-mediated degradation of cyclophilin D that activates PTP. Therefore, the regulation of lipids by GPD2 may also be tissue specific, employing different pathways.
As the NAD+/NADH ratio is directly related to the function of GPD2 as one of the two NADH shuttle systems, its physiological involvement has been studied where the GPD2 level is high. One such tissue is pancreatic beta cells, which secrete insulin with high aerobic glycolysis activity79,80. As early as the 1980s, GPD2-mediated bioenergetic metabolism was implicated in glucose-mediated insulin secretion, linking GPD2 to diabetes81,82,83. Later, a study with pancreatic islets from GPD2 KO mice revealed that blocking both NADH shuttles (GPS and MAS) is required for the inhibition of glucose-induced insulin secretion84, revealing the redundant roles of GPS and MAS. Additionally, overexpression of GPD2 could not rescue the impairment in glucose-induced insulin secretion in GPD2-low GK rats85 or GPD2-low rodent cells86. Compared to these genetic studies performed in rodent systems, GPD2 mutations in human patients were linked to type 2 diabetes87,88, and autoantibodies against GPD2 were detected in insulin-dependent diabetic patients89,90. Therefore, there might be differences between rodent and human systems, and it will be interesting to determine whether there are coexisting mutations or malfunctions in MAS in GPD-mutant human patients.
Another tissue with relatively high GPD2 activity is the brain. The absence of an increase in lactate in brains with ARALAR deficiency (a component of the MAS)91, in contrast to the increase in lactate in the brain in most cases of mitochondrial dysfunction92,93,94, was attributed to GPS being a major NADH shuttle in astrocytes, not in neurons. Another study of the brain showed that high GPD2 activity was present in areas with high synaptic density in the mouse brain, such as the hippocampal stratum oriens, suggesting a role for GPD2 in neurotransmission95. However, there is controversy regarding the presence of GPD1 or GPD2 in different types of brain cells. For example, GPD1 is selectively expressed in oligodendroglial cells96,97, in contrast to the selective presence of GPD2 in neurons, making the role of GPS as a NADH shuttling machinery irrelevant in the brain95. Nevertheless, the GPS might function with another GPD1 isotype, GPD1L, and the actual activities of cytosolic and mitochondrial GPD were observed at similar levels91. Therefore, histological (antibody staining), pharmacological (inhibitors), and biochemical (enzyme activity) experiments have provided somewhat discordant views on the roles of GPS in different brain cell types, and studies incorporating different approaches in the same setting should be performed to obtain better insight. Apart from GPS activity, targeting GPD2 with metformin in brain abnormalities has been studied98,99, but the effect may not be specifically due to GPD2 inhibition, considering the various effects of metformin.
NAD+/NADH shuttling by GPD2 transfers reducing equivalents to the mitochondrial ETC, and overactivation of this process can cause reactive oxygen species (ROS) generation through reverse electron transport (RET)100,101,102. Interestingly, this phenomenon has been found in immune cell modulation. During T-cell activation, T-cell receptor (TCR) signaling shifted glycolytic flux from the GAPDH direction to the GPD1 direction using DHAP as the substrate103. The subsequent activation of GPS led to RET through the hyperreduction of ubiquinone and the generation of ROS at CI. This was followed by ROS-induced NF-kB-dependent gene expression, such as that of IL-2 and IL-8. A slightly more intricate role of GPD2-mediated RET was also reported in macrophages under LPS stimulation104. The enhanced activity of GPD2 was responsible for boosting glucose oxidation to support acetyl-CoA production and thus to provide materials for histone acetylation for the induction of pro-inflammatory genes in the acute phase. However, prolonged exposure to LPS and long-lasting GPD2 activation led to RET and a reduction in oxidative metabolism, reversing histone acetylation and initial macrophage activation. Thus, GPD2 was proposed to be a critical switch in the time-dependent activation and tolerance of macrophages to LPS stimulation.
The GPD2-ROS relationship also seems to be relevant in rather unrelated tissues, including sperm, placenta, and heart, and even in some tissues where GPD2 activity and expression are low, such as the kidney105. During sperm capacitation, GPD2 was reported to be phosphorylated, and its activity was correlated with the acrosome reaction106. Using GPD2 KO mice, it was further shown that sperm capacitation requires GPD2 activity for ROS generation in spermatozoa107. The placenta has disproportionately higher GPD2 levels among mitochondrial respiratory enzymes than other tissues108,109 and exhibits high GPD2-dependent generation of hydrogen peroxide (H2O2)110, the physiological meaning of which is not clear at this point. More relevant to pathological conditions is probably the role of GPD2-mediated ROS in ischemia‒reperfusion injury (IRI), where resumed blood flow after blood vessel blockage induces paradoxical tissue damage111. GPD2-mediated ROS seem to be at least partly responsible for cell death during IRI, which is reduced by miR-210, which inhibits GPD2112. In comparison, a protective role of GPD2 in ischemic disease was also reported, where GPD2 deficiency exacerbated cardiac dysfunction during myocardial infarction (MI)113. The activation of GPD2 under ischemic conditions, which might result from the increase in intracellular Ca2+ in MI, was necessary for ATP synthesis from glycerol as an adaptation to the limited oxygen supply. Another interesting ROS relationship was found in kidney podocytes during diabetic kidney disease114, where GPD2 was found to inhibit the receptor-for-advanced-glycation-end-product (RAGE) pathway. RAGE inhibition protected podocytes by enhancing mitochondrial biogenesis/metabolism and lowering ROS, the co-occurrence of which is interesting. Hence, the GPD2-ROS relationship can be either physiological (protective) or pathological (destructive) and, therefore, should be understood in specific contexts.
Many of the roles of GPD2 in pathophysiological conditions have been studied using mice with genetic deletion of GPD2 (GPD2 KO). Several different breeds of KO strains (including one liver-specific KO78 and another podocyte-specific KO114) have been generated, and some of those have been used in multiple studies by different research groups72,77,84,104. For example, the mice generated by Eto et al. were used in studies by DosSantos73, Ishihama113, and Kota107 to address phenotypes in different tissues. Additionally, the two tissue-specific KO mice used the same background GPD2flox/flox mice78,114. As not all the phenotypes were reproduced in different breeds, it might be important to consider the lineage of the KO breeds. Additionally, some studies have focused on particular tissues, and these tissue-specific phenotypes may need to be further confirmed in tissue-specific KO mice in the future. Furthermore, species considerations may need to be taken when interpreting GPD2 KO phenotypes. For example, the citrin KO mouse model failed to exhibit symptoms of human citrin deficiency, which required additional GPD2 KO115,116, despite expected changes in some of the molecular metabolic phenotypes117. This was suggested to occur due to higher GPD2 activity in the mouse liver than in the human liver, which could have compensated for the phenotypes of suppressed MAS activity in citrin KO mice115,118. GPD2 mutations linked to diabetes87,88 and haploinsufficiency of GPD2 in mild mental retardation119 are other examples not observed in mouse systems. Overall, these studies demonstrate interesting and complex roles of GPD2 in surprisingly diverse tissues of endocrine, nervous, reproductive, immune, adipose, muscular, and cardiovascular origin.
Glycerol phosphate dehydrogenases in cancer
Reprogramming of cellular metabolism is one of the hallmarks of cancer120,121. Well-known oncogenes or tumor suppressor genes, such as KRAS, P53, MYC, and EGFR, have profound effects on metabolism122,123,124. Additionally, growth factor signaling pathways critical for tumor growth, such as the PI3K-AKT and MEK/ERK signaling pathways, directly engage metabolic pathways through mTOR or c-myc in cancer125,126. Furthermore, glycolysis and lipid metabolism are widely altered in cancer127,128,129. Therefore, it should not be surprising that the components of GPS, which are at the crossroads of glycolysis and lipid metabolism, are abnormally modulated in cancer (Fig. 2). Interestingly, GPD1 and GPD2 expression is negatively correlated in most cancer types130, even though they are components of the GPS. Supporting this is that the roles of each GPD are mostly opposite in cancer, with GPD1 generally acting as a tumor suppressor and GPD2 acting as a tumor promoter. Individual examples and possible mechanisms for regulating cancer cell proliferation by altering cellular metabolism are presented below.

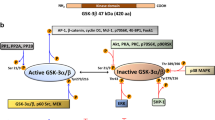
GPD1 and GPD2 affect cancer cell growth through several mechanisms. GPD1 has mostly tumor-suppressive functions, while GPD2 has mostly tumor-promoting functions, with some studies suggesting otherwise. GPD1 mediates the production of G3P, which has been shown to directly hinder cancer cell growth in some studies or to exert indirect anticancer effects by being a precursor of pro-apoptotic LysoPC. In other cases, G3P formation might enhance cancer growth by contributing to membrane formation through GPLs. Several mechanisms have been reported for the cancer-promoting role of GPD2. GPD2 produces DHAP, which acts, along with fatty alcohols, as an important substrate for ether lipid (plasmalogen) biosynthesis. Ether lipids promote AKT and downstream mTOR signaling by enhancing lipid raft localization and activation of AKT, ultimately supporting cancer growth via a non-bioenergetic mechanism. The bioenergetic contribution of GPD2 to cancer growth has also been reported, wherein it transports reducing equivalents to CoQ, a component of the electron transport chain for ATP generation. Elevated ATP levels may directly fuel cancer cell growth and indirectly impact proliferation by inhibiting AMPK activity and sequentially activating mTOR signaling. The reducing power of GPD2 may also neutralize lipid peroxyl radicals, protecting cancer cells from ferroptosis. Overactivation of GPD2 may trigger reverse electron transport (RET), which results in the production of ROS. Found mostly in immune cells, this RET-driven ROS either activates NF-kB-dependent gene expression or inhibits oxidative metabolism in different cell types. Although not explicitly studied in cancer cells, these immune-related activities may also have implications for cancer cell growth. Dashed arrows and a brown star denote electron transfer and ROS, respectively. See the text for details. AMPK AMP-activated protein kinase, DHAP dihydroxyacetone phosphate, DHODH dihydroorotate dehydrogenase, FAR1 fatty acyl-CoA reductase 1, G3P glycerol 3-phosphate, GNPAT glyceronephosphate O-acyltransferase, GPL glycerophospholipid, PLOO phospholipid hydrogen peroxide radical, Q coenzyme Q/ubiquinone, LysoPC lysophosphatidylcholine, QH2 reduced form of coenzyme Q/ubiquinol.
GPD1 acts as a tumor suppressor in most cases
It was initially demonstrated that GPD1 activity is markedly lower in malignant tissues of the colon and rectum than in their normal counterparts131. Subsequently, consistent studies have reported that GPD1 expression is significantly reduced in several other cancers, such as breast, lung, bladder, and kidney cancer, and that a low expression level of GPD1 is significantly correlated with a poor survival rate17,132,133. Particularly for breast cancer, a proteomics-based study revealed the lowest expression of GPD1 protein in triple-negative breast cancer (TNBC), suggesting a possible relationship between GPD1 and hormone receptors in breast cancer134. Interestingly, a decreased GPD1 level was also found in serum samples of TNBC patients, suggesting the possible use of GPD1 as a diagnostic biomarker135. In kidney cancer, overexpression of GPD1 downregulated various lipid synthesis genes, leading to a decrease in lipid droplets17, the enrichment of which is a characteristic and cancer-requiring feature of ccRCC136,137,138. In addition to these lipid metabolic changes, decreased cell proliferation, migration, and invasion, as well as the inhibition of xenografted tumor growth, were also observed, consistent with the correlation between high GPD1 protein levels and better ccRCC patient survival.
It should also be mentioned that some reports have suggested that GPD1 plays a tumor-promoting role. For example, a significantly higher GPD1 level was found in dormant brain tumor stem cells than in normal neuronal stem cells, which can contribute to differences in glycerophospholipid (GPL) metabolism and tumor relapse after chemotherapy139. KO of GPD1 in these cells downregulated genes related to stem cell identity, cell cycle progression, and the mTOR pathway, as well as the phosphorylation of S6. In another study under hypoxic conditions, GPD1-driven G3P synthesis was shown to maintain 143B cancer cell proliferation38, and GPD1/GPD1L double knockdown (KD) in mouse kidney cancer cells inhibited lipid synthesis and in vitro/in vivo tumor growth130. An observational bladder cancer study also suggested correlated increases in GPD1 and fatty acid synthetic enzyme activities in tumor tissues140. With more clinical studies showing lower GPD1 in cancer tissues, it seems that the tumor-promoting mechanism of GPD1 may operate in particular contexts. These may include cancer types (e.g., ccRCC vs. non-ccRCC kidney cancer or bladder vs. kidney cancer), species (mouse vs. human), or experimental conditions (e.g., hypoxia, glycolysis, or dual knockdown of GPD1 and GPD1L). The exact extent of these GPD1 tumor-promoting roles and the context may require further studies.
Mechanistic aspects of GPD1 in cancer cell proliferation
As many studies have demonstrated the low expression of GPD1 in various types of cancer and have suggested that it is a tumor suppressor, most of the mechanistic studies have investigated the effects of GPD1 overexpression on cancer cell proliferation and growth. There seem to be several mechanisms through which it either is regulated or regulates cell survival/growth pathways. In breast cancer cells, Zhou et al. showed that GPD1 is a direct target of a microRNA (miRNA), miR-370, which downregulates it post-transcriptionally132. As miR-370 is known to be upregulated in breast cancer cells and related to tumor progression, this study revealed an upstream regulator of GPD1 in breast cancer. Regarding the downstream mechanisms by which GPD1 regulates cancer growth, two studies proposed the common involvement of the reaction product G3P in different contexts. In one study, GPD1 and metformin synergized to increase G3P, which exhibited a direct cancer-inhibitory effect in several cancer types141. For the cancer-inhibition mechanism, the authors specifically excluded possible pathways of mTOR or methylglyoxal toxicity by increased G3P and, instead, proposed the inhibition of mitochondrial respiration and ATP generation by G3P. Notably, the increase in G3P induced by GPD1 overexpression (OE) was not statistically significant, but GPD1 OE alone, without metformin, reduced in vivo tumor xenograft growth and mitochondrial oxygen consumption. In another study on bladder cancer133, GPD1 OE significantly increased G3P and NAD+ levels, as well as that of lysophosphatidylcholine (LysoPC). LysoPC activated its receptor (platelet-activating factor receptor; PAFR)-mediated transient receptor potential vanilloid 2 (TRPV2) channel opening, ultimately leading to Ca2+ influx and apoptosis. Therefore, the downstream pathways from G3P in the two studies seemed to diverge to mitochondrial respiration and to membrane channel-mediated Ca2+ influx. Future studies should investigate whether these differences are due to different cancer types or whether there are commonalities in these mechanisms. Additionally, the use of several tens or hundreds of millimolar concentrations of G3P for observing cancer cell phenotypes in both studies might need to be considered in future studies for any effect of osmolarity.
However, another study employing the GPD1 OE approach revealed interesting mutual upregulation between HIF1α and GPD1, which affects lipid metabolism in kidney cancer17. GPD1 OE suppressed mitochondrial basal respiration and ATP synthesis, activating AMPK and inhibiting mTOR. This, in turn, decreased the levels of lipid droplets, which are primarily composed of neutral triacylglycerol and cholesterol esters142, although it increased the total phospholipid content. It would be worthwhile to compare these findings with those of another study on mouse kidney cancer, which suggested apparently different roles for GPD1 in lipid synthesis130, as lipid accumulation is a characteristic phenotype in ccRCC143,144, the most common type of kidney cancer145. Using GPD1/GPD1L double KD in mouse renal adenocarcinoma Renca cells, Yao et al. observed a decrease in lipid synthesis, particularly GPL, and mitochondrial respiration. From a lipid metabolic perspective, the increased synthesis of phospholipids by cytosolic GPD was identical in both studies, but the characteristic features of ccRCC, the increase in neutral lipids and its role in tumors, seem to have been better addressed in the former study. Yao et al. also suggested that GPL synthesis and redox homeostasis for tumor growth are driven by efficient G3P production by GPD1/GPD1L, which is different from the inhibition of tumor growth by G3P and GPD1 reported by two other studies in different cancers, as stated above133,141. The tumor promotion by GPD1, as shown in Yao et al.’s study, was also mechanistically suggested with several models under hypoxia or dysfunctional ETC conditions38. GPD1 KO significantly abolished G3P synthesis and increased the NADH/NAD+ ratio when the ETC was inhibited in 143B (osteosarcoma) and HeLa (adenocarcinoma) cells, consistent with all other studies showing G3P generation by GPD1. However, GPD1 KO increased the sensitivity of cancer cells to the antiproliferative effects of ETC inhibitors and suppressed xenograft tumor growth under ETC inhibition, indicating that GPD1 promoted tumor growth. Moreover, GPD1 OE promoted the activation of GPS to rescue cancer cell proliferation under CI inhibition, demonstrating that GPS can compensate for mitochondrial dysfunction in ATP production and redox homeostasis.
Overall, GPD1-mediated G3P production was consistently observed in all the cited studies employing both the GPD1 KO/KD and OE approaches. The downstream effects of increased G3P include different mechanisms for the tumor-promoting and tumor-suppressing activities of GPD1. In this respect, the results from the GPD1 OE and GPD1 KO/KD setups may not necessarily be opposite, as compensation mechanisms may differ and, therefore, should be compared with the particular approach in mind. For the direct effect of G3P, the compound concentration, hence osmolarity, and its permeability across the cell membrane should be carefully considered in future studies. These findings suggest that despite the availability of an atomic-level structure of human GPD1, the functions of GPD1 in cancer have yet to be fully elucidated.
GPD2 is upregulated in many cancer types
In contrast to the generally lower expression of GPD1 in cancer, an early study showed that patients with hepatocellular carcinoma (HCC) possess higher GPD2 activity than normal controls146. Subsequently, GPD2 activity has been shown to be higher than succinate dehydrogenase activity in various cancer types belonging to the amine precursor uptake decarboxylation system79. Moreover, GPD2 expression and activity were higher in prostate and liver cancer cell lines and tissues than in their normal counterparts147,148. Additionally, in prostate cancer, a recent study revealed an interesting posttranslational processing of GPD2 in the formation of a functional dimer (see above)49. As the proposed processing enzyme IMMP2L, along with GPD2, inhibits senescence149, it may be interesting to study whether GPD2 also inhibits oncogene-induced senescence that suppresses cancer initiation and progression150.
The effect of GPD2 on drug sensitivity and tumor grade/prognosis was also investigated. The expression of GPD2 was higher in thyroid cancer tissues than in normal thyroid tissues151. More importantly, thyroid cancers with higher GPD2 levels and metastatic tumors derived from them responded better to metformin than did those with lower GPD2 levels. Posttranslational modification may be related to GPD2 involvement in tumor grade in glioma69. The level of the phosphorylated form of GPD2 at threonine 10 (GPD2 pT10), which has a higher substrate affinity, was higher in high-grade (grade IV) glioblastoma than in grade I/II astrocytoma despite no difference in the unmodified GPD2 levels. Additionally, patients with an above-median GPD2 pT10 status had worse survival than those with a below-median GPD2 status. Like GPD1139, GPD2 is related to cancer cell stemness. In the Huh-7 HCC cell line, a subpopulation with the stem cell marker CD133 exhibited increased GPD2 levels152 and in vivo tumorigenicity153, and KD of GPD2 decreased anchorage-independent cell proliferation152. An analogous involvement of GPD2 in neuronal cancer cells was reported by the same group154. A broader investigation of the expression of GPD2 revealed that its level is higher in tumors than in normal tissues in most cancer types and that higher GPD2 expression is correlated with poorer survival in some cancers155. Notably, when all the cancer types were combined, the expression levels were higher in tumor tissues, and higher GPD2 levels were correlated with poorer survival.
Not surprisingly, there may be some cases in which the above cancer-promoting roles do not apply. Highlighting the anti-correlation of GPD1 and GPD2 in cancer, a recent study showed that GPD1 has tumor-promoting effects and that GPD2 has tumor-suppressing effects130. They showed that GPD2 KD in mouse renal adenocarcinoma Renca cells enhanced tumorigenicity in the syngeneic graft setting, which was recapitulated in human ccRCC cell lines (786-O and Caki-1) in vitro. Another study reported that GPD2 inhibits melanoma metastasis in vitro and in vivo through downregulation of NRF2156. Interestingly, NRF2 also has well-known dual roles in tumor initiation, progression, and chemotherapy157. Overall, GPD2 seems to have generally tumor-promoting effects, with higher expression in tumor tissues. Nevertheless, as in the GPD1 case, there can be some specific contexts where GPD2 may have tumor-suppressive roles, and the extent of these effects should be addressed in future studies.
GPD2 regulates cancer progression via various mechanisms
As stated above, there are still apparent discrepancies in the role of GPD2 in cancer, and therefore, it is crucial to study the detailed mechanism by which GPD2 regulates cancer growth. Along with the increased activity of GPD2 in prostate cancer cells, prostate cancer cells produce 2- to 3-fold more H2O2147 and express higher levels of antioxidant enzymes, including catalase, MnSOD, and CuZnSOD, than normal prostate epithelial cells148. COX levels were low in a subset of the cell lines. Increased ROS are observed in many cancers158, and ROS can cause DNA mutations that can initiate carcinogenesis159. Nevertheless, to protect against too much damage, cancer cells also engage in detoxifying mechanisms160. As GPD2-specific superoxide production is comparable to that at other major production sites in mitochondria161 and G3P can be a significant contributor to cellular H2O2162, the above results in prostate cancer seem to point toward GPD2-driven ROS in cancer. Despite evidence of GPD2-driven ROS generation in different tissues and its roles in tissue functions105,161,163, its contribution to cancer still requires more evidence in terms of the involvement of different ROS forms and regulators of GPD2-driven ROS generation. Additionally, how ROS is increased may need to be considered, as the glycolysis-driven increase in ROS reported in prostate cancer148 contradicts the observation that prostate cancer cells tend to have less glycolytic flux than normal prostate cells49,164.
In other cancers, enhanced glycolytic flux is a well-established phenomenon (Warburg effect), and the ensuing bioenergetic metabolism is known to be important for cancer cell growth127,129. In fact, the physiological relevance of GPD2 has been ascribed to its involvement in bioenergetics through glucose metabolism. In cancer, GPD2-driven bioenergetic mechanisms involving glucose have also been described in several cancer types. For example, a decrease in G3P in human liver cancer tissue compared to normal tissue was described with increased glycolytic flux165, suggesting increased GPD2 activity. Enhanced GPD2-mediated G3P consumption might occur in para-preneoplastic hepatocytes, rather than in preneoplastic cells themselves, to supply glucose in the early stage of rat liver cancer166. In glioma, macrophage-derived IL-1β induces the activation of GPD2 through interaction with PKCδ, ultimately leading to enhanced glycolysis and proliferation69. Although not explicitly stated, an increase in the DHAP/G3P ratio due to the activation of GPD2 and hence a decrease in glycolysis may contribute to increased glycolytic flux, as measured by 13C-lactate generation from 13C-glucose. Among the available studies, studies on the effect of metformin on thyroid cancer have focused on the bioenergetic contribution of GPD2 to cancer growth151. In line with the finding that GPD2 is a target of metformin in gluconeogenesis167,168, metformin was shown to lower GPD2 expression for its suppressive activity on thyroid cancer. This inhibitory activity resulted in decreased oxidative phosphorylation (OXPHOS), leading to decreased growth of thyroid cancer cells. In further support of the bioenergetic role of GPD2, the overexpression of GPD2 in thyroid cancer cells increased mitochondrial respiration and ATP production, resulting in increased cell growth. They also showed that metformin inhibited the metastasis of thyroid cancer cells with high GPD2 and OXPHOS levels but not those with lower GPD2 and OXPHOS levels. The potential of GPD2 as a target of metformin for its anticancer activity is also notable considering that both CI-AMPK-mTOR pathway modulation and IGF pathway modulation through lower blood insulin have been conventionally suggested to explain the mechanism underlying the anticancer activity of metformin169,170. The involvement of GPD2-mediated oxidative metabolism, e.g., oxygen consumption and ROS production, has also been described in prostate cancer cells compared to normal epithelial prostate cells49. Both fully fledged cancer cells and cancer stem cells may depend on GPD2-mediated ATP synthesis for sphere formation and growth, as shown for liver and neuroblastoma cancer cells by the same authors152,154.
Although the ROS or bioenergetic mechanism of GPD2 in cancer growth is related to well-established functions of GPD2 in normal physiology, recent reports have studied less-explored aspects of GPD2-mediated metabolism for cancer growth or survival: ether lipids or lipid peroxidation. Although GPD2 is considered the crossroad of glucose and lipid metabolism, its role is mainly attributed to G3P, the substrate of GPD2 and the rate-limiting metabolite in glycerolipid synthesis171,172. In comparison, DHAP is the product of the GPD2-mediated reaction and the starting substrate for ether-linked lipids173,174,175. With 4T1 mouse breast cancer cells, it was shown that GPD2 is causally involved in tumorigenesis both in vitro and in vivo155. Importantly, it was not GPD2-mediated bioenergetics, e.g., respiration or ATP generation, but GPD2-derived DHAP and the resulting ether lipid biosynthesis that were critical for the cancer growth. Further mechanistic investigation revealed the role of the GPD2-DHAP-ether lipid-AKT axis in tumor cell growth. Another study related to lipids suggested that GPD2-mediated reduction of CoQ to CoQH2 protects against mitochondrial lipid peroxidation, preventing ferroptosis in cancer cells both in vitro and in vivo176. Interestingly, they showed that G3P supplementation can rescue RSL3-induced cell death in a GPD2-dependent manner. Nevertheless, the contribution of GPD2 to CoQ reduction seems smaller than that of DHODH.
Overall, several mechanisms may influence the role of GPD2 in cancer cell growth. Among them, ROS and bioenergetics, along with CoQ reduction, are connected with GPD2’s established role in the mitochondrial ETC. DHAP-mediated ether lipid synthesis and its activation of the AKT-mTOR pathway seem to be the only route that does not directly involve the mitochondrial electron transport mechanism. Additionally, this DHAP-related mechanism seems to be the first by which GPD2 modulates one of the most altered signaling pathways in cancer, the PI3K/AKT/mTOR pathway. As many metabolic enzymes have been shown to have non-metabolic roles, e.g., functioning as transcription factors and signaling molecules177,178,179, future research may add additional roles to the list.
Bioenergetic contribution of GPD2
As a well-recognized function, GPD2 transfers reducing equivalents to the ETC, specifically to complex III (CIII), it may be worth discussing the bioenergetic contributions of GPD2 in both cancer and non-cancer contexts. In addition to CI and complex II (CII), additional proteins, such as electron transfer flavoprotein dehydrogenase and dihydroorotate dehydrogenase (DHODH), are known to transport electrons to CIII. Although detailed studies comparing the exact contributions of each of these components have been scarce, the involvement of ETC components other than CI and CII seems to be substantial only under some specific conditions rather than being universally present. For instance, the deletion of GPD2 in mouse bone marrow-derived macrophages had minimal effects on basal and maximal respiration but significantly decreased the oxygen consumption rate (OCR) upon short-term LPS stimulation104. Interestingly, after long-term LPS stimulation, which induces tolerance, the LPS-mediated decrease in the OCR in the WT was attenuated in the GPD2-deleted cells, suggesting a negative effect of GPD2 on oxygen consumption under these LPS-tolerant conditions. Similarly, Bajzikova et al. reported that the contribution of DHODH to oxygen consumption was less than 10% of total respiration (sum of CI, CII, and DHODH) and that DHODH KO did not affect overall respiration in murine mammary carcinoma 4T1 cells180. A similar quantitative contribution of GPD2 to overall cellular respiration was reported in the same 4T1 cells155. Additionally, several lines of evidence revealed only minor contributions of GPD2 to the mitochondrial bioenergetics of cancer cells, such as in mouse kidney cancer cells (Renca)130 or in human kidney cancer cells, including Caki1 and 769P cell lines17. On the other hand, notable contributions have also been reported under other conditions38,49,176. For example, a study focusing on metabolism under CI-compromised conditions showed that GPD2 was responsible for the majority of CI-independent OCRs in cancer cell lines that generally have high CI-independent OCRs, such as OVCAR438. In contrast, GPD2 KO had no such effect on cell lines with a low CI-independent OCR. Nevertheless, it should be noted that the effect of GPD2 KO on basal respiration in this study was only modest (~12%) and that the CI-dependent respiration of many cancer cells, as measured with piericidin A, was much larger than the CI-independent respiration, which includes GPD2-dependent respiration. In another study with prostate cancer cell lines, cancer cells exhibited a higher OCR than did normal epithelial cells, which could be associated with a higher presence of GPD2, despite the equal levels of other ETC components, such as CI through complex IV (CIV)49. A substantial decrease in the basal OCR (~40%) in HCT116 cells upon GPD2 KO, accompanied by an increase in the CoQ/CoQH2 ratio, was also reported176, although this might be at odds with the very little CI-independent respiration (~5% of the CI-dependent respiration) reported by Liu et al. in the same cells38. This finding, along with further experiments, indicated the involvement of GPD2 in the ferroptosis defense mechanism in cancer cells, specifically through the CoQ system, suggesting the bioenergetic contribution of GPD2. Although DHODH was also proposed to have a similar ferroptosis defense function181, it is interesting that the overexpression of DHODH completely recovered ferroptosis sensitization in GPD2 KO cells, whereas the overexpression of GPD2 in DHODH KO cells only partially rescued the ferroptosis sensitization phenotype. These data suggest that GPD2 plays a narrower role in regulating ferroptosis than DHODH. Taken together, the overall contribution of GPD2 to cellular respiration seems to be minimal to modest but may be important under specific conditions, such as in prostate cells, during ferroptosis, or under CI-compromised conditions. Other enzymes, such as DHODH, may play similar roles under these conditions, which warrant further investigation.
Therapeutic targeting of GPD1 and GPD2 in cancer
Compared to other components of the bioenergetic machinery, e.g., the TCA cycle, glycolysis, and the ETC, much less is known about the roles of GPS or its components GPD1 and GPD2 in cancer. Therefore, the number of modulators targeting GPD1 and GPD2 is relatively small but holds high potential given the wealth of new studies mentioned above.
For GPD1, no inhibitor studies have been reported, probably because it is known for its tumor-suppressive activity. Interestingly, an activator of GPD1, wedelolactone, exhibited anticancer activity. In bladder cancer, the activation of GPD1 by wedelolactone successfully decreased cancer cell viability by ~50% and reduced tumor weight by ~70% in xenograft models133. As this natural product also has other activities, further studies are needed to understand the contribution of wedelolactone-mediated GPD1 activation to its anticancer activity.
Currently, there are only a few inhibitors specifically developed for GPD2; the iGP series182 and KM04416183 were identified from small molecule screening. KM04416, with an isothiazolone moiety, was discovered based on its ability to inhibit H2O2 generation from G3P (with an EC50 of ~1 μM) and prostate cancer cell growth. The iGP-series containing the benzimidazole moiety was also discovered by G3P-based H2O2 generation screening. Among those, iGP-1 and iGP-5 inhibited actual GPD2 activity (G3P-mediated reduction in DCPIP), and the latter exhibited an order of magnitude higher activity (Ki ~1 μM). Additionally, iGP-1 exhibited negligible activity on GPD1. As these compounds were derived from a single-round screening, further medicinal chemistry-based optimization may lead to more potent inhibitors. It is also worth noting that the activity of iGP-1 may not be achievable under all experimental conditions, as it neither increased the G3P level nor sensitized colon cancer HCT116 cells to ferroptosis, which was observed in GPD2 KO cells176. Moreover, iGP-1 did not suppress the growth of cancer cells in our hands either (data not shown), whereas KM04416 did155. This may be due to the use of different cell lines and the fact that the iGP series were tested for GPD2 activity per se but not for its anticancer activity. Generally, the potencies of both the KM04416 and iGP series are in the medium range, and more studies are needed for inhibitors with stronger specificity and potency.
Although it was not specifically developed as a GPD2 inhibitor, metformin is a well-known antidiabetic drug that has been shown to inhibit GPD2 in two ways. First, in diabetes, metformin was shown to inhibit GPD2 non-competitively, reducing the conversion of G3P to DHAP and changing the cellular redox state, which ultimately lowered the gluconeogenic flux from glycerol to glucose167,168. Second, metformin was shown to lower the expression of GPD2 and thus OXPHOS activity in thyroid cancer, which was responsible for its antithyroid cancer effect151. Notably, 50 μM metformin inhibited recombinant GPD2 and mitochondrial oxygen consumption in gluconeogenesis studies167; this dose was much lower than the concentrations required for metformin to exhibit in vitro anticancer activity or CI inhibition (≥1 mM184,185,186), as used in thyroid cancer studies. Interestingly, metformin exhibited an enhanced anticancer effect in the GPD1-overexpressing background141. An in vivo synergistic effect between GPD1 and metformin was observed even though the G3P level was not significantly enhanced by GPD1 overexpression in vitro. Therefore, synergistic effects may occur regardless of the somewhat controversial supraphysiological G3P concentrations used to address the mechanism involved. Additionally, it would be interesting to determine whether patients with high GPD1 and/or GPD2 levels in their tumor tissues might respond better to metformin treatment. In future mechanistic studies, it may be worth noting that metformin inhibits only GPD2 without altering the activity of GPS or GPD1167.
Other molecules with GPD2-inhibiting activity have been reported, including several from natural product sources. As such, these molecules may not be specific to GPD2 but are expected to affect other enzymes. α-tocopheryl succinate, a mitochondrial CII inhibitor, was found to inhibit GPD2 more efficiently than CII in terms of substrate-mediated oxygen consumption (IC50 of ~10 μM) and H2O2 generation187. For natural products, scopolin, esculetin, and taraxasterol inhibited tumor growth by suppressing GPD2-related glycolysis188,189,190. Scopolin and esculin were shown to bind to GPD2 using the monolith nanotemp fluorescence method, but there were significant discrepancies in concentrations for GPD2 inhibition and cell phenotype inhibition, which may require caution in interpreting the results. Taraxasterol inhibited GPD2 inhibition at 15 μM and its apoptosis-inducing effect could be partially reversed by overexpression of GPD2.
Conclusions and future perspectives
GPD1 and GPD2 have been implicated in three major processes: glucose metabolism (glycolysis and gluconeogenesis), bioenergetics (NAD+ recycling, ATP production, and ROS generation), and lipid metabolism (G3P-derived glycerolipids and DHAP-derived ether lipids). These processes are interrelated, and GPS component enzymes are at key crossroads between energy-consuming and energy-generating pathways. Due to their key roles in metabolism, alterations in GPD activity have been associated with various (patho)physiological conditions, such as diabetes, obesity, muscle regeneration, brain neurotransmission, immune regulation, and cancer, and the causative mechanisms involved are being investigated. Nevertheless, there are some apparent discrepancies among studies, including but not limited to, the pro- vs. anti-tumorigenic roles of GPD. The differences might be context-dependent and might not have been clearly defined, such as cancer types, or due to the limitations of the methodological approaches, e.g., supraphysiological concentrations of G3P.
There are also several points that future research should address based on recent advancements in the knowledge of GPDs. First, more studies are needed to explore the non-conventional or non-bioenergetic roles of GPD2, such as ether lipid-related functions. This is becoming an important issue with three recent independent reports on the absence of ATP production and/or changes in basal oxygen consumption in GPD2 KO or KD systems78,130,155. Therefore, it will be interesting to explore whether the ether lipid synthesis observed in cancer is also relevant in other systems, including muscle, adipose tissues, or immune cells, in which GPD is reported to play important roles. Additionally, the role of GPD1 in ether lipid synthesis should be an interesting topic given that it consumes DHAP. Second, the role of the GPS component as a signaling molecule should be explored. A recent study on a cell line suggested that DHAP activates mTORC1 signaling independently of energy stress or growth factor signaling191. Due to the importance of mTORC1 in cell survival and growth, studies in other cells or in vivo systems are highly anticipated. Additionally, GPD1 was shown to affect HIF1α levels in glioblastoma. HIF1α signaling is also important in many different systems, and therefore, the GPD1-HIF1α relationship should be studied in many other cancers and non-cancer systems. These signaling roles of GPD-related metabolites may be another example of metabolic enzymes’ second function (moonlighting). Third, as GPDs are enzymes, specific inhibitors are needed for both mechanistic studies and practical use. Currently available inhibitors are not satisfactory in terms of potency and specificity. The activator of GPD1 may also be relevant because it is known to be involved in tumor suppression. Fourth, the reported negative correlation between GPD1 and GPD2 is intriguing from both mechanistic and functional perspectives. The two components of the GPS are expected to correlate with each other, but they do not in cancer. A possible mechanism was proposed in ccRCC, where GPD1-mediated stabilization of HIF1α transcriptionally represses GPD2 expression, which resulted in tumor suppression17. Despite showing the same negative correlation, another study in mouse kidney cancer cells showed GPD1-mediated lipid synthesis and tumor growth130. Therefore, the functional consequences of these negative correlations need to be clarified. Additionally, the existence of any mechanistic factors other than HIF1α and the (patho)physiological contexts in which these factors are involved, other than kidney cancer, should be investigated. Notably, increased and decreased activities of GPD1 and GPD2, respectively, in all tissues of the jerboa during hibernation have been reported, except skeletal muscles for GPD2192, thus indicating the existence of other conditions for the negative correlation.
Overall, decades of research on GPS have provided insights into the functions of its components across diverse conditions, and future studies are expected to address important mechanistic and functional questions.
References
Meyerhof, O. Über die Atmung der Froschmuskulatur. Pflug. Arch. Gesamt. Physiol. Menschen Tiere 175, 20–87 (1919).
Estabrook, R. W. & Sacktor, B. α-glycerophosphate oxidase of flight muscle mitochondria. J. Biol. Chem. 233, 1014–1019 (1958).
Sacktor, B. & Dick, A. Pathways of hydrogen transport in the oxidation of extramitochondrial reduced diphosphopyridine nucleotide in flight muscle. J. Biol. Chem. 237, 3259–3263 (1962).
Borst, P. The malate-aspartate shuttle (Borst cycle): how it started and developed into a major metabolic pathway. IUBMB Life 72, 2241–2259 (2020).
Klingenberg, M. Localization of the glycerol-phosphate dehydrogenase in the outer phase of the mitochondrial inner membrane. Eur. J. Biochem 13, 247–252 (1970).
Boxer, G. E. & Shonk, C. E. Low levels of Soluble DPN-linked α-glycerophosphate dehydrogenase in tumors*. Cancer Res. 20, 85–91 (1960).
Ohkawa, K. I., Vogt, M. T. & Farber, E. Unusually high mitochondrial alpha glycerophosphate dehydrogenase activity in rat brown adipose tissue. J. Cell Biol. 41, 441–449 (1969).
Houstĕk, J., Cannon, B. & Lindberg, O. Gylcerol-3-phosphate shuttle and its function in intermediary metabolism of hamster brown-adipose tissue. Eur. J. Biochem. 54, 11–18 (1975).
Greenhouse, W. V. & Lehninger, A. L. Occurrence of the malate-aspartate shuttle in various tumor types. Cancer Res. 36, 1392–1396 (1976).
Greenhouse, W. V. & Lehninger, A. L. Magnitude of malate-aspartate reduced nicotinamide adenine dinucleotide shuttle activity in intact respiring tumor cells. Cancer Res. 37, 4173–4181 (1977).
Brown, L. J., Koza, R. A., Marshall, L., Kozak, L. P. & MacDonald, M. J. Lethal hypoglycemic ketosis and glyceroluria in mice lacking both the mitochondrial and the cytosolic glycerol phosphate dehydrogenases. J. Biol. Chem. 277, 32899–32904 (2002).
Mráček, T., Drahota, Z. & Houštěk, J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochim. Biophys. Acta 1827, 401–410 (2013).
Ou, X. et al. Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1). J. Mol. Biol. 357, 858–869 (2006).
Patsouris, D. et al. PPARalpha governs glycerol metabolism. J. Clin. Investig. 114, 94–103 (2004).
Bogacka, I., Xie, H., Bray, G. A. & Smith, S. R. The effect of pioglitazone on peroxisome proliferator-activated receptor-gamma target genes related to lipid storage in vivo. Diabetes Care 27, 1660–1667 (2004).
Chen, K. W., Chen, Y. S., Chen, P. J. & Yeh, S. H. Androgen receptor functions in pericentral hepatocytes to decrease gluconeogenesis and avoid hyperglycemia and obesity in male mice. Metabolism 135, 155269 (2022).
Liu, R. et al. A HIF1alpha-GPD1 feedforward loop inhibits the progression of renal clear cell carcinoma via mitochondrial function and lipid metabolism. J. Exp. Clin. Cancer Res. 40, 188 (2021).
Musri, M. M., Corominola, H., Casamitjana, R., Gomis, R. & Parrizas, M. Histone H3 lysine 4 dimethylation signals the transcriptional competence of the adiponectin promoter in preadipocytes. J. Biol. Chem. 281, 17180–17188 (2006).
Zhang, S. et al. Identification of potential key genes associated with adipogenesis through integrated analysis of five mouse transcriptome datasets. Int. J. Mol. Sci. 19, 3557 (2018).
Lee, Y. J., Jeschke, G. R., Roelants, F. M., Thorner, J. & Turk, B. E. Reciprocal phosphorylation of yeast glycerol-3-phosphate dehydrogenases in adaptation to distinct types of stress. Mol. Cell Biol. 32, 4705–4717 (2012).
Swierczynski, J. et al. Enhanced glycerol 3-phosphate dehydrogenase activity in adipose tissue of obese humans. Mol. Cell Biochem. 254, 55–59 (2003).
Sledzinski, T. et al. Association between cytosolic glycerol 3-phosphate dehydrogenase gene expression in human subcutaneous adipose tissue and BMI. Cell Physiol. Biochem. 32, 300–309 (2013).
Park, J. J., Berggren, J. R., Hulver, M. W., Houmard, J. A. & Hoffman, E. P. GRB14, GPD1, and GDF8 as potential network collaborators in weight loss-induced improvements in insulin action in human skeletal muscle. Physiol. Genom. 27, 114–121 (2006).
Basel-Vanagaite, L. et al. Transient infantile hypertriglyceridemia, fatty liver, and hepatic fibrosis caused by mutated GPD1, encoding glycerol-3-phosphate dehydrogenase 1. Am. J. Hum. Genet. 90, 49–60 (2012).
Joshi, M. et al. A compound heterozygous mutation in GPD1 causes hepatomegaly, steatohepatitis, and hypertriglyceridemia. Eur. J. Hum. Genet. 22, 1229–1232 (2014).
Li, J. Q. et al. A novel homozygous mutation in the glycerol-3-phosphate dehydrogenase 1 gene in a Chinese patient with transient infantile hypertriglyceridemia: a case report. BMC Gastroenterol. 18, 96 (2018).
Lin, H. et al. Case report: identification of a novel homozygous mutation in GPD1 gene of a Chinese child with transient infantile hypertriglyceridemia. Front. Genet. 12, 726116 (2021).
Polchar, L. & Vallabhaneni, P. Case of GPD1 deficiency causing hypertriglyceridaemia and non-alcoholic steatohepatitis. BMJ Case Rep. 15, e246369 (2022).
MacDonald, M. J. & Marshall, L. K. Mouse lacking NAD+-linked glycerol phosphate dehydrogenase has normal pancreatic beta cell function but abnormal metabolite pattern in skeletal muscle. Arch. Biochem. Biophys. 384, 143–153 (2000).
MacDonald, M. J. & Marshall, L. K. Survey of normal appearing mouse strain which lacks malic enzyme and Nad+-linked glycerol phosphate dehydrogenase: normal pancreatic beta cell function, but abnormal metabolite pattern in skeletal muscle. Mol. Cell Biochem. 220, 117–125 (2001).
Hilgers, J. et al. Genetic differences in BALB/c sublines. Curr. Top. Microbiol. Immunol. 122, 19–30 (1985).
Prochazka, M., Kozak, U. C. & Kozak, L. P. A glycerol-3-phosphate dehydrogenase null mutant in BALB/cHeA mice. J. Biol. Chem. 264, 4679–4683 (1989).
Sato, T., Morita, A., Mori, N. & Miura, S. Glycerol 3-phosphate dehydrogenase 1 deficiency enhances exercise capacity due to increased lipid oxidation during strenuous exercise. Biochem. Biophys. Res. Commun. 457, 653–658 (2015).
Sato, T., Sayama, N., Inoue, M., Morita, A. & Miura, S. The enhancement of fat oxidation during the active phase and suppression of body weight gain in glycerol-3-phosphate dehydrogenase 1 deficient mice. Biosci. Biotechnol. Biochem. 84, 2367–2373 (2020).
Sato, T. et al. EID1 suppresses lipid accumulation by inhibiting the expression of GPDH in 3T3-L1 preadipocytes. J. Cell Physiol. 235, 6725–6735 (2020).
Hwang, K. et al. Alteration of the NAD+/NADH ratio in CHO cells by stable transfection with human cytosolic glycerol-3-phosphate dehydrogenase: resistance to oxidative stress. Mol. Cells 9, 429–435 (1999).
Jeong, D. W., Kim, T. S., Cho, I. T. & Kim, I. Y. Modification of glycolysis affects cell sensitivity to apoptosis induced by oxidative stress and mediated by mitochondria. Biochem. Biophys. Res. Commun. 313, 984–991 (2004).
Liu, S. et al. Glycerol-3-phosphate biosynthesis regenerates cytosolic NAD(+) to alleviate mitochondrial disease. Cell Metab. 33, 1974–1987.e1979 (2021).
Feng, Z. E. et al. The prognostic value of glycerol-3-phosphate dehydrogenase 1-like expression in head and neck squamous cell carcinoma. Histopathology 64, 348–355 (2014).
Moras, E. et al. Genetic and molecular mechanisms in brugada syndrome. Cells. 12, 1791 (2023).
London, B. et al. Mutation in glycerol-3-phosphate dehydrogenase 1-like gene (GPD1-L) decreases cardiac Na+ current and causes inherited Arrhythmias. Circulation 116, 2260–2268 (2007).
Van Norstrand, D. W. et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation 116, 2253–2259 (2007).
Valdivia, C. R., Ueda, K., Ackerman, M. J. & Makielski, J. C. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am. J. Physiol. Heart Circ. 297, H1446–H1452 (2009).
Liu, T. et al. GPD1L inhibits renal cell carcinoma progression by regulating PINK1/Parkin-mediated mitophagy. J. Cell Mol. Med. 27, 2326–2339 (2023).
Brown, L. J., MacDonald, M. J., Lehn, D. A. & Moran, S. M. Sequence of rat mitochondrial glycerol-3-phosphate dehydrogenase cDNA. Evidence for EF-hand calcium-binding domains. J. Biol. Chem. 269, 14363–14366 (1994).
Parsonage, D., Luba, J., Mallett, T. C. & Claiborne, A. The soluble alpha-glycerophosphate oxidase from Enterococcus casseliflavus. Sequence homology with the membrane-associated dehydrogenase and kinetic analysis of the recombinant enzyme. J. Biol. Chem. 273, 23812–23822 (1998).
Claiborne, A. Studies on the structure and mechanism of Streptococcus faecium L-alpha-glycerophosphate oxidase. J. Biol. Chem. 261, 14398–14407 (1986).
Yeh, J. I., Chinte, U. & Du, S. Structure of glycerol-3-phosphate dehydrogenase, an essential monotopic membrane enzyme involved in respiration and metabolism. Proc. Natl Acad. Sci. USA 105, 3280–3285 (2008).
Pecinová, A. et al. Role of mitochondrial glycerol-3-phosphate dehydrogenase in metabolic adaptations of prostate cancer. Cells. 9 (2020).
Syngkli, S. & Das, B. Purification and characterization of human glycerol 3-phosphate dehydrogenases (mitochondrial and cytosolic) by NAD(+)/NADH redox method. Biochimie 214, 199–215 (2023).
Koza, R. A. et al. Sequence and tissue-dependent RNA expression of mouse FAD-linked glycerol-3-phosphate dehydrogenase. Arch. Biochem. Biophys. 336, 97–104 (1996).
Weitzel, J. M., Grott, S., Radtke, C., Kutz, S. & Seitz, H. J. Multiple promoters direct the tissue-specific expression of rat mitochondrial glycerol-3-phosphate dehydrogenase. Biol. Chem. 381, 611–614 (2000).
Muller, S. & Seitz, H. J. Cloning of a cDNA for the FAD-linked glycerol-3-phosphate dehydrogenase from rat liver and its regulation by thyroid hormones. Proc. Natl Acad. Sci. USA 91, 10581–10585 (1994).
Weitzel, J. M., Kutz, S., Radtke, C., Grott, S. & Seitz, H. J. Hormonal regulation of multiple promoters of the rat mitochondrial glycerol-3-phosphate dehydrogenase gene: identification of a complex hormone-response element in the ubiquitous promoter B. Eur. J. Biochem. 268, 4095–4103 (2001).
Gong, D. W., Bi, S., Weintraub, B. D. & Reitman, M. Rat mitochondrial glycerol-3-phosphate dehydrogenase gene: multiple promoters, high levels in brown adipose tissue, and tissue-specific regulation by thyroid hormone. DNA Cell Biol. 17, 301–309 (1998).
Urcelay, E. et al. Cloning and functional characterization of the 5’ regulatory region of the human mitochondrial glycerol-3-phosphate dehydrogenase gene. Lack of 3,5,3’-triiodothyronine responsiveness in adipose tissue. Eur. J. Biochem. 267, 7209–7217 (2000).
Singh, A. et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J. Clin. Investig. 123, 2921–2934 (2013).
MacDonald, M. J. & Brown, L. J. Calcium activation of mitochondrial glycerol phosphate dehydrogenase restudied. Arch. Biochem. Biophys. 326, 79–84 (1996).
Fisher, A. B., Scarpa, A., LaNoue, K. F., Bassett, D. & Williamson, J. R. Respiration of rat lung mitochondria and the influence of Ca 2+ on substrate utilization. Biochemistry 12, 1438–1445 (1973).
Hansford, R. G. & Chappell, J. B. The effect of Ca2+ on the oxidation of glycerol phosphate by blowfly flight-muscle mitochondria. Biochem. Biophys. Res. Commun. 27, 686–692 (1967).
Dhoundiyal, A., Goeschl, V., Boehm, S., Kubista, H. & Hotka, M. Glycerol-3-phosphate shuttle is a backup system securing metabolic flexibility in neurons. J. Neurosci. 42, 7339–7354 (2022).
Llorente-Folch, I. et al. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 593, 3447–3462 (2015).
Denton, R. M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316 (2009).
Bukowiecki, L. J. & Lindberg, O. Control of sn-glycerol 3-phosphate oxidation in brown adipose tissue mitochondria by calcium and acyl-CoA. Biochim. Biophys. Acta 348, 115–125 (1974).
Rauchova, H. & Drahota, Z. Inhibition of the glycerol 3-phosphate oxidation by free fatty acids. Int. J. Biochem. 16, 243–245 (1984).
Amler, E., Rauchova, H., Svobodova, J. & Drahota, Z. Regulation of glycerol 3-phosphate oxidation in mitochondria by changes in membrane microviscosity. FEBS Lett. 206, 1–3 (1986).
Nałecz, M. J., Zborowski, J., Famulski, K. S. & Wojtczak, L. Effect of phospholipid composition on the surface potential of liposomes and the activity of enzymes incorporated. Eur. J. Biochem. 112, 75–80 (1980).
Meng, J., Zhang, C., Wang, D., Zhu, L. & Wang, L. Mitochondrial GCN5L1 regulates cytosolic redox state and hepatic gluconeogenesis via glycerol phosphate shuttle GPD2. Biochem. Biophys. Res. Commun. 621, 1–7 (2022).
Lu, J. et al. Tumor-associated macrophage interleukin-beta promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci. 111, 1979–1990 (2020).
Garrib, A. & McMurray, W. C. Purification and characterization of glycerol-3-phosphate dehydrogenase (flavin-linked) from rat liver mitochondria. J. Biol. Chem. 261, 8042–8048 (1986).
Rauchová, H., Beleznai, Z. & Drahota, Z. Effect of chemical modification in situ on L-glycerol-3-phosphate dehydrogenase in brown adipose tissue mitochondria. J. Bioenerg. Biomembr. 20, 623–632 (1988).
Brown, L. J. et al. Normal thyroid thermogenesis but reduced viability and adiposity in mice lacking the mitochondrial glycerol phosphate dehydrogenase. J. Biol. Chem. 277, 32892–32898 (2002).
DosSantos, R. A., Alfadda, A., Eto, K., Kadowaki, T. & Silva, J. E. Evidence for a compensated thermogenic defect in transgenic mice lacking the mitochondrial glycerol-3-phosphate dehydrogenase gene. Endocrinology 144, 5469–5479 (2003).
Arsenijevic, D. et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 26, 435–439 (2000).
Gong, D. W. et al. Lack of obesity and normal response to fasting and thyroid hormone in mice lacking uncoupling protein-3. J. Biol. Chem. 275, 16251–16257 (2000).
Alfadda, A., DosSantos, R. A., Stepanyan, Z., Marrif, H. & Silva, J. E. Mice with deletion of the mitochondrial glycerol-3-phosphate dehydrogenase gene exhibit a thrifty phenotype: effect of gender. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R147–R156 (2004).
Liu, X. et al. Mitochondrial glycerol 3-phosphate dehydrogenase promotes skeletal muscle regeneration. EMBO Mol. Med. 10, e9390 (2018).
Zheng, Y. et al. Deficiency of mitochondrial glycerol 3-phosphate dehydrogenase contributes to hepatic steatosis. Hepatology 70, 84–97 (2019).
MacDonald, M. J., Warner, T. F. & Mertz, R. J. High activity of mitochondrial glycerol phosphate dehydrogenase in insulinomas and carcinoid and other tumors of the amine precursor uptake decarboxylation system. Cancer Res. 50, 7203–7205 (1990).
MacDonald, M. J. High content of mitochondrial glycerol-3-phosphate dehydrogenase in pancreatic islets and its inhibition by diazoxide. J. Biol. Chem. 256, 8287–8290 (1981).
Meglasson, M. D., Smith, K. M., Nelson, D. & Erecinska, M. alpha-Glycerophosphate shuttle in a clonal beta-cell line. Am. J. Physiol. 256, E173–E178 (1989).
Ferrer, J. et al. Mitochondrial glycerol-3-phosphate dehydrogenase. Cloning of an alternatively spliced human islet-cell cDNA, tissue distribution, physical mapping, and identification of a polymorphic genetic marker. Diabetes 45, 262–266 (1996).
Sener, A., Herberg, L. & Malaisse, W. J. FAD-linked glycerophosphate dehydrogenase deficiency in pancreatic islets of mice with hereditary diabetes. FEBS Lett. 316, 224–227 (1993).
Eto, K. et al. Role of NADH shuttle system in glucose-induced activation of mitochondrial metabolism and insulin secretion. Science 283, 981–985 (1999).
Ueda, K. et al. Overexpression of mitochondrial FAD-linked glycerol-3-phosphate dehydrogenase does not correct glucose-stimulated insulin secretion from diabetic GK rat pancreatic islets. Diabetologia 41, 649–653 (1998).
Ishihara, H. et al. Effect of mitochondrial and/or cytosolic glycerol 3-phosphate dehydrogenase overexpression on glucose-stimulated insulin secretion from MIN6 and HIT cells. Diabetes 45, 1238–1244 (1996).
Novials, A. et al. Mutation in the calcium-binding domain of the mitochondrial glycerophosphate dehydrogenase gene in a family of diabetic subjects. Biochem. Biophys. Res. Commun. 231, 570–572 (1997).
Gudayol, M. et al. Identification and functional analysis of mutations in FAD-binding domain of mitochondrial glycerophosphate dehydrogenase in caucasian patients with type 2 diabetes mellitus. Endocrine 16, 39–42 (2001).
Fabregat, M. E. et al. Enzyme-linked immunosorbent assay of autoantibodies against mitochondrial glycerophosphate dehydrogenase in insulin-dependent and non-insulin-dependent diabetic subjects. Biochem. Mol. Med. 62, 172–177 (1997).
Fabregat, M. E. et al. Autoantibodies against mitochondrial glycerophosphate dehydrogenase in patients with IDDM. Diabetes Res. Clin. Pract. 38, 115–121 (1997).
Juaristi, I. et al. ARALAR/AGC1 deficiency, a neurodevelopmental disorder with severe impairment of neuronal mitochondrial respiration, does not produce a primary increase in brain lactate. J. Neurochem. 142, 132–139 (2017).
Bianchi, M. C., Sgandurra, G., Tosetti, M., Battini, R. & Cioni, G. Brain magnetic resonance in the diagnostic evaluation of mitochondrial encephalopathies. Biosci. Rep. 27, 69–85 (2007).
Finsterer, J. Leigh and Leigh-like syndrome in children and adults. Pediatr. Neurol. 39, 223–235 (2008).
Haas, R. H. et al. The in-depth evaluation of suspected mitochondrial disease. Mol. Genet. Metab. 94, 16–37 (2008).
Nguyen, N. H. T., Brathe, A. & Hassel, B. Neuronal uptake and metabolism of glycerol and the neuronal expression of mitochondrial glycerol-3-phosphate dehydrogenase. J. Neurochem. 85, 831–842 (2003).
Leveille, P. J., McGinnis, J. F., Maxwell, D. S. & de Vellis, J. Immunocytochemical localization of glycerol-3-phosphate dehydrogenase in rat oligodendrocytes. Brain Res. 196, 287–305 (1980).
Montz, H. P., Althaus, H. H., Gebicke-Haerter, P. J. & Neuhoff, V. Glycerol phosphate dehydrogenase activity of oligodendrocytes isolated from adult pig brain: its inducibility by hydrocortisone. J. Neurochem. 45, 1201–1204 (1985).
Giulivi, C., Wang, J. Y. & Hagerman, R. J. Artificial neural network applied to fragile X-associated tremor/ataxia syndrome stage diagnosis based on peripheral mitochondrial bioenergetics and brain imaging outcomes. Sci. Rep. 12, 21382 (2022).
Li, W. et al. Metformin alters locomotor and cognitive function and brain metabolism in normoglycemic mice. Aging Dis. 10, 949–963 (2019).
Votyakova, T. V. & Reynolds, I. J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 79, 266–277 (2001).
Chavda, V. & Lu, B. Reverse electron transport at mitochondrial complex I in ischemic stroke, aging, and age-related diseases. Antioxidants 12, 895 (2023).
Barrientos, A. & Moraes, C. T. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J. Biol. Chem. 274, 16188–16197 (1999).
Kaminski, M. M. et al. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2, 1300–1315 (2012).
Langston, P. K. et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 20, 1186–1195 (2019).
Mracek, T., Pecinova, A., Vrbacky, M., Drahota, Z. & Houstek, J. High efficiency of ROS production by glycerophosphate dehydrogenase in mammalian mitochondria. Arch. Biochem. Biophys. 481, 30–36 (2009).
Kota, V., Dhople, V. M. & Shivaji, S. Tyrosine phosphoproteome of hamster spermatozoa: role of glycerol-3-phosphate dehydrogenase 2 in sperm capacitation. Proteomics9, 1809–1826 (2009).
Kota, V. et al. Role of glycerol-3-phosphate dehydrogenase 2 in mouse sperm capacitation. Mol. Reprod. Dev. 77, 773–783 (2010).
Swierczyński, J., Scislowski, P. & Aleksandrowicz, Z. High activity of alpha-glycerophosphate oxidation by human placental mitochondria. Biochim. Biophys. Acta 429, 46–54 (1976).
Olivera, A. A. & Meigs, R. A. Mitochondria from human term placenta. II. Characterization of respiratory pathways and coupling mechanisms. Biochim. Biophys. Acta 376, 436–445 (1975).
Honzík, T. et al. Specific properties of heavy fraction of mitochondria from human-term placenta—glycerophosphate-dependent hydrogen peroxide production. Placenta 27, 348–356 (2006).
Algoet, M. et al. Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc. Med. 33, 357–366 (2023).
Song, R., Dasgupta, C., Mulder, C. & Zhang, L. MicroRNA-210 controls mitochondrial metabolism and protects heart function in myocardial infarction. Circulation 145, 1140–1153 (2022).
Ishihama, S. et al. LPL/AQP7/GPD2 promotes glycerol metabolism under hypoxia and prevents cardiac dysfunction during ischemia. FASEB J. 35, e22048 (2021).
Qu, H. et al. Deficiency of mitochondrial glycerol 3-phosphate dehydrogenase exacerbates podocyte injury and the progression of diabetic kidney disease. Diabetes 70, 1372–1387 (2021).
Saheki, T. et al. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double knock-out mice recapitulate features of human citrin deficiency. J. Biol. Chem. 282, 25041–25052 (2007).
Saheki, T. et al. Metabolomic analysis reveals hepatic metabolite perturbations in citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mice, a model of human citrin deficiency. Mol. Genet. Metab. 104, 492–500 (2011).
Sinasac, D. S. et al. Slc25a13-knockout mice harbor metabolic deficits but fail to display hallmarks of adult-onset type II citrullinemia. Mol. Cell Biol. 24, 527–536 (2004).
Moriyama, M. et al. Mechanism for increased hepatic glycerol synthesis in the citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mouse: Urine glycerol and glycerol 3-phosphate as potential diagnostic markers of human citrin deficiency. Biochim. Biophys. Acta 1852, 1787–1795 (2015).
Daoud, H. et al. Haploinsufficiency of the GPD2 gene in a patient with nonsyndromic mental retardation. Hum. Genet. 124, 649–658 (2009).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Finley, L. W. S. What is cancer metabolism? Cell 186, 1670–1688 (2023).
Faubert, B., Solmonson, A. & DeBerardinis, R. J. Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020).
DeBerardinis, R. J. & Chandel, N. S. Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016).
Mossmann, D., Park, S. & Hall, M. N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 18, 744–757 (2018).
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A. L. & Dang, C. V. MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 (2015).
Hay, N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat. Rev. Cancer 16, 635–649 (2016).
Munir, R., Lisec, J., Swinnen, J. V. & Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 120, 1090–1098 (2019).
Kwon, H., Oh, S., Jin, X., An, Y. J. & Park, S. Cancer metabolomics in basic science perspective. Arch. Pharm. Res. 38, 372–380 (2015).
Yao, C. H. et al. Uncoupled glycerol-3-phosphate shuttle in kidney cancer reveals that cytosolic GPD is essential to support lipid synthesis. Mol. Cell 83, 1340–1349.e1347 (2023).
Shonk, C. E., Arison, R. N., Koven, B. J., Majima, H. & Boxer, G. E. Enzyme patterns in human tissues. 3. Glycolytic enzymes in normal and malignant tissues of the colon and rectum. Cancer Res. 25, 206–213 (1965).
Zhou, C. F. et al. Identification of glycerol-3-phosphate dehydrogenase 1 as a tumour suppressor in human breast cancer. Oncotarget 8, 101309–101324 (2017).
Zhang, W. et al. Allosteric activation of the metabolic enzyme GPD1 inhibits bladder cancer growth via the lysoPC-PAFR-TRPV2 axis. J. Hematol. Oncol. 15, 93 (2022).
Yoneten, K. K. et al. Comparative proteome analysis of breast cancer tissues highlights the importance of glycerol-3-phosphate dehydrogenase 1 and monoacylglycerol lipase in breast cancer metabolism. Cancer Genom. Proteom. 16, 377–397 (2019).
Karaosmanoglu Yoneten, K., Kasap, M., Arga, K. Y., Akpinar, G. & Utkan, N. Z. Decreased serum levels of glycerol-3- phosphate dehydrogenase 1 and monoacylglycerol lipase act as diagnostic biomarkers for breast cancer. Cancer Biomark. 34, 67–76 (2022).
Sanchez, D. J. & Simon, M. C. Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim. Biophys. Acta Rev. Cancer 1870, 23–31 (2018).
Hakimi, A. A. et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 29, 104–116 (2016).
Nguyen, T. T. M. et al. GPX8 regulates clear cell renal cell carcinoma tumorigenesis through promoting lipogenesis by NNMT. J. Exp. Clin. Cancer Res. 42, 42 (2023).
Rusu, P. et al. GPD1 specifically marks dormant glioma stem cells with a distinct metabolic profile. Cell Stem Cell 25, 241–257.e248 (2019).
Turyn, J. et al. Increased activity of glycerol 3-phosphate dehydrogenase and other lipogenic enzymes in human bladder cancer. Horm. Metab. Res. 35, 565–569 (2003).
Xie, J. et al. GPD1 enhances the anticancer effects of metformin by synergistically increasing total cellular glycerol-3-phosphate. Cancer Res. 80, 2150–2162 (2020).
Petan, T. Lipid droplets in cancer. Rev. Physiol. Biochem. Pharm. 185, 53–86 (2020).
Wettersten, H. I. Reprogramming of metabolism in kidney cancer. Semin. Nephrol. 40, 2–13 (2020).
Tan, S. K. & Welford, S. M. Lipid in renal carcinoma: queen bee to target? Trends Cancer 6, 448–450 (2020).
Hsieh, J. J. et al. Renal cell carcinoma. Nat. Rev. Dis. Prim. 3, 17009 (2017).
Hunt, S. M., Osnos, M. & Rivlin, R. S. Thyroid hormone regulation of mitochondrial α-glycerophosphate dehydrogenase in liver and hepatoma. Cancer Res. 30, 1764–1768 (1970).
Chowdhury, S. K., Gemin, A. & Singh, G. High activity of mitochondrial glycerophosphate dehydrogenase and glycerophosphate-dependent ROS production in prostate cancer cell lines. Biochem. Biophys. Res. Commun. 333, 1139–1145 (2005).
Chowdhury, S. K., Raha, S., Tarnopolsky, M. A. & Singh, G. Increased expression of mitochondrial glycerophosphate dehydrogenase and antioxidant enzymes in prostate cancer cell lines/cancer. Free Radic. Res. 41, 1116–1124 (2007).
Yuan, L. et al. Switching off IMMP2L signaling drives senescence via simultaneous metabolic alteration and blockage of cell death. Cell Res. 28, 625–643 (2018).
Kaplon, J. et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112 (2013).
Thakur, S. et al. Metformin targets mitochondrial glycerophosphate dehydrogenase to control rate of oxidative phosphorylation and growth of thyroid cancer in vitro and in vivo. Clin. Cancer Res. 24, 4030–4043 (2018).
Mikeli, M. et al. Contribution of GPD2/mGPDH to an alternative respiratory chain of the mitochondrial energy metabolism and the stemness in CD133-positive HuH-7 cells. Genes Cells 25, 139–148 (2020).
Suetsugu, A. et al. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 351, 820–824 (2006).
Mikeli, M., Fujikawa, M. & Tanabe, T. GPD2: the relationship with cancer and neural stemness. Cells Dev. 173, 203824 (2023).
Oh, S. et al. Non-bioenergetic roles of mitochondrial GPD2 promote tumor progression. Theranostics 13, 438–457 (2023).
Li, X. et al. mGPDH deficiency leads to melanoma metastasis via induced NRF2. J. Cell Mol. Med. 25, 5305–5315 (2021).
Gallorini, M., Carradori, S., Panieri, E., Sova, M. & Saso, L. Modulation of NRF2: biological dualism in cancer, targets and possible therapeutic applications. Antioxid. Redox Signal. https://doi.org/10.1089/ars.2022.0213 (2023).
Reuter, S., Gupta, S. C., Chaturvedi, M. M. & Aggarwal, B. B. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 49, 1603–1616 (2010).
Sabharwal, S. S. & Schumacker, P. T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 14, 709–721 (2014).
Hayes, J. D., Dinkova-Kostova, A. T. & Tew, K. D. Oxidative stress in cancer. Cancer Cell 38, 167–197 (2020).
Orr, A. L., Quinlan, C. L., Perevoshchikova, I. V. & Brand, M. D. A refined analysis of superoxide production by mitochondrial sn-glycerol 3-phosphate dehydrogenase. J. Biol. Chem. 287, 42921–42935 (2012).
Wong, H. S., Dighe, P. A., Mezera, V., Monternier, P. A. & Brand, M. D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809 (2017).
Mracek, T. et al. ROS generation and multiple forms of mammalian mitochondrial glycerol-3-phosphate dehydrogenase. Biochim. Biophys. Acta 1837, 98–111 (2014).
Dueregger, A. et al. Differential utilization of dietary fatty acids in benign and malignant cells of the prostate. PLoS One 10, e0135704 (2015).
Beyoglu, D. et al. Tissue metabolomics of hepatocellular carcinoma: tumor energy metabolism and the role of transcriptomic classification. Hepatology 58, 229–238 (2013).
Lorenzetti, F., Capiglioni, A. M., Marinelli, R. A., Carrillo, M. C. & Alvarez, M. L. Hepatic glycerol metabolism is early reprogrammed in rat liver cancer development. Biochimie 170, 88–93 (2020).
Madiraju, A. K. et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546 (2014).
Madiraju, A. K. et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 24, 1384–1394 (2018).
Mallik, R. & Chowdhury, T. A. Metformin in cancer. Diabetes Res Clin. Pract. 143, 409–419 (2018).
Hua, Y. et al. Metformin and cancer hallmarks: shedding new lights on therapeutic repurposing. J. Transl. Med. 21, 403 (2023).
Bell, R. M. & Coleman, R. A. Enzymes of glycerolipid synthesis in eukaryotes. Annu. Rev. Biochem. 49, 459–487 (1980).
Yu, J. et al. Update on glycerol-3-phosphate acyltransferases: the roles in the development of insulin resistance. Nutr. Diabetes 8, 34 (2018).
Hajra, A. K. Dihydroxyacetone phosphate acyltransferase. Biochim. Biophys. Acta 1348, 27–34 (1997).
Brites, P., Waterham, H. R. & Wanders, R. J. Functions and biosynthesis of plasmalogens in health and disease. Biochim. Biophys. Acta 1636, 219–231 (2004).
Jiménez-Rojo, N. & Riezman, H. On the road to unraveling the molecular functions of ether lipids. FEBS Lett. 593, 2378–2389 (2019).
Wu, S. et al. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc. Natl Acad. Sci. USA 119, e2121987119 (2022).
Richard, A. J., Hang, H. & Stephens, J. M. Pyruvate dehydrogenase complex (PDC) subunits moonlight as interaction partners of phosphorylated STAT5 in adipocytes and adipose tissue. J. Biol. Chem. 292, 19733–19742 (2017).
Shegay, P. V., Shatova, O. P., Zabolotneva, A. A., Shestopalov, A. V. & Kaprin, A. D. Moonlight functions of glycolytic enzymes in cancer. Front. Mol. Biosci. 10, 1076138 (2023).
Castello, A., Hentze, M. W. & Preiss, T. Metabolic enzymes enjoying new partnerships as RNA-binding proteins. Trends Endocrinol. Metab. 26, 746–757 (2015).
Bajzikova, M. et al. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab. 29, 399–416.e310 (2019).
Mao, C. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021).
Orr, A. L. et al. Novel inhibitors of mitochondrial sn-glycerol 3-phosphate dehydrogenase. PLoS One 9, e89938 (2014).
Singh, G. Mitochondrial FAD-linked glycerol-3-phosphate dehydrogenase: a target for cancer therapeutics. Pharmaceuticals 7, 192–206 (2014).
Lee, J. et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 568, 254–258 (2019).
Owen, M. R., Doran, E. & Halestrap, A. P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614 (2000).
Ashton, T. M., McKenna, W. G., Kunz-Schughart, L. A. & Higgins, G. S. Oxidative phosphorylation as an emerging target in cancer therapy. Clin. Cancer Res. 24, 2482–2490 (2018).
Rauchová, H., Vokurková, M. & Drahota, Z. Inhibition of mitochondrial glycerol-3-phosphate dehydrogenase by α-tocopheryl succinate. Int. J. Biochem. Cell Biol. 53, 409–413 (2014).
Wang, C., Zhou, Q. & Wu, S. T. Scopolin obtained from Smilax china L. against hepatocellular carcinoma by inhibiting glycolysis: a network pharmacology and experimental study. J. Ethnopharmacol. 296, 115469 (2022).
Zhao, Y., Zhang, L., Guo, M. & Yang, H. Taraxasterol suppresses cell proliferation and boosts cell apoptosis via inhibiting GPD2-mediated glycolysis in gastric cancer. Cytotechnology 73, 815–825 (2021).
Wu, S. T. et al. Esculetin inhibits cancer cell glycolysis by binding tumor PGK2, GPD2, and GPI. Front. Pharm. 11, 379 (2020).
Orozco, J. M. et al. Dihydroxyacetone phosphate signals glucose availability to mTORC1. Nat. Metab. 2, 893–901 (2020).
Berrada, W., Naya, A., Ouafik, L. & Bourhim, N. Effect of hibernation, thyroid hormones and dexamethasone on cytosolic and mitochondrial glycerol-3-phosphate dehydrogenase from jerboa (Jaculus orientalis). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 125, 439–449 (2000).
Acknowledgements
This work was supported by the Basic Science Research Program (Grant NRF-2018R1A3B1052328 to S.P.) funded by the Ministry of Science, Information and Communication Technology and by Future Planning through the National Research Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oh, S., Mai, X.L., Kim, J. et al. Glycerol 3-phosphate dehydrogenases (1 and 2) in cancer and other diseases. Exp Mol Med (2024). https://doi.org/10.1038/s12276-024-01222-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s12276-024-01222-1
