
Abstract
Oxygen is crucial for life and acts as the final electron acceptor in mitochondrial energy production. Cells adapt to varying oxygen levels through intricate response systems. Hypoxia-inducible factors (HIFs), including HIF-1α and HIF-2α, orchestrate the cellular hypoxic response, activating genes to increase the oxygen supply and reduce expenditure. Under conditions of excess oxygen and resulting oxidative stress, nuclear factor erythroid 2-related factor 2 (NRF2) activates hundreds of genes for oxidant removal and adaptive cell survival. Hypoxia and oxidative stress are core hallmarks of solid tumors and activated HIFs and NRF2 play pivotal roles in tumor growth and progression. The complex interplay between hypoxia and oxidative stress within the tumor microenvironment adds another layer of intricacy to the HIF and NRF2 signaling systems. This review aimed to elucidate the dynamic changes and functions of the HIF and NRF2 signaling pathways in response to conditions of hypoxia and oxidative stress, emphasizing their implications within the tumor milieu. Additionally, this review explored the elaborate interplay between HIFs and NRF2, providing insights into the significance of these interactions for the development of novel cancer treatment strategies.
Similar content being viewed by others

Introduction
Oxygen (O2) is the most fundamental element for most living organisms on Earth and serves as the final electron acceptor in the mitochondrial energy production process1. The concentration of O2 encountered by cells undergoes significant changes due to environmental perturbations and the transport and distribution processes within living organisms. Hypoxic conditions, characterized by an insufficient O2 supply relative to the required amount, are prevalent in various pathological conditions, such as chronic obstructive pulmonary disease and ischemic heart disease. These conditions are particularly noticeable in the tumor microenvironment (TME), where vasculature is limited2. However, although O2 is a relatively stable molecule, O2-derived free radical formation is proportional to the O2 concentration. Inhalation of O2 concentrations higher than the normal partial pressure can lead to high reactive oxygen species (ROS) levels. Under these conditions, cells experience oxidative stress, where oxidant production significantly outweighs that of antioxidants and the capacity to repair damaged cellular components by oxidants3. Accordingly, cells have evolved sophisticated response systems to acclimatize to variable O2 availability. The primary effectors of the cellular hypoxic response are hypoxia-inducible factors (HIFs), including HIF-1α and HIF-2α. These transcription factors activate the expression of an array of genes involved in increasing O2 supply (genes regulating erythropoiesis and angiogenesis) and decreasing O2 expenditure (genes for O2-independent glycolytic metabolism), eventually triggering a cellular adaptive response to an O2-restricted environment2,4. The opposite of hypoxia, known as hyperoxia, is also a critical condition that induces cellular macromolecule dysfunction and, ultimately, cell death. In response, the expression levels of genes involved in hyperoxia-derived ROS removal and damaged cell repair increase. Nuclear factor erythroid 2-related factor 2 (NRF2) is the primary transcription factor mediating this response and promotes the adaptation and survival of cells under oxidative stress conditions5.
Hypoxia and oxidative stress are core hallmarks of solid tumors6. During the rapid tumor growth process, insufficient angiogenesis creates a hypoxic environment within the tumor tissue. Indeed, the average pO2 is reportedly 10 mmHg (1.4% O2) for breast, head, neck, and cervical cancers7. Under these conditions, HIFs are activated and promote the expression of various genes that lead to tumor vascularization, epithelial–mesenchymal transition (EMT), invasion/metastasis, cancer-specific metabolism, immune escape, and cancer stem cell (CSC) trait acquisition. Consistent with this, HIF levels in tumor biopsy samples have been associated with high mortality rates in patients with cancer; therefore, chemical HIF inhibitors are under investigation as a therapeutic option for cancer treatment2. Compared with noncancer cells, cancer cells exhibit high ROS levels due to oncogene activation, a high metabolic rate, and hypoxic stress8. To cope with oxidative stress, cancer cells aberrantly activate NRF2, promoting the expression of genes involved in ROS removal, cell proliferation, apoptotic resistance, and metabolic reprogramming, which ultimately favors cancer growth and progression9. In addition, hypoxia and oxidative stress in the TME are connected factors rather than independent factors; therefore, their effects on the HIF and NRF2 signaling systems are notably complex. For instance, hypoxia and oxidative stress often coexist within the TME. Hypoxia can lead to temporally increased ROS production through mitochondrial complex III dysfunction10. Notably, oxidative stress also serves as a signaling component of the hypoxic response. ROS can stabilize HIF-1α by inhibiting prolyl hydroxylase domain protein (PHD) activity11. Moreover, when hypoxic stress is sustained, mitochondrial ROS generation is reduced through a mechanism involving the substitution of the cytochrome c oxidase subunit and subsequent improvement in electron transfer efficiency12. Given the pivotal role of HIFs and NRF2 signaling in the context of tumor growth and progression, gaining a comprehensive understanding of how these systems respond to unique environmental stresses and alterations within the TME is imperative. This review aimed to elucidate the dynamic changes and functions of the HIF and NRF2 signaling pathways, particularly in response to hypoxia and oxidative stress conditions, with a specific emphasis on their implications within the tumor milieu. Furthermore, this review examines the intricate interplay between HIFs and NRF2 in the context of cancer.
Adaptive response systems to variable O2 tensions
Low O2 tension and HIFs
The discovery of HIFs and their regulation by the von Hippel–Lindau tumor suppressor protein (pVHL) and PHDs represents a significant breakthrough in our understanding of cellular responses to hypoxia. HIFs are heterodimeric transcription factor proteins composed of two subunits of the basic helix–loop–helix-Per–Arnt–Sim family: the O2-sensitive α subunit and the constitutively active β subunit13,14. Among the α subunits, HIF-1α (encoded by HIF1A) and HIF-2α (encoded by EPAS1) are well-characterized proteins that govern the transcriptional activity of numerous genes and contribute to adaptive strategies in low-O2 environments. In contrast, HIF-3α (encoded by HIF3A) has been relatively understudied13. The HIF-1β subunit (encoded by aryl hydrocarbon nuclear translocator; ARNT) heterodimerizes with HIF-α, stabilizing the HIF complex and enabling HIF target gene transcription14.
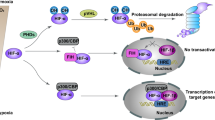
O2 plays an inevitable role in the regulation of HIF-α protein stability. Under normal O2 concentrations, O2 atoms are inserted into specific proline residues of HIFs (P402/P564 of HIF-1α and P405/P531 of HIF-2α) by PHDs. These hydroxylated HIF-α subunits are subsequently selectively recognized and ubiquitylated by the pVHL-elongin BC-Cullin 2 (CUL2) complex, which induces proteasomal degradation (Fig. 1, left panel)15. Notably, mammals possess three characterized PHDs, namely, PHD1–3, which catalyze the hydroxylation of HIF-α subunits in the presence of Fe2+ and α-ketoglutarate16. These PHDs exhibit varying affinities for HIF-α subunits, with PHD2 primarily targeting HIF-1α, while PHD3 displays greater activity toward HIF-2α than toward HIF-1α17. Under hypoxic conditions (0.5–2% O2), PHD-driven hydroxylation is inhibited due to a lack of O2 as a substrate, resulting in HIF-α accumulation. The translocation of stabilized HIF-α subunits into the nucleus facilitates their dimerization with HIF-1β, leading to the formation of a complex capable of recognizing and binding to the hypoxia response element (HRE; 5′-(A/G)CGTG-3′) in the promoter regions of target genes13. In addition to the canonical PHD/pVHL-mediated pathway, another regulatory mechanism involving factor-inhibiting HIF-1 (FIH-1) exists. FIH-1 modulates HIF activity by hydroxylating the asparagine 803 residues within the HIF-α subunits, thereby inhibiting the interaction of HIF-1 with transcription coactivators, i.e., CREB-binding protein and p300 (Fig. 1, left panel)18.

[Left] In hypoxic conditions, HIFs stabilize, translocate into the nucleus, and bind to the HRE, thereby inducing the expression of their target genes. In the presence of oxygen, HIFs are continuously degraded by the canonical PHD/pVHL-mediated pathway. The regulatory mechanism of FIH-1 also contributes to the inhibition of HIF activity. [Right] NRF2 maintains cellular redox homeostasis in response to oxidative stress, which can be induced by an excess oxygen environment. ROS/electrophiles and nonelectrophiles, such as p62, can inhibit NRF2-KEAP1 interactions to activate NRF2 target gene expression. This inhibition occurs through the KEAP1 Cys modification and competition with KEAP1 for binding to NRF2. Additionally, as a KEAP1-independent regulatory pathway, NRF2 phosphorylation by GSK-3β leads to β-TrCP-mediated proteasomal degradation of NRF2.
Although HIF-1α and HIF-2α share structural similarities and the ability to bind to the same HRE, they exhibit distinct functional characteristics. HIF-1α is ubiquitously expressed in hypoxic tissues, whereas HIF-2α is more selectively expressed in specific tissues, such as the vascular endothelium19. HIF-1α is rapidly activated in response to acute hypoxia, leading to the upregulation of genes associated with glycolytic metabolic shift and cell cycle arrest. In contrast, HIF-2α gradually becomes activated under persistent hypoxic conditions, elevating the expression levels of genes involved in erythropoiesis and tumor stemness19,20,21.
HIFs have been shown to upregulate the expression of numerous genes in response to low O2 concentrations. Microarray analysis of human pulmonary endothelial cells revealed the upregulation of 245 genes, including those encoding growth factors, cytokines, receptors, signal transduction molecules, and transcription factors, as commonly elevated genes in response to both HIF-1α overexpression and hypoxic incubation22. The HIF-1α-regulated genes included vascular endothelial growth factor (VEGF), glucose transporters (GLUTs), pyruvate dehydrogenase kinase 1 (PDK1), erythropoietin, insulin-like growth factor 2 (IGF-2), transforming growth factor α, B-cell lymphoma 2 (BCL-2), BCL-2 interacting protein 3 (BNIP3), and heme oxygenase-1 (HO-1)23. In neuroblastoma cells, HIF-1α and HIF-2α reportedly share several target genes, such as VEGF, tyrosine hydroxylase, and N-myc downstream regulated 120. In addition to these shared targets, specific target genes have been reported. For instance, BNIP3 expression solely depends on HIF-1α, while octamer-binding transcription factor 4 (OCT4) expression relies on HIF-2α24,25.
O2 excess, oxidative stress, and NRF2
The transcription factor NRF2, encoded by the NFE2L2 gene, plays a crucial role as a master regulator in maintaining cellular redox homeostasis in response to oxidative stress, which can be induced by excess O25,26. Upon activation, NRF2 triggers the expression of hundreds of genes involved in various cellular processes, including antioxidant and xenobiotic responses, cell proliferation and survival, and metabolism, among others9,27. Although H2O2 production increases in the lungs of rats exposed to hyperoxia, increased antioxidant enzyme expression delays H2O2-induced lung damage28. Subsequently, NRF2 was revealed to be the primary transcription factor responsible for this enhanced response system29. Similarly, hyperoxic incubation of pulmonary epithelial cells activated the NRF2 pathway via the ROS-epidermal growth factor receptor-phosphoinositide-3-kinase (PI3K) signaling axis30.
NRF2 is a cap’n’collar (CNC) leucine zipper (bZIP) transcription factor consisting of seven Neh domains, each with distinct functions5. The Neh1 domain contains the CNC-bZIP region for DNA binding and heterodimerization with small MAF (sMAF) proteins. The Neh2 domain of NRF2 contains highly conserved DLG and ETGE motifs, which bind to its negative regulator Kelch-like ECH-associated protein 1 (KEAP1)31. The binding of the Neh2 domain of NRF2 to the Kelch domain of KEAP1 is the primary mechanism of NRF2 activity regulation5,9. The Neh3–5 domains play a role in transactivation activity through binding to multiple components of the transcriptional machinery. The Neh6 domain mediates KEAP1-independent negative regulation of NRF2 stability through binding to β-transducing repeat-containing protein (β-TrCP) (Fig. 1, right panel)32.
NRF2 activity is tightly regulated by the KEAP1-mediated turnover process. Under normal conditions, NRF2 is bound to two molecules of KEAP1, an adapter protein for CUL3-based E3 ubiquitin ligase, and is continuously degraded by the 26S proteasome5. Under oxidative stress conditions, ROS or electrophiles interact with or modify KEAP1 cysteine (Cys) residues, resulting in conformational changes in the KEAP1 protein and subsequent disruption of the NRF2 DLG motif-KEAP1–CUL3 complex. This process eventually leads to the nuclear translocation of newly synthesized NRF2 and the binding of the NRF2–sMAF complex to antioxidant response elements (AREs; 5′-A/GTGACnnnGC-3′), resulting in the transactivation of target genes (Fig. 1, right panel)33. The interaction between NRF2 and KEAP1 involves two models based on KEAP1 responses. The first model is the Cys sensor-dependent mechanism, where Cys residues in KEAP1 sense oxidizing or electrophilic signals34. For example, Cys151 in the BTB domain responds to sulforaphane, while Cys226/613/622/624 are identified as H2O2-responding sensors for NRF2 activation33,35. The second model is the Cys-sensor-independent mechanism, where KEAP1 function is hindered by protein–protein interactions rather than being dependent on changes in Cys residues. The ETGE motif in NRF2 strongly binds to KEAP1, acting as a hinge, while the DLG motif functions as a latch due to its decreased binding affinity. P62, which has a greater affinity for KEAP1 than the DLG motif in NRF2, acts as a protein–protein interaction inhibitor. Consequently, increased p62 levels can disrupt NRF2 DLG-KEAP1 binding, leading to the activation of NRF2 target gene expression36.
NRF2 is involved in the regulation of more than 200 genes. These genes encode several phase I- and II-metabolizing enzymes, including NAD(P)H: quinone oxidoreductase 1 (NQO1), aldo-keto reductases, glutathione (GSH) S-transferases, and UDP glucuronosyltransferases. They also encompass phase III drug efflux transporters and components of the GSH-based system, including glutamate-cysteine ligase catalytic (GCLC) and modifier subunits (GCLM), GSH reductase, and GSH peroxidases (GPXs). Additionally, NRF2 regulates genes related to thiol proteins, including thioredoxins (TRXs) and peroxiredoxins, as well as genes involved in carbohydrate metabolism and NADPH-generating enzymes such as glucose-6-phosphate dehydrogenase (G6PD) and malic enzyme 1. NRF2 also influences genes associated with heme and iron metabolism, such as HO-1 and ferritins, as well as genes encoding signaling molecules and transcription factors37. This regulatory mechanism is crucial for maintaining the cellular redox balance and defending against oxidative stress-related damage.
Cross response: hypoxia-NRF2 and oxidative stress-HIFs
In real-world situations, the coexistence and interplay of hypoxia and oxidative stress result in a more complex response of HIFs and NRF2. It has long been established that hypoxic conditions lead to an increase in oxidative stress markers, including oxidized GSH. Consistent with this, increases in antioxidants, such as GPX and superoxide dismutase, have been reported in various animal models of hypoxia10. These observations suggest that ROS production increases under hypoxic conditions; therefore, determining both the stage of hypoxia and the mechanisms through which this increase occurs has been of interest. In most experimental hypoxia settings, ROS elevation has been a rapid response10; superoxide levels transiently increase within 10 min following acute hypoxia in human cells38. The involvement of mitochondria in ROS generation has been confirmed in several studies. Mitochondria-depleted Hep3B cells cannot produce ROS during hypoxia39. The transient superoxide burst induced by hypoxia also relies on functional mitochondria38. In particular, mitochondrial complex III has been identified as a mechanism for transient mitochondrial ROS generation under hypoxic conditions. Silencing of the Rieske iron-sulfur protein of complex III reduced hypoxia-induced ROS generation, suggesting the potential role of complex III as a hypoxic sensor40. Hypoxia-induced transient mitochondrial ROS are known to function as signaling molecules for cell proliferation and HIF activation41.
Under sustained hypoxic conditions, excess ROS accumulation can be harmful to cells. To protect mitochondria from oxidative stress, HIF-1α increases PDK1 expression levels, thereby activating mitochondria-independent metabolism. Moreover, HIF-1α enhances electron transfer efficiency by replacing the complex IV subunit and reducing ROS generation through the inhibition of complex I12,42. These actions help to prevent excessive mitochondrial ROS production during hypoxia. Despite the transient increase in ROS and antioxidants under hypoxic conditions, NRF2 activation has not been consistently observed. For instance, NRF2 levels in pulmonary epithelial and colorectal cancer cells and the rat retina remain unchanged or are reduced during hypoxia43,44,45, suggesting that other transcription factors activated by hypoxia, such as nuclear factor kappa B (NF-κB) and forkhead box O3 (FoxO), primarily lead to increased antioxidant protein expression levels10. Additionally, activation of HIF-1α reduces NRF2 transcriptional activity in ischemic mouse kidneys and vascular endothelial cells46,47.
Moreover, oxidative stress serves as an additional stimulator of HIF activation. Specifically, PHDs function as ROS sensors. PHD2, the primary HIF-1α hydroxylase, retains several Cys residues in its catalytic domain, and oxidative stress induces the dimerization of PHD2 through Cys oxidation, leading to the inhibition of PHD2 activity11. PHDs require Fe2+ as a cofactor, and the redox status of this cofactor is also crucial for their enzymatic activity. Depletion of the cellular antioxidant ascorbate leads to the oxidation of Fe2+ to Fe3+ in PHDs, resulting in the inactivation of these enzymes48. Additionally, the formation of an oxidant-induced GSH-Cys520 adduct in the HIF-1α protein promotes HIF-1α stabilization49. These lines of evidence collectively suggest HIF activation occurs in response to oxidative stress. Consistent with these findings, mitochondrial ROS, which increases during hypoxia, reportedly contributes to HIF upregulation39,50. These results showed that hypoxia and oxidative stress are linked factors rather than independent factors. Therefore, the effects of these two systems on HIFs and NRF2 signaling systems are complex, implying the presence of a network connecting these two systems.
The interplay between HIFs and NRF2
Considerable evidence suggests a correlation between the function and regulation of HIFs and NRF2. As indicated in Table 1, multiple reports suggest a positive association between HIFs and NRF2, which is often observed in cancer cells. In multiple cancer cell models, NRF2 silencing has been shown to reduce HIF-1α levels, resulting in the suppression of HIF-1α-mediated cell proliferation, angiogenesis, tumor growth, and migration/invasion45,51,52,53,54,55. The positive relationship between HIF-1α and NRF2 has been explained through several mechanisms. First, as a direct regulatory mechanism, a conserved functional ARE was identified in the promoter region of the HIF1A gene (Fig. 2a). In this context, NRF2-mediated elevation of HIF-1α has been linked to increased levels of both NRF2 and HIF-1α in breast and bladder cancers, as well as cisplatin resistance in hepatocarcinoma cells (HCCs)56,57. Second, NRF2 target genes have been proposed to constitute a molecular link between HIF-1α and NRF2 (Fig. 2b). For example, NQO1 can directly bind to HIF-1α and subsequently prevent PHD binding and proteasomal degradation, leading to HIF-1α accumulation58. The NRF2 target TRX1 has been shown to increase HIF-1α activity59. In adenocarcinoma cells following intermittent hypoxia, NADPH oxidase 1 (NOX1)-induced ROS increases NRF2/TRX1 levels and thereby increase HIF-1α protein levels60. Carbon monoxide (CO), a major metabolite of HO-1, has been reported to stabilize the HIF-1α protein via translational suppression of HIF-1α ubiquitination61,62. Third, several signaling molecules have been suggested to act as intermediaries in the relationship between HIF-1α and NRF2. In glioblastoma, mitochondrial Nip3-like protein X (NIX), whose expression levels are increased by NRF2, promotes HIF elevation, resulting in the maintenance of glioblastoma stem cells63. In breast and colorectal cancer cells, NRF2 silencing increased miR-181c-5p expression levels, subsequently suppressing mitochondrial respiration and O2 consumption by targeting the complex IV subunit, ultimately preventing HIF-1α accumulation under 24-h hypoxic conditions (Fig. 2c)64,65. Fourth, a direct interaction between the HIF-1α and NRF2 proteins has been reported (Fig. 2d). NRF2 directly binds to HIF-1α through the O2-dependent degradation domain; this interaction prevents PHD association, resulting in HIF-1α accumulation in pseudohypoxic HCC55. Most studies on the correlation between HIFs and NRF2 have focused primarily on HIF-1α, but an association with HIF-2α has been recently reported. HIF-2α levels, which gradually increase under continuous hypoxic conditions (72 h), decline following NRF2 silencing and treatment with a chemical NRF2 inhibitor. In this context, NRF2 inhibition leads to the upregulation of miR-181a-2-3p, which directly targets HIF-2α, consequently resulting in the attenuation of the HIF-2α-mediated CSC phenotype in colorectal cancer cells (Fig. 2c)66.

[Top] Positive associations between HIFs and NRF2. a As a direct regulatory mechanism, NRF2 binds to the ARE located in the promoter region of the HIF1A gene, inducing the expression of HIF-1α. b The NRF2 targets TRX1 and NQO1 enhance the accumulation and stabilization of HIF-1α. Carbon monoxide produced from HO-1 also contributes to HIF-1α stabilization. c Inhibition of NRF2 increases the expression levels of miR-181c-5p and miR-181a-2-3p, suppressing HIF-1α and HIF-2α levels, respectively. d NRF2 can directly bind to the oxygen-dependent degradation domain of HIF-1α and prevent the interaction of HIF-1α with PHD2. [Bottom] Negative associations between HIFs and NRF2. e Increased NRF2 levels can suppress HIF-α through reduced ROS levels. f HIF-1α can repress the expression of NRF2 and HO-1 through the elevation of BACH1, a repressive partner of NRF2. g UBXN7 is a cofactor for the ubiquitination of both HIFs by CUL2- and NRF2 via CUL3-based complexes. UBXN7 has opposing effects on HIFs and NRF2. For instance, when UBXN7 is knocked out, NRF2 levels increase while simultaneously leading to a decrease in HIF-1α levels.
HIFs and NRF2 do not always positively correlate. For instance, under certain experimental conditions or in specific cell types, their relationship exhibits an inverse association. For example, there are reports describing the regulation of HIFs and NRF2 in terms of changes in ROS (Fig. 2e). For instance, in fumarate hydratase-deficient cancer cells, accumulated fumarate results in the production of succinate GSH, resulting in increased mitochondrial ROS and subsequent HIF-1α activation. In this context, fumarate-induced NRF2 contributes to HIF-1α inhibition; therefore, NRF2 knockdown in these cells further increases HIF-1α levels67. The molecular clock protein BMAL1 directly upregulates NRF2 expression via E-box binding. Therefore, BMAL1-deficient cells exhibit increased HIF-1α expression levels due to ROS accumulation, which results from a reduction in NRF2 expression levels68. More specific causal relationships have been reported. HIF-1α induction repressed the NRF2 transcription of HO-1 and interleukin-8 in endothelial cells; the decreased NRF2 activity was found to be due to the elevation of BTB and CNC homology 1 (BACH1), a repressive partner of NRF2 (Fig. 2f)47. The reciprocal regulation of HIFs and NRF2 is reportedly attributed to a ubiquitination process involving these proteins (Fig. 2g). UBXN7 is a cofactor that provides a scaffold for both the ubiquitination of HIFs by a CUL2-based E3 ligase and that of NRF2 by a CUL3-based E3 ligase. Elevated UBXN7 levels are associated with HIF-1α accumulation, while UBXN7 knockout results in increased NRF2 levels and HIF-1α downregulation69. In line with these reports of a negative association, NRF2 silencing reinforced HIF-1α accumulation in alcohol-treated liver tissue70 and ischemia‒reperfusion-treated lung tissue71. In this context, treatment with several types of NRF2-activating agents suppresses HIF levels. Treating human endothelial cells with andrographolide suppressed HIF-1α, and NRF2 activation was involved in this process72.
O2 response systems and cancer
Elevated HIFs and NRF2 in cancer
In advanced solid tumors, hypoxic regions arise due to abnormalities in the tumor vasculature; hence, intratumoral hypoxia is the principal stimulus for HIF activation2. In support of this, immunohistological analysis of human cancer biopsies has shown that cancer cells exhibit higher HIF expression levels than their surrounding normal cells23,73. Tumor cells express HIF-regulated target genes that are critical for cancer progression, including those involved in tumor angiogenesis, metabolic shifts, metastasis/invasion, therapeutic resistance, and immune invasion74. Recent findings have also revealed the significant role of HIF-2α in cancer stemness19.
In some tumor tissues, HIFs are evenly expressed regardless of blood vessel formation, suggesting the occurrence of an O2-independent mechanism for HIF elevation. One well-characterized example is a loss-of-function mutation of the pVHL gene and subsequent elevation of HIF-1α and HIF-2α in clear cell renal cell carcinomas75. Additional associations between tumor suppressor gene deletion and HIF elevation have been identified. Loss of the p53 tumor suppressor impairs MDM2-mediated HIF-1α degradation by the proteasome, thus enhancing the transcriptional activation of VEGF76. Dominant-negative mutations in the tumor suppressor phosphatase and tensin homolog (PTEN) increase HIF-1α expression levels, leading to target gene expression in prostate cancer77. Fumarate, an oncometabolite that accumulates in fumarate hydratase-deficient renal cell carcinoma, induces HIF upregulation by competing at the PHD78. In addition, hypoxia-induced miRNAs, termed hypoxamiRs, regulate HIFs and subsequent cell responses to hypoxia79.
NRF2 prevents cancer initiation by orchestrating protective mechanisms against potential carcinogens and other hazards in normal and healthy cells. However, in cancer cells, NRF2 has oncogenic effects, contributing to tumor growth and progression9,80. Within the TME characterized by oxidative conditions, the delicate balance of NRF2 regulation is frequently disrupted, resulting in aberrant NRF2 activation9,31,80. Dysfunction of the NRF2 pathway leads to the activation of cellular defense systems and subsequent target gene upregulation, thereby promoting the survival and proliferation of cancer cells.
NRF2 mutations are classified as gain-of-function mutations and are concentrated in the DLG and ETGE motifs responsible for KEAP1 binding. These mutations disrupt the interaction between NRF2 and KEAP1. This prevents the formation of the NRF2-KEAP1-CUL3 complex required for NRF2 degradation, resulting in NRF2 accumulation and constitutive activation of its target genes80,81. Mutations in KEAP1 are classified as loss-of-function mutations and are distributed throughout the entire KEAP1 genome, with particular enrichment observed within the Kelch domain, which interacts with the DLG and ETGE motifs of NRF29,35,80. In these mutant cancer cells, NRF2 is constitutively activated, upregulating target genes that promote cancer cell survival, proliferation, progression, and therapeutic resistance81,82. Mutations in NRF2 and KEAP1 have been identified in multiple cancers with varying prevalence. Somatic mutations in NRF2 were found in 6% of patients with HCC83, and KEAP1 mutations have been reported to be present in 30% and 20% of patients with gallbladder and lung adenocarcinomas, respectively84,85. In addition to somatic mutations in NRF2 and KEAP1, several molecular mechanisms have been elucidated for NRF2 overactivation in cancers, including KEAP1 silencing through promoter methylation86, transcriptional NRF2 activation by oncogenes such as KRAS87, NRF2 stabilization by p62 accumulation88, and NRF2 activation by the oncometabolite fumarate89.
Activated HIFs and NRF2 in cancer cells can cooperatively promote tumor growth and progression by upregulating common target genes (Table 2). Consistent with these findings, HIF-1α and NRF2 expression levels are mutually associated, and high levels of these transcription factors correlate with poor outcomes in glioblastoma patients90. In the following sections, we investigate the specific roles of HIFs and NRF2 in the fundamental characteristics of cancer, including cancer growth and survival, therapeutic resistance, angiogenesis, metastasis, CSCs, and ferroptosis resistance (Fig. 3). Specifically, our emphasis will be on scrutinizing the interactions between HIFs and NRF2 concerning these fundamental aspects of cancer.

In the tumor microenvironment characterized by hypoxia and oxidative stress, HIFs, and NRF2 are aberrantly activated, promoting core cancer phenotypes, such as cancer proliferation and survival, therapeutic resistance, angiogenesis, EMT/metastasis, CSC trait acquisition, and resistance to ferroptosis. HIFs respond to both hypoxia and oxidative stress, which are likely inevitable conditions under low-oxygen conditions. In contrast, changes in NRF2 under hypoxic conditions vary in a context-dependent manner, indicating that hypoxia-associated oxidative stress does not necessarily accompany NRF2 activation.
Cancer proliferation and survival
HIF-1α plays a critical role in cancer survival and proliferation in hypoxic environments by regulating a wide range of genes. First, HIF-1α promotes glycolysis by upregulating key glycolytic enzymes, including aldolase A, phosphoglycerate kinase 1, and pyruvate kinase M. Similarly, HIF-1α inhibits mitochondrial oxidative phosphorylation through the upregulation of lactate dehydrogenase A and PDK191,92. Additionally, HIF-1α elevates GLUT4 levels to increase cellular glucose uptake93. This metabolic shift enables cancer cells to produce ATP with reduced ROS generation under O2-deprived conditions, ultimately promoting cancer proliferation. Second, as an adaptive stress resistance mechanism, HIF-1α activates autophagy by upregulating BNIP3, facilitating the removal of damaged organelles such as mitochondria94. Third, HIF-1α suppresses protein synthesis by modulating multiple genes involved in the translation process, thereby reducing energy expenditure and preventing unfolded protein accumulation95.
In parallel, NRF2 participates in cancer proliferation and survival through downstream target genes. First, antioxidant and detoxification systems, such as GSH synthesis and regenerating enzymes, protect cancer cells against the oxidizing tumor environment and cytotoxic anticancer drug treatment96. Second, the elevation of pentose phosphate pathway genes, including G6PD and 6-phosphogluconate dehydrogenase, facilitates purine biosynthesis and NAD(P)H generation, resulting in enhanced cell proliferation37. Third, NRF2 has been shown to regulate the expression of genes associated with cell growth, including IGF-1 and bone morphogenetic protein receptor 1 (BMPR1), as revealed by ChIP-seq profiling analysis97.
Multiple lines of evidence support the collaborative interplay between NRF2 and HIF in promoting cancer proliferation and survival. Research conducted on breast cancer cells demonstrated the cooperative effect of upregulated NRF2 and HIF-1α on cell proliferation, which led to the enhancement of glycolytic genes, including hexokinase 2, pyruvate kinase M2, and lactate dehydrogenase A98. Targeting NRF2 with shRNA resulted in the downregulation of HIF-1α and glycolytic genes, ultimately leading to the suppression of cancer proliferation. The upregulation of NRF2 stimulates the expression of G6PD and activates HIF-1α, resulting in NOTCH1 signaling activation and breast cancer proliferation99. Similarly, in HCC, elevated NRF2 and HIF-1α levels contribute to cancer growth and progression. NRF2 knockdown mitigated HIF-1α accumulation, while HIF-1α silencing did not affect NRF2 status. Mechanistically, a direct interaction between NRF2 and HIF-1α could impede PHD-VHL-mediated degradation, thus contributing to HIF-1α stabilization55. In tissue specimens from patients with gastric cancer, high NRF2 expression levels were positively correlated with HIF-1α and HO-1 expression100. In vitro, hypoxic conditions increased NRF2 and HO-1 expression levels, as did HIF-1α. NRF2 knockdown blocked HIF-1α and HO-1 upregulation, further suppressing gastric cancer survival. Mechanistically, hypoxia-driven VEGF-A activates the nuclear translocation of NRF2 through the RAP1B/ERK/AKT signaling cascade100. The significance of AKT signaling in the regulation of HIFs and NRF2 has also been demonstrated in ERBB3-induced breast cancer growth101. The ERBB3 mutant protein (ERBB3 N418Q) blocked HIF-1α and NRF2 accumulation by inhibiting PI3K-AKT signaling activation, which resulted in the suppression of cancer growth and migration. These results demonstrate that in many tumors, HIFs and NRF2 jointly contribute to cancer cell proliferation and survival, with increases in NRF2 often directly or indirectly linked to increases in HIFs.
Therapeutic resistance
Increased anticancer drug efflux is one of the major mechanisms by which cancer cells mediate multidrug resistance and therapy refractoriness. HIF-1α promotes the expression of ATP-binding cassette (ABC) transporters, including multidrug resistance 1 (MDR1), multidrug resistance-associated protein 1 (MRP1), and breast cancer resistance protein (BCRP), by acting on HREs102,103. Additionally, HIF-1α induces the expression of antiapoptotic proteins, such as BCL-2 and BCL-XL, while inhibiting the expression of proapoptotic proteins, such as BCL-2-associated X protein (BAX) and BH3 interacting domain death agonist (BID)104,105,106. These changes consequently result in coordinated regulation that promotes cancer cell survival by preventing apoptotic cell death, further contributing to drug resistance.
The constitutive activation of NRF2 in cancer is a significant factor that enhances the elimination of anticancer drugs by increasing the expression levels of phase II metabolizing enzymes, leading to chemotherapy resistance107. In addition, NRF2 promotes drug efflux by upregulating multiple ABC transporters, such as MDR1 and BCRP107,108. Functional AREs have been characterized in the promoter regions of these ABC transporters109,110. Furthermore, NRF2-mediated elevations in the levels of the antiapoptotic protein BCL-2 are involved in resistance to etoposide-induced cell death111.
Several studies have shed light on the link between NRF2 and HIFs in conferring resistance to anticancer therapy. Under mildly hypoxic conditions (5% O2) in HCC, NRF2 can directly interact with the ARE located at the 5′ upstream region of the HIF1A gene56. This interaction leads to the upregulation of HIF-1α expression, thereby resulting in resistance to cisplatin treatment. In 5-fluorouracil-resistant HCC, NRF2 knockdown decreases HIF-1α expression levels, while its overexpression increases HIF-1α expression levels112. In this context, the administration of siRNA targeting NRF2 enhances the anticancer effect of As2O3 by inhibiting HIF-1α/HSP70 signaling and subsequently inhibiting BCL-2. These studies suggest that NRF2 and HIF-1α cooperate in the development of chemotherapy resistance.
Tumor angiogenesis
Angiogenesis occurs as tumors grow. It involves the release of proangiogenic factors that stimulate tumors to progress from dormancy and lead to the formation of new blood vessels, inducing rapid growth113. One well-established proangiogenic factor is VEGF. Under hypoxic conditions, tumor cells invade neighboring tissues from primary sites and metastasize through tumor neovascularization, which is regulated by VEGF. A HIF-1 binding site has been identified and characterized in the VEGF gene, and ARNT-deleted cells failed to increase VEGF expression levels upon exposure to hypoxic stimuli114. Additionally, other angiogenic growth factors, including stromal-derived factor 1, stem cell factor, and angiopoietin family members, are also regulated by HIFs and participate in tumor vascularization115,116,117.
NRF2 has been reported to increase the expression of several angiogenic target genes, including HO-1, VEGF, and IGF-1; therefore, its activity can influence the blood vessel formation process9,41. The association between NRF2 and angiogenesis regulation has been demonstrated in studies using Nrf2−/− mice118,119. In these studies, revascularization was promoted in Nrf2−/− mice compared with wild-type mice in a model of hindlimb ischemia. However, in vitro, angiogenic cytokine treatment activates the nuclear translocation of NRF2, which stimulates the tube formation of endothelial cells. In addition, the survival and angiogenic response of bone marrow-derived proangiogenic cells from Nrf2−/− mice were significantly reduced114. These discrepancies between in vitro cell systems and in vivo animal models can be attributed to the potential proangiogenic role of ROS and the increased inflammatory response in Nrf2-deleted mice118. Additional studies also suggest a positive role for NRF2 in angiogenesis. The specific deletion of NRF2 in endothelial cells results in decreased vascular density and angiogenic sprouting by increasing delta-like ligand 4 expression and NOTCH activity120. Loss of Nrf2 inhibited VEGF elevation in the brains of venous hypertensive rats, and Nrf2 knockout suppressed vascular tube formation in primary brain microvascular endothelial cells121. A coculture model of HCC with monocytes revealed that NRF2 activation in macrophages induces an M2-like phenotype, promoting VEGF elevation in cancer cells and subsequent EMT122.
In this context, evidence suggests that NRF2 silencing blocks HIF-1α signaling to suppress blood vessel formation and xenograft tumor growth45,52,54. In colon cancer cells, NRF2 silencing impaired HIF-1α elevation, leading to angiogenesis suppression both in vitro and in vivo45,64. In addition to these molecular events, elevated miR-181c expression levels and a subsequent reduction in O2 consumption were suggested to promote HIF-1α protein degradation in NRF2-inhibited cells. Similarly, NRF2 knockdown in glioblastoma and ovarian cancer cells led to reduced HIF-1α levels, with a concurrent reduction in VEGF expression levels52,54. However, curcumin supplementation upregulated NRF2 and GSH, thus inhibiting HIF-1α accumulation and ultimately suppressing HCC angiogenesis and invasion123.
EMT and cancer metastasis
Hypoxia is a strong driving force for EMT, a process in which cancer cells undergo phenotypic conversion to a mesenchymal phenotype, enhancing their migration and invasion. The signaling pathways and transcription factors associated with EMT, including NF-κB, transforming growth factor β (TGF-β), SNAIL, and TWIST, are enhanced by hypoxia124. A preliminary study analyzing HIF-1α levels in tumor specimens showed that HIF-1α levels were elevated in 69% of metastatic breast cancers, while only 29% of primary breast cancers exhibited elevations in HIF-1α levels125. Consistent with these findings, the proximal promoter regions of SNAIL, TWIST, and ZEB1 contain HREs that directly interact with HIF-1α under hypoxic conditions126,127,128. In breast cancer cells, elevated expression levels of a disintegrin and metalloproteinase 12, mediated by HIF-1α and HIF-2α, induce hypoxia-driven migration and invasion, resulting in breast cancer metastasis to the lung129.
Despite the clear involvement of HIFs in EMT and cancer migration, the impact of NRF2 on these aspects is not consistent. Cancer cells with constitutively high NRF2 levels exhibit activated EMT and cancer migration130,131. Overactivation of NRF2 by deleting the KEAP1 gene in lung squamous cell carcinoma facilitates tumor metastasis132. NRF2 has also been linked to TGF-β1-induced EMT by directly upregulating NOTCH4 expression133. In this context, NRF2 knockdown or ROS inhibition attenuated NOTCH signaling and subsequently suppressed EMT in lung cancer cells. Moreover, NRF2 was identified as a phenotypic stability factor for hybrid epithelial/mesenchymal and activated NOTCH signaling for cancer migration134. However, a negative role of NRF2 in EMT and cancer metastasis has been suggested. In HCC, loss of NRF2 increases cancer plasticity and motility by enhancing SMAD signaling135. NRF2-silenced lung cancer cells exhibit an increase in both basal and TGF-β-induced cell motility through increased ROS levels136.
Several studies have suggested a cooperative link between HIFs and NRF2 in cancer metastasis. NRF2 overexpression and KEAP1 knockdown promoted the expression of G6PD and HIF-1α/NOTCH1 to induce EMT and breast cancer migration99. Hypoxic conditions increase NRF2, HIF-1α, and HO-1 expression in gastric cancer cells, whereas NRF2 inhibition suppresses cancer invasion via concomitant reductions in HIF-1α expression100. In a hypoxia-mimicking model using CoCl2, NRF2 knockdown reduced HIF-1α, HO-1, and matrix metalloproteinase 2 expression levels, resulting in the suppression of cancer migration and invasion in esophageal squamous cell carcinoma53.
Cancer stem cell traits
CSCs are a small subset of cells within a tumor that possess self-renewal and tumor-initiating capacities. The presence of CSCs is thought to cause cancer treatment failure, as they drive malignant properties, including metastasis, invasion, therapeutic resistance, and immune escape137. Several embryonic pluripotency markers, including SRY-box 2 (SOX2), Krüppel-like factor 4 (KLF4), OCT4, and NANOG, have been identified as CSC markers138. Specific CSC markers have been identified for each tumor type and are commonly used for the isolation and characterization of CSCs. For example, CD44+, CD24−, and CD133+ have been used to characterize breast CSCs139, while CD44+, epithelial cell adhesion moleculehigh, and CD166+ are used as colorectal CSC markers140.
Restricted O2 availability induces enrichment and dependency of CSCs on HIFs for survival, self-renewal, and growth141. In triple-negative breast cancer, chemotherapy increases the expression levels of HIF-1α and its target genes, subsequently leading to an increased number of breast CSCs through elevations in the levels of interleukins 6 and 8142. This study showed that HIF inhibitors can restore the sensitivity of breast CSCs to chemotherapy. Antiangiogenic agents, such as bevacizumab, induce CSC enrichment in breast cancer xenografts by creating an intratumoral hypoxic environment and upregulating HIF-1α expression143. As factors contributing to underlying molecular mechanisms, HIFs activate transcription factors and signaling pathways involved in CSC self-renewal and pluripotency, including OCT4 and NOTCH144. HIF-2α directly binds to the OCT4 promoter and increases OCT4 expression levels, promoting teratoma growth145. Chemotherapy-induced HIF-1α upregulates the expression of S100A10, which forms a transcription complex at the OCT4 binding site to facilitate the expression of other pluripotency genes, including NANOG, SOX2, and KLF4146. In ovarian cancer, the HIF-1α-driven NF-κB pathway is responsible for inducing CSC properties through SIRT1 upregulation147. Moreover, evidence suggests that HIF-2α plays a role in maintaining CSC traits through the activation of WNT and NOTCH signaling148 by elevating superoxide dismutase (SOD) levels and by reducing mitochondrial reactive oxygen species (ROS) levels149. Consistent with these findings, HIF-2α knockdown in renal cell carcinoma inhibited the expression of CXCR4, a chemokine receptor used as a renal cell carcinoma stem cell marker, resulting in the inhibition of spheroid and tumor growth150. These studies collectively underscore the critical role of the HIF pathway in regulating CSC traits.
CSCs reportedly exhibit higher antioxidant gene expression levels and lower ROS levels than normal cells151. This finding suggests that NRF2 is involved in the development of CSC traits. Currently, accumulating evidence suggests that NRF2 signaling is involved in maintaining the stemness phenotype of CSCs152. Head and neck cancer cells displaying low ROS levels exhibit more CSC traits, such as stemness and chemoresistance. Elevated NRF2 levels are responsible for maintaining low-ROS CSCs by increasing the expression of antioxidant and glycolytic enzymes and stemness markers153,154. In sorafenib-resistant HCC, NRF2 signaling promoted CSC traits and increased ABC transporter expression levels155. In glioblastoma stem cells (GSCs) isolated from surgical specimens, NRF2 silencing decreased the proportion of GSCs and the expression levels of self-renewal markers, including SOX2156. Chemotherapy-induced breast CSCs exhibit CD44 enrichment and CSC traits, including spheroid growth and increased tumorigenesis. These changes are mediated by p62-associated NRF2 activation157. Similarly, additional experiments involving the isolation of CSC fractions using aldehyde dehydrogenase, epithelial cell adhesion molecule, or CD133 revealed that increased NRF2 levels play a critical role in developing CSC traits and maintaining and promoting CSC growth158,159,160. In addition to its role in antioxidant and drug efflux systems, NRF2 supports CSC stemness traits through the upregulation of NOTCH, FOXO3, and β-catenin expression161,162,163. Although the exact underlying mechanism by which NRF2 expression increases in CSCs has not been fully elucidated, it has been suggested that increases in the levels of p62 or the endoplasmic reticulum stress-activated protein kinase R-like ER kinase (PERK) may be involved153,157,159,160.
Several reports have demonstrated an association between HIFs and NRF2 in CSCs. Lung epithelial cells exposed to inorganic arsenic develop CSC-like properties with increased expression levels of stemness markers. During this process, NRF2 activation stimulates increases in HIF-1α levels, allowing metabolic reprogramming toward glycolysis51. In this context, NRF2 knockout diminished inorganic arsenic-induced HIF-1α accumulation and stemness marker expression. In clinical samples from patients with glioblastoma, elevated mitochondrial NIX expression levels were identified as a marker of GSCs, and hypoxia-induced elevations in NIX levels were linked to increased ROS-driven NRF2 expression, further contributing to CSC stemness through HIF/mTOR activation63. Hypoxia-induced M2 phenotype macrophages promote the stemness traits of glioblastoma through VEGF secretion, which is mediated by HIF-1α activation. PI3K/AKT-mediated NRF2 activation is involved in this VEGF-GSC axis164. In a study involving spheroid culture of colon cancer cells transfected with a reporter monitoring the transcriptional activity of HIFs and NRF2, increases in the expression levels of these transcription factors were observed within the spheroid cores, suggesting coordinated HIF and NRF2 regulation in a 3D culture system165. Additionally, in colorectal cancer cells, prolonged hypoxia-induced elevations in the levels of OCT4 and CSC traits are suppressed by NRF2 inhibition, and the underlying mechanism of this effect is the repression of HIF-2α accumulation66.
Resistance to ferroptosis
Ferroptosis is an iron-dependent lipid peroxidation-mediated cell death pathway that involves characteristics distinct from those of apoptosis, such as an intact cell membrane and nucleus, increased bilayer membrane density, and reduced mitochondrial volume and cristae166. Increased intracellular free iron causes ROS generation through the Fenton reaction, resulting in lipid peroxide accumulation and subsequent ferroptosis167. In this context, the iron chelating agent RSL3 inhibits ferroptosis168. The removal of H2O2 by GPX4 is essential for preventing ferroptosis. Gpx4 knockout in mice leads to ferroptosis-induced cell death in the kidney169. Emerging studies on ferroptosis in various diseases have revealed the association of this novel cell death system with pathological processes, including cancer. Treatment-resistant mesenchymal-type cancer cells are strongly dependent on GPX4, and lipoxygenase inhibition can block ferroptotic cell death via GPX4 inhibition170.
Hypoxia and HIFs are associated with ferroptosis in a context-dependent manner. Hypoxia decreases cellular free iron levels and increases ferritin expression levels, resulting in the protection of hypoxic macrophages against RSL3-induced ferroptosis171. In clear cell renal cell carcinoma, elevated HIF levels lead to the suppression of ferroptosis in response to erastin or GSH synthesis inhibition172. The pharmacological or siRNA-mediated inhibition of iron-sulfur cluster assembly 2 in mitochondria decreases HIF-1α and HIF-2α levels, resulting in subsequent iron overaccumulation and ferroptosis-mediated cell death in clear cell renal cell carcinoma173. Aryl hydrocarbon receptor nuclear translocator-like protein inhibits ferroptosis by activating HIF-1α in non-small cell lung cancer cells174. In contrast, some reports suggest that HIFs induce ferroptosis under certain conditions. HIF-2α activation in colorectal cancer upregulates genes involved in lipid and iron metabolism, resulting in enhanced susceptibility to dimethyl fumarate-induced ferroptosis175. Treatment of glioblastoma cells with the PHD inhibitor roxadustat results in the accumulation of lipid peroxides and iron, leading to ferroptosis176; in this case, HIF-2α contributes to ferroptosis by upregulating lipid regulatory genes.
NRF2 directly upregulates genes associated with iron metabolism (HO-1, ferritin light and heavy chain) and antioxidant activity (GPX4, thioredoxin reductase 1, GCLC, GCLM, and the cystine/glutamate antiporter SLC7A11), which prevent free iron accumulation and lipid peroxidation. Therefore, NRF2 has been recognized as a negative regulator of ferroptosis177. In glioblastoma cells, forced NRF2 expression upregulates SLC7A11 (also known as xCT) and promotes ferroptosis resistance178. NRF2 inhibition augments erastin-induced ROS generation via SLC7A11 reduction, thereby sensitizing glioma cells to ferroptosis. Another study involving head and neck cancer cells demonstrated that RSL3 resistance is associated with increased expression levels of NRF2 and p62; therefore, NRF2 or p62 inhibition reverses ferroptosis resistance179. Similarly, cetuximab treatment facilitates RSL3-induced ferroptosis through the inhibition of NRF2/HO-1 signaling in KRAS-mutant colorectal cancer cells180. Recently, ferroptosis suppressor protein 1 (FSP1) was identified as a novel target of NRF2. KEAP1 mutations in lung cancer cells upregulate FSP1 expression, leading to resistance to ferroptosis and radiotherapy181. Additionally, NRF2 directly regulates the E3 ubiquitin ligases HERC2 and VAMP3. Therefore, NRF2 knockout in ferroptosis-resistant ovarian cancer cells elevates apoferritin in the autophagosome and intracellular labile iron pool, ultimately leading to enhanced ferroptosis sensitivity182. These studies indicate that the NRF2 pathway plays a crucial role in the antiferroptosis mechanism.
Considering the role of both the HIF and NRF2 pathways in ferroptosis, associations between these signaling pathways and cancer are anticipated. However, comprehensive studies on this relationship are currently lacking. In noncancer cells, hypoxia-reperfusion induces ferroptosis, whereas NRF2 activation prevents ferroptotic cell death71,183. In gastric cancer cells, silencing of the transient receptor potential melastatin-2 (TRPM2) increases the levels of ROS, iron, and lipid peroxides, resulting in ferroptosis. In this study, TRPM-2 silencing-mediated ferroptosis was attributed to HIF-1α and NRF2 destabilization184. Elucidation of the roles of HIF and NRF2 in regulating ferroptosis will enable the development of novel cancer treatment strategies through the induction of ferroptosis.
Concluding remarks
In the characteristic hypoxic and oxidative stress environment of tumors, increases in the levels of HIFs and NRF2 play a crucial role in tumor growth and progression. Elevations in the levels of these two transcription factors can occur not only due to environmental stimuli but also in response to changes in specific signaling pathways within the tumor. For instance, KRAS mutations increase NRF2 expression levels, promoting tumor growth87. When mutant KRAS was silenced in colorectal cancer cells, hypoxia-induced HIF-1α accumulation was suppressed185. Additionally, PTEN loss-mediated AKT activation and subsequent GSK-3β inhibition lead to an increase in NRF2186 and HIF-1α transcriptional activity77. Similarly, activation of human epidermal growth factor receptor 2 (HER2) results in an increase in HIF-1α expression levels via mTOR activation77. HER2 has also been reported to induce the stabilization of NRF2 through direct protein binding187. An oncometabolite, fumarate, simultaneously induces elevations in NRF2 levels through KEAP1 modification and activates HIF-1α through an increase in mitochondrial ROS67,89. These reports indicate a profound interconnection between tumor signaling pathways and the promotion of tumor growth and progression through increases in HIF and NRF2 levels, regardless of whether the tumor microenvironment is hypoxic or oxidative stress-enriched.
The control of the HIF and NRF2 signaling pathways in tumors has emerged as a critical issue. Current reports suggest that, during the interplay between these two transcription factors, NRF2 appears to be an upstream regulator of HIF. Inhibition of NRF2 in cancer cells has been reported to suppress HIF activity through multiple molecular events, including direct regulation of HIF-1α expression by NRF2, HIF-1α stabilization by the NRF2 target NQO1 and TRX1, and HO-1-driven CO and miRNA expression58,59,61,65,66. The relative dominance of NRF2 in the regulation of these two factors implies that HIFs evolved as a system responding to both hypoxic and oxidative stress, which is likely inevitable under low-oxygen conditions. In contrast, changes in NRF2 under hypoxic conditions vary in a context-dependent manner, indicating that hypoxia-associated oxidative stress does not necessarily accompany NRF2 activation. This relationship suggests that inhibiting NRF2 in tumors with both HIF and NRF2 upregulation could be a more efficient strategy considering the response of HIFs to hypoxia and oxidative stress. For instance, treatment with brusatol, an inhibitor of NRF2 protein synthesis, prevents hypoxia-induced HIF-1α accumulation and reduces glucose consumption in colorectal cancer cells188. Moreover, under sustained hypoxic conditions, brusatol inhibited the increase in HIF-2α and suppressed the development of hypoxia-induced CSC traits66.
On the other hand, reports indicate that a single substance can inhibit both HIFs and NRF2 through independent mechanisms. Triptolide, a diterpenoid isolated from Tripterygium wilfordii, has been shown to inhibit NRF2 and HIF-1α expression in myeloid leukemia cells189. In more detail, in doxorubicin-resistant or imatinib-resistant leukemic cells that highly express both NRF2 and HIF-1α, treatment with triptolide alone or in combination with the respective resistant drugs inhibited NRF2 and its target genes, as well as HIF-1α and its target genes. This inhibitory effect enhances the apoptotic ratio within cells, indicating that triptolide can restore drug sensitivity by suppressing the NRF2 and HIF-1α pathways. Furthermore, combined treatment with triptolide and idarubicin has synergistic effects on leukemia stem cells, enhancing cell apoptosis through the reduced expression of NRF2, HIF-1α, and their target genes190. Cardamonin, a natural chalcone from Alpiniae katsumadai, inhibits breast cancer cell growth by suppressing HIF-1α through blockade of the mTOR pathway and inducing metabolic reprogramming characterized by reduced glucose uptake and lactate transport191. Cardamonin also inhibited NRF2, NQO1, and HO-1 expression, thereby leading to increased ROS-induced apoptosis.
Overall, this review explored the intricate interplay between HIFs and NRF2, providing insights into the relevance of these interactions for the development of novel cancer treatment strategies. In particular, considering that both of these transcription factors also play critical roles in normal cell physiology, elucidating the common signaling pathways associated with their increase in tumors and revealing the molecular mechanisms underlying the correlation between these factors are expected to enable the development of the selective regulation of HIFs and NRF2 in cancers.
References
Raymond, J. & Segrè, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 311, 1764–1767 (2006).
Wicks, E. E. & Semenza, G. L. Hypoxia-inducible factors: cancer progression and clinical translation. J. Clin. Investig. 132, e159839 (2022).
Halliwell, B. Biochemistry of oxidative stress. Biochem Soc. Trans. 35, 1147–1150 (2007).
Semenza, G. L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29, 625–634 (2010).
Yamamoto, M., Kensler, T. W. & Motohashi, H. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 98, 1169–1203 (2018).
O’Malley, J., Kumar, R., Inigo, J., Yadava, N. & Chandra, D. Mitochondrial stress response and cancer. Trends Cancer 6, 688–701 (2020).
Vaupel, P., Höckel, M. & Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal 9, 1221–1235 (2007).
Nakamura, H. & Takada, K. Reactive oxygen species in cancer: current findings and future directions. Cancer Sci. 112, 3945–3952 (2021).
Rojo de la Vega, M., Chapman, E. & Zhang, D. D. NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43 (2018).
Hermes-Lima, M. et al. Preparation for oxidative stress under hypoxia and metabolic depression: revisiting the proposal two decades later. Free Radic. Biol. Med. 89, 1122–1143 (2015).
Lee, G. et al. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1α activation. Sci. Rep. 6, 18928 (2016).
Fukuda, R. et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122 (2007).
Semenza, G. L. The genomics and genetics of oxygen homeostasis. Annu. Rev. Genomics Hum. Genet. 21, 183–204 (2020).
Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl Acad. Sci. USA 92, 5510–5514 (1995).
Schofield, C. J. & Ratcliffe, P. J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 5, 343–354 (2004).
Bruick, R. K. & McKnight, S. L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340 (2001).
Appelhoff, R. J. et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor*. J. Biol. Chem. 279, 38458–38465 (2004).
Mahon, P. C., Hirota, K. & Semenza, G. L. FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 15, 2675–2686 (2001).
Cowman, S. J. & Koh, M. Y. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer 8, 28–42 (2022).
Holmquist-Mengelbier, L. et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 10, 413–423 (2006).
Koh, M. Y. & Powis, G. Passing the baton: the HIF switch. Trends Biochem. Sci. 37, 364–372 (2012).
Manalo, D. J. et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105, 659–669 (2005).
Semenza, G. L. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharm. Sci. 33, 207–214 (2012).
Covello, K. L. et al. HIF-2α regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557–570 (2006).
Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H. & Harris, A. L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673 (2001).
Dodson, M. et al. Modulating NRF2 in disease: timing is everything. Annu. Rev. Pharm. Toxicol. 59, 555–575 (2019).
Torrente, L. & DeNicola, G. M. Targeting NRF2 and its downstream processes: opportunities and challenges. Annu. Rev. Pharm. Toxicol. 62, 279–300 (2022).
Ho, Y. S., Dey, M. S. & Crapo, J. D. Antioxidant enzyme expression in rat lungs during hyperoxia. Am. J. Physiol. 270, L810–L818 (1996).
Cho, H. Y., Reddy, S. P., Debiase, A., Yamamoto, M. & Kleeberger, S. R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 38, 325–343 (2005).
Papaiahgari, S., Zhang, Q., Kleeberger, S. R., Cho, H. Y. & Reddy, S. P. Hyperoxia stimulates an Nrf2-ARE transcriptional response via ROS-EGFR-PI3K-Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid. Redox Signal. 8, 43–52 (2006).
Taguchi, K. & Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 7, 85 (2017).
Rada, P. et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/β-TrCP axis. Mol. Cell Biol. 32, 3486–3499 (2012).
Suzuki, T., Takahashi, J. & Yamamoto, M. Molecular basis of the KEAP1-NRF2 signaling pathway. Mol. Cells 46, 133–141 (2023).
Iso, T., Suzuki, T., Baird, L. & Yamamoto, M. Absolute amounts and status of the Nrf2-Keap1-Cul3 complex within cells. Mol. Cell Biol. 36, 3100–3112 (2016).
Baird, L. & Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell Biol. 40, e00099–20 (2020).
Horie, Y. et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 4, 576 (2021).
Hayes, J. D. & Dinkova-Kostova, A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218 (2014).
Hernansanz-Agustín, P. et al. Acute hypoxia produces a superoxide burst in cells. Free Radic. Biol. Med. 71, 146–156 (2014).
Chandel, N. S. et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl Acad. Sci. USA 95, 11715–11720 (1998).
Guzy, R. D. et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408 (2005).
Toth, R. K. & Warfel, N. A. Strange bedfellows: nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants 6, 27 (2017).
Tello, D. et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 14, 768–779 (2011).
Xin, X., Li, Y. & Liu, H. Hesperidin ameliorates hypobaric hypoxia-induced retinal impairment through activation of Nrf2/HO-1 pathway and inhibition of apoptosis. Sci. Rep. 10, 19426 (2020).
Potteti, H. R. et al. Nrf2-AKT interactions regulate heme oxygenase 1 expression in kidney epithelia during hypoxia and hypoxia-reoxygenation. Am. J. Physiol. Ren. Physiol. 311, F1025–f1034 (2016).
Kim, T.-H. et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 71, 2260–2275 (2011).
Bondi, C. D. et al. Suppression of NRF2 Activity by HIF-1⍺ promotes fibrosis after ischemic acute kidney injury. Antioxidants 11, 1810 (2022).
Loboda, A. et al. HIF-1 induction attenuates Nrf2-dependent IL-8 expression in human endothelial cells. Antioxid. Redox Signal. 11, 1501–1517 (2009).
Pan, Y. et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 27, 912–925 (2007).
Watanabe, Y. et al. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc. Natl Acad. Sci. USA 113, 6011–6016 (2016).
Hamanaka, R. B., Weinberg, S. E., Reczek, C. R. & Chandel, N. S. The mitochondrial respiratory chain is required for organismal adaptation to hypoxia. Cell Rep. 15, 451–459 (2016).
Bi, Z. et al. Nrf2 and HIF1α converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells. Theranostics 10, 4134–4149 (2020).
Ji, X. et al. Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int. J. Cancer 135, 574–584 (2014).
Shen, H. et al. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis. Esophagus 27, 685–692 (2014).
Zhang, Z. et al. Reactive oxygen species regulate FSH-induced expression of vascular endothelial growth factor via Nrf2 and HIF1α signaling in human epithelial ovarian cancer. Oncol. Rep. 29, 1429–1434 (2013).
Zheng, J. et al. Overactivated NRF2 induces pseudohypoxia in hepatocellular carcinoma by stabilizing HIF-1alpha. Free Radic. Biol. Med. 194, 347–356 (2023).
Jin, X., Gong, L., Peng, Y., Li, L. & Liu, G. Enhancer-bound Nrf2 licenses HIF-1alpha transcription under hypoxia to promote cisplatin resistance in hepatocellular carcinoma cells. Aging (Albany NY) 13, 364–375 (2020).
Lacher, S. E., Levings, D. C., Freeman, S. & Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox. Biology 19, 401–411 (2018).
Oh, E. T. et al. NQO1 inhibits proteasome-mediated degradation of HIF-1α. Nat. Commun. 7, 13593 (2016).
Csiki, I. et al. Thioredoxin-1 modulates transcription of cyclooxygenase-2 via hypoxia-inducible factor-1alpha in non-small cell lung cancer. Cancer Res. 66, 143–150 (2006).
Malec, V. et al. HIF-1 alpha signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 48, 1626–1635 (2010).
Chin, B. Y. et al. Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc. Natl Acad. Sci. USA 104, 5109–5114 (2007).
Choi, Y. K. et al. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J. Biol. Chem. 285, 32116–32125 (2010).
Jung, J. et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 79, 5218–5232 (2019).
Jung, K. A., Lee, S. & Kwak, M. K. NFE2L2/NRF2 activity is linked to mitochondria and AMP-activated protein kinase signaling in cancers through miR-181c/mitochondria-encoded cytochrome c oxidase regulation. Antioxid. Redox Signal 27, 945–961 (2017).
Lee, S., Hallis, S. P., Jung, K.-A., Ryu, D. & Kwak, M.-K. Impairment of HIF-1α-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 24, 101210 (2019).
Hallis, S. P., Kim, S. K., Lee, J.-H. & Kwak, M.-K. Association of NRF2 with HIF-2α-induced cancer stem cell phenotypes in chronic hypoxic condition. Redox Biol. 60, 102632 (2023).
Sullivan, L. B. et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236–248 (2013).
Early, J. O. et al. Circadian clock protein BMAL1 regulates IL-1β in macrophages via NRF2. Proc. Natl Acad. Sci. USA 115, E8460–E8468 (2018).
Di Gregorio, J. et al. UBXN7 cofactor of CRL3KEAP1 and CRL2VHL ubiquitin ligase complexes mediates reciprocal regulation of NRF2 and HIF-1α proteins. Biochim. Biophys. Acta 1868, 118963 (2021).
Lu, C. et al. Nrf2 activation is required for ligustrazine to inhibit hepatic steatosis in alcohol-preferring mice and hepatocytes. Toxicol. Sci. 155, 432–443 (2017).
Li, Y. et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 27, 2635–2650 (2020).
Lin, H. C. et al. Andrographolide inhibits hypoxia-induced HIF-1α-driven endothelin 1 secretion by activating Nrf2/HO-1 and promoting the expression of prolyl hydroxylases 2/3 in human endothelial cells. Environ. Toxicol. 32, 918–930 (2017).
Talks, K. L. et al. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 157, 411–421 (2000).
Schito, L. & Semenza, G. L. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2, 758–770 (2016).
Krieg, M. et al. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19, 5435–5443 (2000).
Ravi, R. et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 14, 34–44 (2000).
Zhong, H. et al. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics1. Cancer Res. 60, 1541–1545 (2000).
Isaacs, J. S. et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 8, 143–153 (2005).
Greco, S., Gaetano, C. & Martelli, F. HypoxamiR regulation and function in ischemic cardiovascular diseases. Antioxid. Redox Signal. 21, 1202–1219 (2014).
Menegon, S., Columbano, A. & Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 22, 578–593 (2016).
Robertson, H., Dinkova-Kostova, A. T. & Hayes, J. D. NRF2 and the Ambiguous Consequences of Its Activation during Initiation and the Subsequent Stages of Tumourigenesis. Cancers (Basel) 12, 3609 (2020).
Kitamura, H. & Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 109, 900–911 (2018).
Guichard, C. et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 44, 694–698 (2012).
Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Shibata, T. et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 135, 1358–1368 (2008).
Muscarella, L. A. et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics 6, 710–719 (2011).
DeNicola, G. M. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011).
Komatsu, M. et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 (2010).
Adam, J. et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537 (2011).
Ji, X. et al. Correlation of Nrf2 and HIF-1alpha in glioblastoma and their relationships to clinicopathologic features and survival. Neurol. Res. 35, 1044–1050 (2013).
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L. & Denko, N. C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197 (2006).
Semenza, G. L., Roth, P. H., Fang, H. M. & Wang, G. L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269, 23757–23763 (1994).
Sakagami, H. et al. Loss of HIF-1α impairs GLUT4 translocation and glucose uptake by the skeletal muscle cells. Am. J. Physiol. Endocrinol. Metab. 306, E1065–E1076 (2014).
Bellot, G. et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 29, 2570–2581 (2009).
Chee, N. T., Lohse, I. & Brothers, S. P. mRNA-to-protein translation in hypoxia. Mol. Cancer 18, 49 (2019).
Singh, A., Bodas, M., Wakabayashi, N., Bunz, F. & Biswal, S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid. Redox Signal 13, 1627–1637 (2010).
Malhotra, D. et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 38, 5718–5734 (2010).
Zhang, H. S. et al. NRF2 facilitates breast cancer cell growth via HIF1a-mediated metabolic reprogramming. Int. J. Biochem. Cell Biol. 95, 85–92 (2018).
Zhang, H. S. et al. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1alpha/Notch1 axis. J. Cell Mol. Med. 23, 3451–3463 (2019).
Yang, Y. et al. Abnormal phenotype of Nrf2 is associated with poor prognosis through hypoxic/VEGF-A-Rap1b/VEGFR2 pathway in gastric cancer. Aging (Albany NY) 14, 3293–3312 (2022).
Takamiya, R. et al. The single N-glycan deletion mutant of soluble ErbB3 protein attenuates heregulin beta1-induced tumor progression by blocking of the HIF-1 and Nrf2 pathway. Biochem. Biophys. Res. Commun. 454, 364–368 (2014).
Kazi, A. A. et al. Nonhypoxic regulation and role of hypoxia-inducible factor 1 in aromatase inhibitor resistant breast cancer. Breast Cancer Res. 16, R15 (2014).
Liu, L. et al. Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced chemoresistance in gastric cancer. Cancer Sci. 99, 121–128 (2008).
Chen, N. et al. BCL-xL is a target gene regulated by hypoxia-inducible factor-1alpha. J. Biol. Chem. 284, 10004–10012 (2009).
Erler, J. T. et al. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell Biol. 24, 2875–2889 (2004).
Yang, T., Xu, F., Sheng, Y., Zhang, W. & Chen, Y. A targeted proteomics approach to the quantitative analysis of ERK/Bcl-2-mediated anti-apoptosis and multi-drug resistance in breast cancer. Anal. Bioanal. Chem. 408, 7491–7503 (2016).
Choi, B.-h & Kwak, M.-K. Shadows of NRF2 in cancer: resistance to chemotherapy. Curr. Opin. Toxicol. 1, 20–28 (2016).
Homma, S. et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 15, 3423–3432 (2009).
Kurz, E. U., Cole, S. P. C. & Deeley, R. G. Identification of DNA–protein interactions in the 5′ Flanking and 5′ Untranslated Regions of the Human Multidrug Resistance Protein (MRP1) gene: evaluation of a putative antioxidant response element/AP-1 binding site. Biochem. Biophys. Res. Commun. 285, 981–990 (2001).
Singh, A. et al. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol. Cancer Ther. 9, 2365–2376 (2010).
Niture, S. K. & Jaiswal, A. K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 287, 9873–9886 (2012).
Duan, X. et al. Nrf2-siRNA enhanced the anti-tumor effects of As(2)O(3) in 5-fluorouracil-resistant hepatocellular carcinoma by inhibiting HIF-1alpha/HSP70 signaling. J. Hepatocell. Carcinoma 9, 1341–1352 (2022).
Hanahan, D. & Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 (1996).
Forsythe, J. A. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613 (1996).
Ceradini, D. J. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10, 858–864 (2004).
Gao, C. et al. SCF, regulated by HIF-1α, promotes pancreatic ductal adenocarcinoma cell progression. PLoS ONE 10, e0121338 (2015).
Simon, M.-P., Tournaire, R. & Pouyssegur, J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J. Cell. Physiol. 217, 809–818 (2008).
Florczyk, U. et al. Nrf2 regulates angiogenesis: effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid. Redox Signal. 20, 1693–1708 (2013).
Ichihara, S. et al. Ablation of the transcription factor Nrf2 promotes ischemia-induced neovascularization by enhancing the inflammatory response. Arterioscler. Thromb. Vasc. Biol. 30, 1553–1561 (2010).
Wei, Y. et al. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc. Natl Acad. Sci. USA 110, E3910–E3918 (2013).
Li, L. et al. Interplay between VEGF and Nrf2 regulates angiogenesis due to intracranial venous hypertension. Sci. Rep. 6, 37338 (2016).
Feng, R. et al. Nrf2 activation drive macrophages polarization and cancer cell epithelial-mesenchymal transition during interaction. Cell Commun. Signal 16, 54 (2018).
Shao, S. et al. Curcumin suppresses hepatic stellate cell-induced hepatocarcinoma angiogenesis and invasion through downregulating CTGF. Oxid. Med. Cell. Longev. 2019, 8148510 (2019).
Hapke, R. Y. & Haake, S. M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 487, 10–20 (2020).
Zhong, H. et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 59, 5830–5835 (1999).
Xu, X. et al. Snail is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells*. J. Biol. Chem. 290, 16653–16664 (2015).
Yang, M.-H. et al. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 10, 295–305 (2008).
Zhang, W. et al. HIF-1α promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS ONE 10, e0129603 (2015).
Wang, R. et al. Hypoxia-inducible factor-dependent ADAM12 expression mediates breast cancer invasion and metastasis. Proc. Natl Acad. Sci. USA 118, e2020490118 (2021).
Wang, H. et al. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 8, 334ra351 (2016).
Zhang, C. et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 7, 73593–73606 (2016).
Jeong, Y. et al. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 7, 86–101 (2017).
Yazaki, K. et al. ROS-Nrf2 pathway mediates the development of TGF-β1-induced epithelial-mesenchymal transition through the activation of Notch signaling. Eur. J. Cell Biol. 100, 151181 (2021).
Vilchez Mercedes, S. A. et al. Nrf2 modulates the hybrid epithelial/mesenchymal phenotype and notch signaling during collective cancer migration. Front. Mol. Biosci. 9, 807324 (2022).
Rachakonda, G. et al. Increased cell migration and plasticity in Nrf2-deficient cancer cell lines. Oncogene 29, 3703–3714 (2010).
Ryu, D., Lee, J.-H. & Kwak, M.-K. NRF2 level is negatively correlated with TGF-β1-induced lung cancer motility and migration via NOX4-ROS signaling. Arch. Pharmacal Res. 43, 1297–1310 (2020).
Batlle, E. & Clevers, H. Cancer stem cells revisited. Nat. Med. 23, 1124–1134 (2017).
Yang, L. et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 5, 8 (2020).
Wright, M. H. et al. Brca1 breast tumors contain distinct CD44+/CD24- and CD133+cells with cancer stem cell characteristics. Breast Cancer Res. 10, R10 (2008).
Dalerba, P. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl Acad. Sci. USA 104, 10158–10163 (2007).
Heddleston, J. M. et al. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 102, 789–795 (2010).
Samanta, D., Gilkes, D. M., Chaturvedi, P., Xiang, L. & Semenza, G. L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl Acad. Sci. USA 111, E5429–E5438 (2014).
Conley, S. J. et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl Acad. Sci. USA 109, 2784–2789 (2012).
Keith, B. & Simon, M. C. Hypoxia-inducible factors, stem cells, and cancer. Cell 129, 465–472 (2007).
Covello, K. L. et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557–570 (2006).
Lu, H. et al. Chemotherapy-induced S100A10 recruits KDM6A to facilitate OCT4-mediated breast cancer stemness. J. Clin. Investig. 130, 4607–4623 (2020).
Qin, J. et al. Hypoxia-inducible factor 1 alpha promotes cancer stem cells-like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci. Rep. 7, 10592 (2017).
Yan, Y. et al. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J. Exp. Clin. Cancer Res. 37, 256 (2018).
Yan, Y. et al. A novel HIF-2α targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPR(ER) axis. Cell Death Differ. 29, 1769–1789 (2022).
Micucci, C., Matacchione, G., Valli, D., Orciari, S. & Catalano, A. HIF2α is involved in the expansion of CXCR4-positive cancer stem-like cells in renal cell carcinoma. Br. J. Cancer 113, 1178–1185 (2015).
Diehn, M. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783 (2009).
Hallis, S. P., Kim, J. M. & Kwak, M.-K. Emerging Role of NRF2 Signaling in Cancer Stem Cell Phenotype. Mol. Cells 46, 153–164 (2023).
Chang, C.-W. et al. ROS-independent ER stress-mediated NRF2 activation promotes warburg effect to maintain stemness-associated properties of cancer-initiating cells. Cell Death Dis. 9, 194 (2018).
Chang, C. W. et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 74, 6291–6305 (2014).
Gao, L. et al. Nrf2 signaling promotes cancer stemness, migration, and expression of ABC transporter genes in sorafenib-resistant hepatocellular carcinoma cells. PLoS ONE 16, e0256755 (2021).
Zhu, J. et al. Nrf2 is required to maintain the self-renewal of glioma stem cells. BMC Cancer 13, 380 (2013).
Ryoo, I.-g, Choi, B.-h, Ku, S.-K. & Kwak, M.-K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: implications for cancer stem cell resistance. Redox Biol. 17, 246–258 (2018).
Park, J., Kim, S. K., Hallis, S. P., Choi, B.-H. & Kwak, M.-K. Role of CD133/NRF2 axis in the development of colon cancer stem cell-like properties. Front. Oncol. 11, 808300 (2022).
Kim, D., Choi, B. H., Ryoo, I. G. & Kwak, M. K. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 9, 896 (2018).
Noman, A. S. M. et al. Chemotherapeutic resistance of head and neck squamous cell carcinoma is mediated by EpCAM induction driven by IL-6/p62 associated Nrf2-antioxidant pathway activation. Cell Death Dis. 11, 663 (2020).
Okazaki, K. et al. Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat. Commun. 11, 5911 (2020).
Kim, D. H. et al. Nuclear factor erythroid-derived 2-like 2-induced reductive stress favors self-renewal of breast cancer stem-like cells via the FoxO3a-Bmi-1 axis. Antioxid. Redox Signal. 32, 1313–1329 (2020).
Fragoulis, A. et al. Nrf2 induces malignant transformation of hepatic progenitor cells by inducing β-catenin expression. Redox Biol. 57, 102453 (2022).
Zhang, G., Tao, X., Ji, B. & Gong, J. Hypoxia-driven M2-polarized macrophages facilitate cancer aggressiveness and temozolomide resistance in glioblastoma. Oxid. Med. Cell. Longev. 2022, 1614336 (2022).
Kipp, A. P., Deubel, S., Arnér, E. S. J. & Johansson, K. Time- and cell-resolved dynamics of redox-sensitive Nrf2, HIF and NF-κB activities in 3D spheroids enriched for cancer stem cells. Redox Biol. 12, 403–409 (2017).
Dixon, ScottJ. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Li, J. et al. Ferroptosis: past, present and future. Cell Death Dis. 11, 88 (2020).
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Fuhrmann, D. C., Mondorf, A., Beifuß, J., Jung, M. & Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36, 101670 (2020).
Miess, H. et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene 37, 5435–5450 (2018).
Green, Y. S. et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene 41, 4709–4723 (2022).
Yang, M. et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5, eaaw2238 (2019).
Singhal, R. et al. HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Invest. 131, e143691 (2021).
Su, X. et al. HIF-α activation by the prolyl hydroxylase inhibitor roxadustat suppresses chemoresistant glioblastoma growth by inducing ferroptosis. Cell Death Dis. 13, 861 (2022).
Dodson, M., Castro-Portuguez, R. & Zhang, D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019).
Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371–e371 (2017).
Shin, D., Kim, E. H., Lee, J. & Roh, J.-L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 129, 454–462 (2018).
Yang, J. et al. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 12, 1079 (2021).
Koppula, P. et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 13, 2206 (2022).
Anandhan, A. et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 9, eade9585 (2023).
Chen, L. D. et al. Nrf2 plays protective role during intermittent hypoxia-induced ferroptosis in rat liver (BRL-3A) cells. Sleep. Breath. 27, 2069–2076 (2023).
Li, D. et al. Silencing TRPM2 enhanced erastin- and RSL3-induced ferroptosis in gastric cancer cells through destabilizing HIF-1α and Nrf2 proteins. Cytotechnology 74, 559–577 (2022).
Kikuchi, H., Pino, M. S., Zeng, M., Shirasawa, S. & Chung, D. C. Oncogenic KRAS and BRAF differentially regulate hypoxia-inducible factor-1alpha and -2alpha in colon cancer. Cancer Res. 69, 8499–8506 (2009).
Cahuzac, K. M. et al. AKT activation because of PTEN loss upregulates xCT via GSK3β/NRF2, leading to inhibition of ferroptosis in PTEN-mutant tumor cells. Cell Rep. 42, 112536 (2023).
Kang, H. J. et al. HER2 confers drug resistance of human breast cancer cells through activation of NRF2 by direct interaction. Sci. Rep. 4, 7201 (2014).
Lu, Y. et al. Brusatol inhibits HIF-1 signaling pathway and suppresses glucose uptake under hypoxic conditions in HCT116 cells. Sci. Rep. 6, 39123 (2016).
Chen, F. et al. Triptolide, a Chinese herbal extract, enhances drug sensitivity of resistant myeloid leukemia cell lines through downregulation of HIF-1alpha and Nrf2. Pharmacogenomics 14, 1305–1317 (2013).
Liu, Y. et al. Low-dose triptolide in combination with idarubicin induces apoptosis in AML leukemic stem-like KG1a cell line by modulation of the intrinsic and extrinsic factors. Cell Death Dis. 4, e948 (2013).
Jin, J. et al. Cardamonin inhibits breast cancer growth by repressing HIF-1alpha-dependent metabolic reprogramming. J. Exp. Clin. Cancer Res. 38, 377 (2019).
Qu, J., Zhang, L., Li, L. & Su, Y. miR-148b functions as a tumor suppressor by targeting endoplasmic reticulum metallo protease 1 in human endometrial cancer cells. Oncol. Res. 27, 81–88 (2018).
Kuper, A. et al. Overcoming hypoxia-induced resistance of pancreatic and lung tumor cells by disrupting the PERK-NRF2-HIF-axis. Cell Death Dis. 12, 82 (2021).
Wakabayashi, N. et al. Regulation of Notch1 signaling by Nrf2: implications for tissue regeneration. Sci. Signal. 3, ra52–ra52 (2010).
He, F., Ru, X. & Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 21, 4777 (2020).
Gustafsson, M. V. et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Develop. Cell 9, 617–628 (2005).
Lv, Y. et al. Hypoxia-inducible factor-1alpha induces multidrug resistance protein in colon cancer. Onco Targets Ther. 8, 1941–1948 (2015).
Lee, P. J. et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia*. J. Biol. Chem. 272, 5375–5381 (1997).
Jang, J. et al. Primary cilium-autophagy-Nrf2 (PAN) axis activation commits human embryonic stem cells to a neuroectoderm fate. Cell 165, 410–420 (2016).
Petruzzelli, R., Christensen, D. R., Parry, K. L., Sanchez-Elsner, T. & Houghton, F. D. HIF-2α regulates NANOG expression in human embryonic stem cells following hypoxia and reoxygenation through the interaction with an Oct-Sox cis regulatory element. PLoS ONE 9, e108309 (2014).
Acknowledgements
The figure was created with BioRender.com. This study was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT; 2022R1A2C2011866, 2018R1A6A1A03025108).
Author information
Authors and Affiliations
Contributions
M.K.K. supervised the overall process and wrote the paper. T.B. and S.P.H. contributed to the writing of the paper and the preparation of the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bae, T., Hallis, S.P. & Kwak, MK. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp Mol Med 56, 501–514 (2024). https://doi.org/10.1038/s12276-024-01180-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-024-01180-8
