
Abstract
Protein lysine methyltransferases (PKMTs) play crucial roles in histone and nonhistone modifications, and their dysregulation has been linked to the development and progression of cancer. While the majority of studies have focused on the oncogenic functions of PKMTs, extensive evidence has indicated that these enzymes also play roles in tumor suppression by regulating the stability of p53 and β-catenin, promoting α-tubulin-mediated genomic stability, and regulating the transcription of oncogenes and tumor suppressors. Despite their contradictory roles in tumorigenesis, many PKMTs have been identified as potential therapeutic targets for cancer treatment. However, PKMT inhibitors may have unintended negative effects depending on the specific cancer type and target enzyme. Therefore, this review aims to comprehensively summarize the tumor-suppressive effects of PKMTs and to provide new insights into the development of anticancer drugs targeting PKMTs.
Similar content being viewed by others

Introduction
Protein methylation refers to the addition of a methyl group to the ε-amino group of proteins, with S-adenosyl-L-methionine (AdoMet) primarily used as the methyl group donor. Protein residues can be methylated on nitrogen (N-methylation), oxygen (O-methylation), sulfur (S-methylation), and carbon (C-methylation) atoms1,2,3,4. Lysine methylation is a form of N-methylation that affects histones, playing a central role in histone–protein interactions5. The lysine residues of histones H3 and H4 are usually methylated, as represented by histone H3 at lysine 4 (H3K4), lysine 9 (H3K9), lysine 27 (H3K27), lysine 36 (H3K36), and lysine 79 (H3K79) and histone H4 at lysine 20 (H4K20)6,7,8,9. These residues can be mono- (Kme1), di- (Kme2), or tri- (Kme3) methylated, and histone lysine methylation activates or represses transcription depending on the position of the methylation site. For example, H3K4me3 is a representative active gene mark that is abundantly detected at active transcription start sites (TSSs) and is positively correlated with gene expression10. In addition, H3K79me3 is found in transcribed regions of active genes and is reported to correlate with the promotion of gene activation11. Conversely, H3K9me3 and H4K20me3 are prominently found in constitutive heterochromatin regions, while H3K27me3 exhibits enrichment in facultative heterochromatin. These epigenetic modifications serve as gene-repressive marks, contributing to the process of gene silencing12,13. Gene activation and repression status are also considerably influenced by the extent of methylation. H3K27me1 is an example of a type of methylation with such effects. Unlike H3K27me3, H3K27me1 is detected in both active and repressed chromatin domains, which implies its versatile role in controlling gene expression14,15. An additional illustration involves H4K20me1, which is notably abundant within actively transcribing gene bodies. This modification enhances the expression of housekeeping genes by promoting chromatin accessibility, and these effects distinctly contrast with those attributed to H4K20me316.
Recently, due to advances in technologies used to identify methylated proteins, such as mass spectrometry, the range of protein methylation is now known to extend beyond histones to nonhistone proteins17,18. According to the PhosphoSitePlus database, 1005 lysine methylation sites in 974 human proteins had been identified as of 201519. The majority of lysine-methylated proteins are transcription factors, such as RelA, STAT, and FOXO3. Kinases or proteins related to phosphorylation also comprise a large proportion of lysine-methylated proteins.
Lysine methylation is a protein lysine methyltransferase (PKMT)-dependent reaction, and lysine residues can be mono-, di-, or trimethylated depending on the type of PKMT. PKMTs play oncogenic roles in various cancers, and preclinical and clinical trials of several PKMT inhibitors for cancer treatment are ongoing20,21. However, growing evidence suggests that some methyltransferases exhibit tumor-suppressive functions in specific cancer types and have both oncogenic and tumor-suppressive functions in other cases. Therefore, obtaining a clear understanding of these functions is critical, particularly for developing cancer therapies that target these enzymes. In this review, we discuss the current understanding of PKMTs as tumor suppressors.
Characteristics of lysine methylation and PKMTs
Lysine methylation
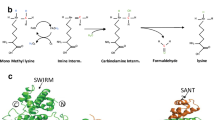
Chemically, lysine methylation can be catalyzed by AdoMet-dependent lysine (K)-specific methyltransferases, resulting in the transfer of methyl groups from AdoMet onto lysine residues of particular substrates. Subsequently, AdoMet is converted to S-adenosyl-L-homocysteine (AdoHcy), the demethylated form of AdoMet. The lysine ε-amino acid groups can receive up to three methyl groups, which results in mono-, di-, or trimethylated lysine (Fig. 1a). These distinct methylation states can have different physiological consequences. Lysine methylation can occur on both histone and nonhistone proteins. Histone methylation is associated with changes in chromatin structure and is closely linked to transcriptional regulation. Methylation of nonhistone proteins affects protein stability, cellular localization, protein‒protein interactions, promoter binding affinity, and the regulation of other posttranslational modifications on the substrates (Fig. 1b).

a The process of protein lysine methylation. b Molecular functions of protein lysine methylation.
Classification of PKMTs
Protein methyltransferases are classified into five classes, I, II, III, IV, and V, according to structural similarities of the catalytic domain22. PKMTs predominantly belong to the class V superfamily, containing a conserved Su(var)3-9, enhancer of zeste, and trithorax (SET) domain. Methyltransferases containing SET domains are distinguished from other AdoMet-dependent methyltransferases in terms of the available binding sites for substrates and AdoMet23. In general, once the substrate binds to the enzyme, the binding of an additional cofactor (e.g., AdoMet) is sterically hindered. However, in methyltransferases with the SET domain, the substrate and AdoMet bind to separate clefts located opposite each other, allowing multiple methylation without dissociation of the substrate24. For this reason, it is thought that PKMTs with the SET domain are suitable for trimethylation. As an exception, histone H3 lysine 79 (H3K79) methyltransferase has a disruptor of telomeric silencing 1-like (DOT1L) domain and is classified in a new category called DOT1L domain-containing lysine methyltransferases25. DOT1L contains a seven β-strand Rossmann-fold domain, which is commonly observed in class I methyltransferases, such as protein arginine methyltransferases (PRMTs), and is phylogenetically closer to PRMTs than PKMTs26.
Oncogenic role of PKMTs
Numerous PKMTs have been reported to promote tumorigenesis. In particular, SETD7, EZH2, G9a, SMYD2 and SUV4-20H2 have been extensively studied in cancer, and these proteins are being pursued as therapeutic targets of anticancer drugs. Their roles in promoting cancers are well summarized in previous literature27. Therefore, we will briefly review their oncogenic functions here.
SETD7 (also known as SET7, SET9, or SET7/9) is highly expressed in hepatocellular carcinoma and colorectal cancer tissues compared to noncancerous tissues, and the increased expression of SETD7 was found to be closely correlated with cancer progression via regulation of cell proliferation and metastasis in these cancers28,29. SETD7 is known to stabilize E2F transcription factor 1 (E2F1) in hepatocellular carcinoma, leading to the expression of cyclin E1, cyclin A2, and CDK1, which triggers the cell cycle in tumorigenesis30. The carcinogenic role of SETD7 was also identified in breast, intestinal, prostate, and ovarian cancers. In breast cancer, SETD7 facilitates the transcription of vascular endothelial growth factor (VEGF), a key regulator of angiogenesis, by associating with GATA131. In contrast, SETD7-dependent Yes-associated protein (YAP) methylation promotes Wnt-mediated β-catenin activation in intestinal tumorigenesis32. Retinoid acid-related orphan receptor α2 (RORα2) is a target of SETD7 in prostate cancer cells33,34. In detail, RORα2 methylation at K87 by SETD7 triggers its transcriptional activities by promoting its interaction with the Tip60 coactivator33. Furthermore, SETD7 selectively protects hypoxia inducible factor-1α (HIF-1α) from proteasomal degradation only under hypoxic conditions and promotes HIF-1α-mediated gene transcription, which is involved in hypoxic glycolysis. As a result, SETD7 facilitates glycolytic adaptation and the survival of cancer cells under hypoxic conditions. In this process, SETD7 selectively increases H3K4me1 levels in the hypoxia response elements (HREs) of glycolytic genes, thereby promoting the formation of transcriptionally active chromatin35. The detailed mechanisms underlying SETD7-mediated HIF-1α stabilization are not yet fully understood.
EZH2 expression is amplified in lymphoma, lung cancer, prostate cancer, breast cancer, colon cancer, melanoma, retinoblastoma, and glioblastoma36,37,38,39,40, and EZH2 is involved in the metastasis of prostate and breast cancers41,42. EZH2 overexpression can drive tumorigenesis through its role as an epigenetic regulator. Several mechanisms have been proposed to explain the oncogenic role of EZH2, including the repression of tumor suppressor genes such as E-cadherin43, CDKN2A/p1644, CDKN1A/p21, and CDKN1B/p2745. EZH2 can also suppress the activation of oncogenic pathways such as the Wnt/β-catenin pathway by epigenetically repressing GSK-3β and TP5346. In addition to its role in tumor initiation and progression, EZH2 has also been implicated in drug resistance. EZH2 can promote drug resistance by enhancing the DNA damage response pathway47. Recently, reduction of EZH2 by miR-138 has been proposed as a strategy to overcome drug resistance in multiple myeloma48.
The pathological function of EZH2 appears to be context-dependent and cancer-specific49. EZH2 cooperates with other oncogenes to accelerate myelodysplastic syndrome, and long-term suppression of EZH2 in glioblastoma causes a significant alteration in cell fate, ultimately leading to tumor progression50. However, the tumor-suppressive effect of EZH2 was also observed in diffuse midline glioma51. Thus, the features of each tumor should be considered in the application of EZH2 inhibitors. For a more comprehensive understanding of the tumor-suppressive function of EZH2, we kindly direct readers to Section “EZH2 (KMT6)”.
G9a, another oncogenic enzyme that methylates p5352, is overexpressed in various cancers, including esophageal squamous cell carcinoma, lung cancer, and aggressive ovarian cancer53,54,55. In addition to p53, elevated G9a under hypoxia has been reported to induce transcriptional repression of runt-related transcription factor 3 (RUNX3), known as a tumor suppressor gene in gastric cancer, via H3K9 dimethylation56. Thus, upregulation of G9a ultimately contributes to the aggressive phenotype of cancer cells.
SMYD2, which is highly expressed in esophageal squamous cell carcinoma and bladder cancer cells, is considered an oncogenic protein57,58. SMYD2 exhibits an oncogenic effect by repressing the activity of important tumor suppressors, such as p53 and Rb, by methylation of these proteins59,60.
SUV4-20H2 favors the mesenchymal state in pancreatic cancer by silencing genes associated with epithelial traits. SUV4-20H2 knockdown induced a transition from a mesenchymal to an epithelial state, reducing stemness and enhancing drug sensitivity. Analysis of pancreatic cancer biopsies confirmed that high SUV4-20H2 levels correlate with loss of epithelial characteristics in invasive cancer. SUV4-20H2 acts as an upstream epigenetic regulator of epithelial/mesenchymal state control, suggesting its potential as a therapeutic target for promoting epithelial identity in cancer61.
Tumor-suppressive role of PKMTs
Extensive literature reviews in the field of protein lysine methylation have reported on its cellular functions, including epigenetic regulation, signaling pathways, and various cellular responses. However, literature reviews on the tumor-suppressive functions of PKMTs are lacking. Eighteen PKMTs have been identified as tumor suppressors, and their conserved domains are depicted in Fig. 2. These PKMTs facilitate the methylation of various substrates, such as p53, β-catenin, α-tubulin, and histone proteins. The tumor-suppressive functions of PKMTs are mediated through methylation of these substrates. Among these PKMTs, SETD2, SETD7, and EZH2 are relatively well characterized with dominant tumor-suppressive functions (Fig. 3a). In this article, we highlight the substrates and tumor-suppressive functions of PKMTs in various human cancers (Table 1).

This illustration portrays the configuration of domains within lysine methyltransferase proteins, acknowledged for its role in suppressing tumors. Each domain is assigned a distinct color to indicate commonalities or differences among various methyltransferases. The universal presence of the red SET domain in all tumor-suppressive lysine methyltransferases, excluding DOT1L, underscores the pivotal role of their catalytic activity in tumor suppression. Fyr-C Phe/Tyr-rich domain C-terminal, Fyr-N Phe/Tyr-rich domain N-terminal, SET Su(var)3-9 Enhancer-of-zeste Trithorax-domain, DHHC Asp-His-His-Cys containing domain, AWS associated with SET domains, MORN membrane occupation and recognition nexus, PWWP Pro-Trp-Trp-Pro containing domain, CXC Cys-rich domain, DOT1L catalytic domain of DOT1L, WW domain with 2 conserved Trp-Trp residues, MBD methyl-CpG binding domain, Tudor tudor domain, Chormo Chromatin organization modifier domain.

a Various tumor types controlled by three lysine methyltransferase variants that are frequently linked to tumor suppressor functions. b Mechanistic role of SETD2 as a tumor suppressor in clear-cell renal cell carcinoma. Reduction in SETD2 levels results in a decrease in H3K36 trimethylation and tubulin α-1a trimethylation, which subsequently initiates genomic instability. c The mechanism of action of SETD7 as a tumor suppressor. Left: SETD7 catalyzes the monomethylation of β-catenin at K180, amplifying its molecular interaction with GSK3β. This prompts GSK3β-mediated phosphorylation, leading to ubiquitination of β-catenin followed by proteasomal degradation in HeLa cells. Consequently, this process leads to diminished levels of c-Myc and cyclin D1, resulting in a decrease in cell proliferation. Right: In an in vivo model, SETD7 initiates monomethylation of p53 at K372, leading to its interaction with Tip1. This interaction subsequently induces acetylation of p53, resulting in the activation of p53 itself. This activation leads to heightened levels of p21 and the preservation of the DNA damage response. d Upper: Structure of EZH2 with domain WDB, WD-40 binding domain; D1, domain 1; D2, domain 2; CXC, Cys-rich domain; SET, catalytic domain of EZH2. Lower: The mechanistic role of EZH2 as a tumor suppressor in lung adenocarcinoma, myelodysplastic disorders, and T-cell acute lymphoblastic leukemia (T-ALL) through its involvement in modulating the trimethylation status of H3K27. Decreased expression and mutation of EZH2 (as depicted) have been identified in individuals with lung adenocarcinoma, myelodysplastic syndromes, and T-ALL. The attenuation of EZH2 expression or activity leads to a reduction in H3K27me3 levels, resulting in enhanced activation of downstream proteins such as AKT, ERK, Nrf2, Myc, and NOTCH1, which are implicated in the development of these conditions.
SETD2 (SET2, KMT3A)
SET domain-containing 2 (SETD2) is part of the SET2 subfamily, histone lysine methyltransferase family, and class V-like SAM binding methyltransferase superfamily. SETD2 consists of several domains, including the associated with SET (AWS) domain; the SET domain; the post-SET domain; the locus control region (LCR); the WW domain, which binds to a proline-rich motif of a protein; and the Set2 Rpb1-interacting (SRI) domain, which is crucial for transcriptional elongation through histone H3 lysine 36 (H3K36) methylation (Fig. 2). SETD2 is located on chromosome 3p21 and facilitates the trimethylation of H3K36 and microtubules at lysine 40 (αTubK40). Nuclear receptor-binding SET domain protein 1 (NSD1) primarily catalyzes the demethylation of H3K36, while NSD2 (also known as WHSC1 or MMSET) acts as a mono- and demethylase of H3K3662,63,64,65. This demethylation provides a substrate for trimethylation by SETD2. In addition, the SETD2 protein level is regulated by miR-106-5p, ubiquitination, and SUMOylation66,67. The R1625 residue in the catalytic domain of SETD2 is important for its interactions with the histone H3 tail and the trimethylation of H3K36 but not for its thermal stability68. Reader proteins that recognize the H3K36me3 modification recruit protein complexes involved in transcription elongation, RNA processing, DNA repair, and other processes69.
In clear cell renal cell carcinoma (ccRCC), SETD2 mediates the trimethylation of α-tubulin at K40, which is required to maintain genomic stability, and loss of SETD2 function results in genomic instability and contributes to ccRCC tumorigenesis. Specifically, SETD2 deficiency and haploinsufficiency led to ccRCC progression through an increase in mitotic defects and micronuclei formation (Fig. 3b)70,71. In addition, loss of function of SETD2 facilitates ccRCC progression through dysregulation of genome integrity-related processes, including nucleosome destabilization, replication stress, and DNA repair72.
SETD2 suppresses ccRCC development through distinct mechanisms mediated by H3K36 trimethylation. SETD2-mediated H3K36me3 promotes ataxia telangiectasia mutated kinase (ATM) activation on DNA double-strand breaks (DSBs) and enhances homologous recombination repair of DSBs by accelerating the formation of RAD51 presynaptic filaments. In contrast, the loss of SETD2 impaired DNA damage signaling, including inactivation of p53, despite the persistence of DNA lesions73. In addition, genetic depletion of SETD2 upregulated oncogenes and inhibited cell metabolic pathways74. This change also enhanced overall metabolic activity, including ATP and glycolytic activity, mitochondrial respiratory capacity, TCA cycle metabolic enzyme activity, and production of TCA metabolites, through PGC1α, a central regulator of mitochondrial oxidative phosphorylation and fatty acid metabolism75.
In addition, SETD2 knockdown prevented cyclin-dependent kinase inhibitor 2A (CDKN2A)-induced cellular senescence of renal primary tubular epithelial cells (PTECs) through activation of E2F signaling pathways76. Finally, inhibition of miR-106-5p-mediated SETD2 downregulation promoted cell cycle arrest at the G0/G1 phases and caspase-3-mediated apoptosis in ccRCC67. Hematopoietic stem cells in Setd2 knockout mice exhibited impaired self-renewal but overcame the growth disadvantage after a latency period and eventually showed characteristics of hematopoietic malignancy. Mechanistically, the loss of SETD2 in hematopoietic stem/progenitor cells triggers the activation of the Klf1-related pathway; Klf1 is an erythroid transcription factor responsible for the development of hematopoietic malignancies. In addition, SETD2 deficiency activates the E2F gene regulatory network and suppresses the expression of the ribonucleotide reductase subunit Rrm2b, leading to DNA replication stress in hematopoietic stem cells, cellular abnormalities, and genomic instability77. These biological functions of SETD2 in hematopoietic stem cells provide insight into the role of SETD2 as a tumor suppressor.
The tumor-suppressive function of SETD2 is observed in various hematopoietic malignancies. SETD2 loss-of-function mutations were found in 22% of patients with leukemia with rearrangements in the mixed-lineage leukemia gene (referred to as MLLr); altered histone methylation due to SETD2 knockdown transcriptionally downregulated tumor suppressors (ASXL1, ASXL2, BCOR) and upregulated oncogenes (ERG, STAT3, FNDC3A, NAMPT, IGF1R)78. In addition, SETD2 deficiency enhanced hematopoietic stem cell signaling and decreased myeloid differentiation pathways in hematopoietic stem and progenitor cells from NUP98-hoxd13 (NDH13) transgenic mice in a myelodysplastic syndrome (MDS) model79. In that study, downregulation of S100a9 by SETD2 contributed to MDS pathology in an H3K36me3-dependent manner79. In chronic lymphocytic leukemia (CLL), SETD2 deletions were related to loss of TP53, genomic complexity, and chromothripsis80. Finally, decreased or absent SETD2 protein expression and reduced levels of H3K36 trimethylation have been observed in systemic mastocytosis (SM), with low expression of SETD2 correlating with disease aggressiveness. Notably, the use of the proteasome inhibitor bortezomib is effective in restoring SETD2 expression and H3K36 trimethylation. Furthermore, restoration of SETD2 expression via bortezomib resulted in the induction of apoptosis and inhibition of colony formation when combined with midostaurin, the primary therapeutic option for advanced SM66.
Loss of SETD2 is associated with chemoresistance in MLLr leukemia, acute lymphocytic leukemia (ALL), and chronic myelogenous leukemia (CML). The incidence of mutations in SETD2 is higher in cases of relapsed ALL and MLLr acute leukemia81,82. Mutations in SETD2 confer resistance to DNA-damaging agents, such as cytarabine, 6-thioguanine, doxorubicin, and etoposide, while not affecting the response to non-DNA-damaging agents82. Additionally, loss of SETD2 leads to decreased activation of the DNA damage response (DDR) to cytotoxic chemotherapy, ultimately resulting in reduced apoptosis82. Consistently, SETD2 loss-of-function mutations cause resistance to conventional cytarabine-based chemotherapy. Specifically, leukemic cells carrying SETD2 mutations exhibit inhibition of signaling pathways involved in cell cycle progression and regulation of the S and G2/M checkpoints83. Consequently, these cells exit the S phase and progress to the G2/M phase after cytarabine treatment83. In addition, SETD2 deficiency enhances resistance against imatinib in CML cells by upregulating novel oncogenes, such as erythroblast transformation‐specific (ETS)‐related gene (ERG) and N‐myc proto-oncogene protein (MYCN)84. In addition, restoration of H3K36me3 expression by the demethylase inhibitor JIB-04 resensitizes CML to imatinib84. Taken together, the findings from these studies indicate that recovery of SETD2 may be a potential therapeutic strategy for overcoming chemotherapy-resistant hematopoietic malignancies.
Deficiency of SETD2 has been implicated in tumorigenesis in several other cancers. In pancreatic ductal adenocarcinoma (PDAC), SETD2 ablation facilitated inflammation and Kras-induced ductal metaplasia by impairing epigenetic regulation of the E3 ligase Fbxw785. In addition, SETD2 loss induced epithelial-mesenchymal transition (EMT) via epigenetic dysregulation of catenin α185. Moreover, SETD2 depletion promoted metastasis through sustained protein kinase B (AKT) activation85. In addition, SETD2 deficiency induced neutrophil reprogramming toward an immunosuppressive phenotype, which facilitated immune evasion of pancreatic cancer progression86.
SETD2 loss promoted progression in early- and late-stage lung adenocarcinoma in a KRASG12D-driven mouse model87. A recent report showed that knockout of SETD2 increases the transcriptional regulation of oncogenes by influencing chromatin accessibility and histone chaperone recruitment, leading to accelerated KRASG12D-driven lung tumorigenesis88.
In non-small cell lung cancer (NSCLC), SETD2-mediated H3K36me3 is essential for cytotoxicity to cisplatin through regulation of the extracellular signal-regulated kinase (ERK) signaling pathways89. Moreover, SETD2 inactivation enhanced mTORC1-associated gene expression in a KRAS-driven lung adenocarcinoma model in vivo, whereas mTORC1 inhibition enhanced therapeutic susceptibilities in SETD2-inactivated cancer90. These findings collectively suggest that mutations in SETD2 confer therapeutic resistance to lung cancer.
SETD2 significantly inhibited cell proliferation, migration, and invasion in gastric cancer91. In gastrointestinal stromal tumors (GISTs), SETD2 inhibited rH2Ax, a DNA damage marker, and its mutations have been linked to upregulation of HOXC cluster genes and hypomethylated heterochromatin92. Additionally, in colorectal cancer (CRC), SETD2 suppressed WNT signal-mediated tissue regeneration and tumorigenesis, including colony formation, cell proliferation, migration, self-renewal, and stemness, through epigenetic regulation of RNA processing93. Furthermore, the majority of SETD2 mutations coincide with mutations in TP53 or KRAS94, which suggests that SETD2 mutations disrupt DNA repair mechanisms by destabilizing TP53, augmenting their oncogenic effects in CRC94.
In an in vivo 7,12-dimethylbenz[a]anthracene (DMBA)-induced breast cancer model, SETD2 gene and protein expression levels were significantly decreased in very early-stage (13 weeks) and full-fledged tumors (6 months)95. In addition, in osteosarcoma, SETD2 significantly inhibited cell growth, cancer stem cell properties, and cisplatin-induced chemoresistance through H3K36me3-mediated transcriptional activation of GSK3β and degradation of β-catenin96.
SETD2 is currently considered a potential tumor suppressor. However, a recent study revealed that artificially inducing excessive accumulation of SETD2 in HEK293T cells triggers the activation of signals associated with the cell cycle, leading to an oncogenic phenotype characterized by increased cell proliferation and migration97. This finding emphasizes the importance of comprehending the regulatory mechanisms concerning SETD2 that govern tumor development and progression in various types of carcinomas. This knowledge is essential to effectively utilize SETD2 as a therapeutic biomarker while mitigating the risk of unforeseen adverse effects. Moreover, this finding indicates that the cellular and molecular environments of a cell, a tissue, and an organ can affect whether a gene is functionally oncogenic or tumor suppressive. Thus, more studies on SETD2 and protein lysine methyltransferase genes are required for their application as therapeutic targets.
SETD7 (SET7/9, KMT7)
SETD7, a histone lysine methyltransferase, is included in the SET7 subfamily, histone lysine methyltransferase family, and class V-like SAM binding methyltransferase superfamily. The SETD7 gene is located on chromosome 4q31.1 and encodes the SETD7 protein, which contains 366 amino acids including MORN1/2/3, SET, and post-SET domains (Fig. 2). This enzyme has been reported to have both histone and nonhistone substrates, including H3K4, p53, and β-catenin.
Multiple reports have indicated that SETD7 exhibits tumor-suppressive functions. In SETD7 knockout mice, SETD7 was found to facilitate the monomethylation of p53 at lysine 372 (Fig. 3b)98. This methylation is critical for activating p53 and enabling its acetylation by Tip60. In addition, defects in SETD7 led to an impaired p53 response to DNA damage98. However, mice lacking SETD7 failed to develop tumors within 1 year, while 74%-83% of p53-deficient mice developed tumors within 6 months99. This observation may be because the SETD7 mutation in mice does not result in complete inactivation of the p53 protein or because SETD7 induces the activation of an alternative compensatory pathway.
In cervical cancer, monomethylation of β-catenin at lysine 180 (β-catenin K180me1) is mediated by SETD7 (Fig. 3c). This modification reduces the stability of β-catenin through ubiquitination induced by GSK3β, which leads to a decrease in cancer cell proliferation. This effect eventually leads to downregulation of Wnt/β-catenin target genes, such as c-Myc and cyclin D1100. In gastric cancer, a reduced expression level of SETD7 was observed in 34.3% (129/376) of patients and was significantly correlated with clinical aggressiveness and a poor prognosis (p < 0.05). SETD7 enhances the gene expression of SREK1IP1, PGC, and CCDC28B through monomethylation of H3K4, significantly decreasing cell proliferation, migration, and invasion101. In multiple myeloma, berberine, a naturally occurring isoquinoline alkaloid, exhibited anticancer activity by mediating the upregulation of SETD7. Elevated SETD7 resulted in proteasome-dependent degradation of NF-κB and promoted berberine-induced apoptosis102. In breast cancer, a negative regulatory loop involving SETD7 and DNA methyltransferase 1 (DNMT1) has been observed. Specifically, SETD7 was found to negatively regulate the stability of the DNMT1 protein, leading to repression of the transcriptional activity of the SETD7 promoter through coordination with Snail. Additionally, SETD7 deficiency enhanced the stability of the E2F1 protein, promoting epithelial mesenchymal and cancer stem cell-like properties103. Furthermore, SETD7 suppressed the proliferation, migration, and invasion of breast cancer cells through regulation of Gli-1 expression104. These findings suggest that loss of SETD7 is an early prognostic marker for metastasis in breast cancer.
SETD7 also exhibits tumor-suppressive effects in lung and bladder cancer. In lung cancer, SETD7 suppresses migration and invasion by downregulating the JAK2/STAT3 pathway105. In bladder cancer, the m6A modification of SETD7 mRNA by METTL3 leads to mRNA decay, which facilitates cell proliferation and metastasis106.
EZH2 (KMT6)
Enhancer of zeste homolog 2 (EZH2) is a subunit of polycomb-repressive complex 2 (PRC2) and belongs to the polycomb complex, SET3 subfamily, histone lysine methyltransferase family, and class V-like SAM binding methyltransferase superfamily. The EZH2 gene is situated on chromosome 7q36 and encodes a protein with CXC and SET domains (Fig. 2). EZH2 catalyzes di- and trimethylation of histone H3 at lysine 27 (H3K27), as well as the monomethylation of GATA binding protein 4 (GATA4) at lysine 299 (GATA4 K299).
In lung adenocarcinoma, loss of function of EZH2 was observed to increase the number of tumor lesions, which was attributed to activation of AKT and ERK through insulin-like growth factor 1 (IGF1) signaling. Furthermore, loss of EZH2 accelerated inflammatory responses, as evidenced by increased infiltration of macrophages and neutrophils, along with secretion of tumor-associated cytokines, such as interleukin (IL)-6 and tumor necrosis factor-alpha (TNF-α)107. Additionally, EZH2 was shown to suppress cell proliferation and colony formation in NSCLC by inhibiting the expression of nuclear factor E2-related factor 2 (Nrf2)108.
Homozygous EZH2 mutations (9/12) are associated with 7q acquired uniparental disomy (aUPD) in myeloid malignancies. The mutations are predominantly present in individuals with MDS/MPNs (12% of 219 individuals) or primary myelofibrosis (PMF) (13% of 30 individuals). These mutations are believed to contribute to the development of myeloid malignancies by causing premature chain termination and direct abrogation of methyltransferase activity109. Loss-of-function mutations in EZH2 are associated with poor prognosis in patients with MDS and MPN110,111. Consistent with clinical outcomes, studies of mice have demonstrated that loss of EZH2 in combination with loss of Tet2 or RUNX1 can promote the development of MDS112,113. Approximately 10% of MPN patients with PMF have EZH2 loss-of-function mutations, while JAK2V617F, a JAK-activating mutation, was identified in 50% of PMF patients with EZH2 mutations111,114. Previous reports have shown that acquisition of the JAK2V617F mutation alone is insufficient for malignancy initiation in vivo115. However, loss of EZH2 in JAK2V617F hematopoietic cells reduced the level of H3K27me3 and resulted in conversion of H3K27 to acetylation. This epigenetic alteration activates Hmga2, a signature gene implicated in PMF pathogenesis116. It is thus hypothesized that EZH2 dysfunction and JAK2 activation together play an essential role in the pathogenesis of PMF.
In addition to its established role in myeloid malignancies, EZH2 has been implicated in the development of T-cell acute lymphoblastic leukemia (T-ALL) and acute myeloid leukemia (AML). EZH2 deletion has been shown to promote the spontaneous development of T-ALL and decrease survival rates117. In T-ALL, EZH2-mediated H3K27me3 was disrupted by NOTCH1 activation118. In contrast, CDK1-mediated phosphorylation of EZH2 at Thr487 led to proteasome-dependent degradation of EZH2, the stability of which is due to HSP90 in AML. Moreover, downregulation of EZH2 induced resistance to tyrosine kinase inhibitors via HOX gene expression119.
In addition to its tumor-suppressive roles, EZH2 has been extensively implicated in the development of cancer. High expression of EZH2 was found to be a feature of several solid cancers, such as prostate cancer37,41, melanoma37, endometrial carcinoma37,120, breast cancer37, esophageal cancer121, gastric cancer122, anaplastic thyroid carcinoma123, and nasopharyngeal carcinoma124. Moreover, the interplay between the expression of EZH2 and BAP-1 (BRCA1-associated protein 1) holds significant implications for epigenetic regulation and regulation of cellular processes. BAP-1 is a deubiquitinating enzyme that participates in histone modification removal, DNA repair, and other essential cellular functions125. Although a direct link between BAP-1 and EZH2 has not been identified, clinical studies have shown that enhanced EZH2 expression is observed in renal clear cell carcinoma with lower BAP-1 expression and poor prognosis126, and loss of BAP1 function increases the efficacy of EZH2 inhibitors127,128. Elevated expression of EZH2 was also detected in rhabdomyosarcoma129 and Ewing sarcoma130 tissues. In the case of lymphomas, the expression level is not significantly altered, likely due to high basal expression in normal proliferating B cells131.
Mutations specifically affecting the tyrosine 641 (Y641) residue within EZH2’s catalytic SET domain have a notable impact on its enzymatic activity in terms of H3K27 methylation. The recurrent heterozygous somatic mutations of EZH2 include Y641F, Y641N, Y641H, and Y641S, which were identified in the germinal center B-cell-like (GCB) subtype of diffuse large B-cell lymphoma and follicular lymphoma132,133. Cells harboring both EZH2 Y641F/N and EZH2 A677G mutations exhibited significantly elevated levels of H3K27me3. Interestingly, this abnormal increase in H3K27me3 was not limited to lymphoma cell lines but was also demonstrated in primary lymphoma specimens. These findings provide additional evidence suggesting the impact of these mutations on the dysregulation of H3K27 methylation in lymphoma development and progression131.
It is crucial to distinguish the context in which EZH2 exerts antitumor or oncogenic effects in tumorigenesis. As shown in Fig. 3a, EZH2 acts as a tumor suppressor in LUAD, MDS, AML, T-ALL, and PMF. The mechanism underlying the tumor-suppressive effect of EZH2 in each of these cancer types is shown in Fig. 3d. Co-occurring EZH2 deletion and KRAS mutations exist in LUAD. Moreover, in blood cancers, inactivating mutations of EZH2 with TET2 have been observed in patients with myelodysplastic disorders, while in vivo deletion of EZH2 could increase the expression of “Myc module” genes and lead to myelodysplastic phenotypes.
The inactivation of EZH2 observed in patients is caused by EZH2 downregulation and mutation. Notably, EZH2 frameshift (fs) or deletion (del) mutations represent prevalent mutation types that prompt EZH2 downregulation and consequent aberrant protein function in patients with myelodysplastic syndrome (EZH2 K718fs) and T-ALL (EZH2 fs; K405fs, L674fs, D183fs, L728fs, K61fs, C606Y, and Q570fs, EZH2 del;(7)(q35q36.1) 142.67-150.97, 142.03-157.56, 143.92-156.84, and 143.33-158.82 Mb) (Fig. 3d). Point mutations within the D2, CXC, and SET domains of EZH2, including D664A and A255T in myelodysplastic syndrome and G266E, T393M, E549X, C606Y, and P577L in T-ALL, have been identified118,134. The enzymatic methyltransferase function of EZH2 primarily relies on its SET domain, although the N-terminal CXC domain adjacent to the SET domain also plays a crucial contributory role in facilitating this activity135. Mutations in the CXC and SET domains, such as D664A, E549X, C606Y, and P577L, disrupt the function of EZH2, contributing to the inhibition of its activity136. In addition, alterations in the D2 region, which overlaps with exon 8, have been associated with exon skipping and the production of out-of-frame EZH2 mRNA. This also underscores the potential impact of D2 mutations on mRNA expression and their significance in EZH2 loss-of-function137.
When EZH2 was inactivated in T-ALL, a proportional decrease in H3K27me3 levels was observed. Consequently, this decrease in H3K27me3 led to an increase in NOTCH1 signaling, which ultimately contributed to oncogenic transformation. The mutational sites associated with loss-of-function in patients with myelodysplastic syndrome or T-ALL do not overlap with the Y641 or A677 mutations previously identified as gain-of-function mutations. This finding suggests a clear distinction between the mutational profiles observed in these diseases.
Other PKMTs exhibiting tumor-suppressive functions
Multiple studies have reported that several PKMTs, including mixed lineage leukemia (MLL)3, PRDM2/RIZ, PRDM5, SMYD4, SUV4-20H2, and EEF1AKMT3, exhibit tumor-suppressive functions rather than oncogenic roles. This section will discuss the tumor-suppressive properties of these PKMTs. In addition to the 8 representative PKMTs with tumor suppressor activity, PRDM16, MLL2, MLL4, SET domain bifurcated histone lysine methyltransferase 1 (SETDB1), SETD3, NSD1, NSD3, DOT1L, SUV39H1, and SUV39H2 have also been suggested to possess tumor suppressor activity138,139,140,141,142,143,144,145,146. However, their predominant function is to promote tumor growth, and the mechanisms underlying their potential tumor suppressor activity remain unclear, limiting their detailed discussion in this review.
MLL3
The MLL3 gene is located on chromosome 7q36 and encodes a protein with a length of 4911 amino acids. The gene includes several conserved domains and regions, such as DHHC, FYR, SET, post-SET domain, 7 coiled coil regions, WIN motif, and 10 zinc fingers (Fig. 2).
MLL3 has been identified as a haploinsufficient tumor suppressor mainly in certain subtypes of leukemia. The Cancer Genome Atlas dataset indicates that approximately 12% of AML patients exhibit a deletion in the MLL3 gene147. Moreover, research has shown that approximately 15% of FLT3-ITD-mutated AML patients exhibit recurrent MLL3 mutations or gene deletions, and patients with MLL3 mutations generally have a poor prognosis148. Consistently, MLL3 knockdown in FLT3-ITD-mutated cell lines or murine models promoted tumor growth148. However, low expression of MLL3 in cytogenetically normal AML does not appear to impact prognosis149. These findings suggest that MLL3 has tumor-suppressing functions that require cooperation with other events, such as TP53 inactivation. In fact, MLL3 deficiency in AML has been found to accelerate the development of myeloid leukemia under p53-deficient conditions and to impair the differentiation of hematopoietic stem and progenitor cells (HSPCs)149. Additionally, MLL3-downregulated leukemia was found to be resistant to conventional chemotherapy but sensitive to the bromodomain and extraterminal (BET) inhibitor JQ1150. Finally, somatic nonsense mutations in the MLL3 gene are accompanied by loss of heterozygosity (LOH) in human T-cell leukemia virus type I (HTLV-I)-associated acute adult T-cell leukemia (ATL). This finding highlights the critical role of MLL3 inactivation in the leukemogenesis of HTLV-1-induced ATL151.
The inactivation of MLL3 through frameshift mutations is common in CRC. Specifically, frameshift mutations in the poly A tract coding region of MLL3 are frequently identified in microsatellite-unstable CRC cells and primary tumors, causing loss of function of the MLL3 protein. According to these observations, it has been postulated that loss of MLL3 function may be an important contributor to the early development of CRC152.
Epigenetic changes due to the loss of MLL3 activity in bladder cancer cells result in the dysregulation of genes associated with the DDR and repair. Consequently, these cells impair the repair process of double-stranded DNA breaks through homologous recombination, leading to the accumulation of endogenous DNA damage and genomic instability. In addition, these cells substantially rely on PARP1/2 for DNA repair. Thus, it has been proposed that utilizing olaparib, a PARP1/2 inhibitor, as a therapeutic intervention can induce synthetic lethality in cells with low MLL3 expression153. Furthermore, inactivation of MLL3 methyltransferase activity in H3K4 in urothelial tumors has been reported to impede the p53-mediated DDR154.
PRDM2/RIZ
The PR domain zinc finger protein 2/RIZ (PRDM2/RIZ) gene is located on chromosome 1p36 and encodes a protein consisting of 1718 amino acids, which includes a PR/SET domain in the N-terminal region, an SH3-binding motif, and eight zinc finger motifs, which function as negative regulators of tumorigenesis (Fig. 2). Via an alternative promoter, the PRDM2/RIZ gene produces two distinct proteins, RIZ1 and RIZ2, with different motif configurations at the N-terminus. RIZ1, but not RIZ2, contains a PR domain155. Notably, RIZ1 knockout mice were shown to exhibit a high incidence of diffuse large B-cell lymphoma and the development of extensive tumors156. Additionally, RIZ1 is frequently expressed at low levels in various cancers, whereas RIZ2 is not. Thus, it is widely accepted that RIZ1 but not RIZ2 plays a crucial tumor-suppressive role157. Indeed, RIZ1 suppresses colony formation, proliferation, migration, and invasion and promotes apoptosis and cell cycle arrest at the G2/M phase through regulation of c-Myc, p53, AKT, and NF-κB in meningiomas, glioma, and breast cancer158,159,160,161,162.
The tumor-suppressive role of RIZ1 has been extensively studied in various types of brain cancers. RIZ1 expression is frequently downregulated in malignant meningioma tissues, while overexpression of RIZ1 suppresses cell proliferation and c-Myc expression, arrests the cell cycle in the G2/M phase, and induces apoptosis158. Furthermore, RIZ1 regulates the expression of UbcH10, a member of the ubiquitin-conjugating enzyme family known to possess oncogenic properties163, in a c-Myc-dependent manner160. Similarly, RIZ1 induces apoptosis and G2/M arrest in somatotroph adenoma cells through c-Myc regulation, and low RIZ1 levels in somatotroph adenoma patients are associated with poor prognosis in terms of tumor size, invasion, and recurrence162. In gliomas, particularly high-grade gliomas, RIZ1 expression is also reduced. RIZ1 expression shows a negative correlation with tumor grade, and high expression of RIZ1 leads to a favorable prognosis. Overexpression of RIZ1 consistently induces apoptosis and cell cycle arrest in the G2/M phase in human malignant glioma. Mechanistically, RIZ1 is thought to exert its tumor-suppressive effects in glioma by increasing p53 expression and suppressing NF-κB and AKT signaling161.
RIZ1 has been found to be genetically altered or downregulated in various types of cancer. For instance, genetic alterations of RIZ1, such as frameshift mutations, LOH, and promoter methylation, have been reported in 37% of primary gastric cancers164. Similarly, RIZ1 mRNA levels are often decreased due to hypermethylation of its promoter CpG island in breast (44%, 11/25) and liver (62%, 20/32) cancer specimens165. In addition, overexpression of RIZ1 in breast, liver, and colon cancer cells has been shown to induce G2/M cell cycle arrest and apoptosis159,166,167.
Nearly 33% (6/18) of endometrial cancer tissues harbor frameshift mutations in RIZ1168. Notably, RIZ1 expression is significantly reduced in estrogen receptor α (ERα)-positive endometrial cancer tissues, indicating a correlation between RIZ1 expression and ERα signaling. Indeed, estrogen reduced RIZ1 expression in an endometrial adenocarcinoma cell line, which facilitated tumor growth and tumor cell proliferation, migration, and invasion169.
The PRDM2/RIZ gene is situated on the 1p chromosomal region that is frequently deleted in testicular germ cell tumors (TGCTs)170. In addition, overexpression of RIZ1 promoted apoptosis and inhibited cell proliferation and colony formation in spermatogonial cells171, indicating a possible tumor suppressor role of RIZ1 in seminoma tumorigenesis. Similar to its effect in endometrial adenocarcinoma, estradiol leads to a decrease in RIZ1 transcript levels in spermatogonial cells. Additionally, RIZ1 binds to ERα in the presence of estradiol, which suggests that RIZ1 may regulate cell growth by mediating ERα signaling171. Finally, in CML, downregulation of PRDM2/RIZ expression is associated with promotion of cell proliferation, inhibition of apoptosis, and reduced differentiation, which are mediated by the dimethylation of H3K9 and subsequent suppression of the IGF-1 signaling pathway172.
PRDM5
The PRDM5 gene is located at chromosome 4q26 and encodes a protein comprising 630 amino acids that includes a PR/SET domain in its N-terminal region, followed by 16 zinc finger motifs158 (Fig. 2). In BRAF-mutated colorectal cancer, suppression of PRDM5 expression is associated with advanced stages across colorectal polyp subtypes of both the serrated and conventional pathways173. In gastric cancer, silencing of PRDM5 mediated by DNA methylation of the 5’ CpG island facilitated cancer cell growth174. Deng et al. demonstrated that PRDM5 induced G2/M-phase arrest and apoptosis in ovarian cancer, breast cancer, and hepatoma cell lines175. Loss of PRDM5 resulted in an increase in the numbers of adenomas and microinvasion foci by impairing gene expression in metabolic pathways in APCmin-driven intestinal adenomas176. Moreover, PRDM5, as a stress-responsive gene, was found to inhibit tumor cell growth and proliferation by aberrantly regulating the Wnt/β-catenin signaling pathway and oncogene expression in multiple tumors177.
SMYD4
SET and MYND domain-containing protein 4 (SMYD4) is composed of 804 amino acids, and the gene is located on chromosome 17p13. SMYD4 possesses a SET domain and a zinc finger motif (Fig. 2). The tumor suppressor function of SMYD4 has been documented in relation to breast cancer development178. SMYD4 suppresses tumorigenesis in breast cancer cell lines by mediating the downregulation of platelet-derived growth factor receptor α (PDGFR-α)178. This finding is consistent with the strong downregulation of SMYD4 in human breast cancer tissues, where its expression is positively correlated with patient relapse-free survival179. In addition, recent reports have shown that miR-1307-3p promotes the tumor response by inhibiting SMYD4 expression. High levels of miR-1307-3p are clinically linked to low SMYD4 expression, which leads to lower overall survival. Moreover, overexpression of miR-1307-3p in an in vivo model reduced SMYD4 expression and promoted the malignant transformation of mammary epithelial cells180. Furthermore, copy number alterations in the SMYD4 gene were observed in renal cell carcinoma181, and copy number alterations in SMYD4 in glioblastoma were associated with a poor prognosis182. However, the molecular mechanism regarding the tumor-suppressive effect of SMYD4 in those tumors is not fully understood.
SUV4-20H2
Suppressor of variegation 4-20 homolog 2 (SUV4-20H2) is a protein consisting of 462 amino acids located on chromosome 19q13 and containing a SET domain (Fig. 2). This protein functions as an H4K20 methyltransferase, predominantly contributing to the trimethylation of H4K20. In the context of cancer, SUV4-20H2 has been reported to act as an epigenetic regulator, promoting EMT in pancreatic cancer61. Despite its oncogenic role, SUV4-20H2 is widely recognized as a tumor suppressor.
The tumor-suppressive role of SUV4-20H2 has been extensively studied in breast cancer. Exogenous expression of SUV4-20H2 in breast cancer cells inhibited the metastatic potential of cancer cells by suppressing the expression of genes involved in cancer migration, such as tensin-3 and focal adhesion183. A recent study demonstrated that miR-29a, which is overexpressed in breast cancer stem cells, accelerates the migration, invasion, and EMT of breast cancer cells by targeting SUV4-20H2. Mechanistically, miR-29a suppresses the trimethylation of H4K20 via suppression of SUV4-20H2, which results in upregulation of connective tissue growth factor (CTGF) and early growth response protein-1 (EGR1)184.
Evidence supporting the tumor-suppressive role of SUV4-20H2 has been documented in various cancer types, including liver cancer, lung cancer, and osteosarcoma. In the context of hepatocarcinogenesis, SUV4-20H2 expression and H4K20 trimethylation levels are significantly reduced185. In lung cancer, loss of SUV4-20H2 and H4K20me3 causes cancer progression and indicates a poor prognosis, potentially by dysregulating telomere length maintenance186. Additionally, reduced expression of SUV4-20H2 has been observed in osteosarcoma cells, and RNA-seq analyses have suggested that the mitogen-activated protein kinase, p53, transforming growth factor, and ErbB pathways are downstream effectors of SUV4-20H2187.
EEF1AKMT3
EEF1AKMT3 is a member of the seven-beta-strand methyltransferase family and family 16 methyltransferases and is a lysine-specific methyltransferase. The EEF1AKMT3 gene is located on chromosome 12q14. EEF1AKMT3 has been identified as a tumor suppressor in gastric cancer. The expression of EEF1AKMT3 is significantly decreased in gastric cancer tissues vs. normal tissues in patients, and a low level of EEF1AKMT3 expression has been linked to a poor prognosis188. EEF1AKMT3 mediates the methylation of lysine residue 296 of the MAP2K7 protein, which has been implicated in tumor suppression. Mechanistically, the methylation of MAP2K7 by EEF1AKMT3 was shown to impede tumor activity by promoting the stabilization of TP53188.
Potential therapeutic approaches targeting tumor-suppressive PKMTs
Several anticancer drugs that target protein lysine methyltransferases have been developed. Notably, chemical inhibitors targeting EZH2, G9a, and DOT1L have been developed. Tazemetostat (EPZ-6438, E7438) is an anticancer drug that targets EZH2 (Table 2). This drug acts as a competitive inhibitor, binding to the SAM-binding site within the SET domain of EZH2. Tazemetostat inhibits the methylation of the lysine 27 residue on histone H3, inducing apoptosis in lymphoma cells, thereby inhibiting its activity and suppressing the growth of non-Hodgkin lymphoma189.
Most EZH2 inhibitors (Table 2) have been studied to demonstrate the suppression of EZH2 oncogenic functions. Notably, GSK126 (one of the EZH2 inhibitors) treatment in the KRAS-mutant NSCLC cell H358 and A549 resulted in an increase in phosphorylated AKT and ERK, potentially promoting cell proliferation190,191,192. However, treatment with GSK126 alone did not impact cell proliferation in these cells, leading the authors to suggest that EZH2 has dual roles at different stages of lung cancer depending on KRAS. Specifically, EZH2 suppressed the initiation of Kras-driven adenocarcinoma but promoted adenocarcinoma progression. Otherwise, it is also possible that each inhibitory chemical has another nonspecific activity affecting other responses to interfere with the tumor-suppressive function of EZH2. Given the pleiotropic role of EZH2 in cancer, it is imperative to develop a clear understanding of the mechanisms of EZH2 activators or inhibitors and to develop methods to increase the efficiency of these drugs.
A drug that targets G9a (histone-lysine N-methyltransferase 2) is A-366, which inhibits the activity of G9a, thereby impeding the proliferation and survival of cancer cells. A-366 inhibits the methylation of the lysine 9 residue of a histone, thereby inhibiting the growth and differentiation of leukemia193,194.
DOT1L, a histone methyltransferase, methylates lysine 79 in histone H3 and has emerged as a critical player in several tumors, including lung cancer, melanoma, neuroblastoma, liver cancer, and head and neck squamous cell carcinomas195,196. EPZ-5676, a potent and selective inhibitor of DOT1L, competes with the methyl-donating cofactor SAM and locks the SAM-binding pocket. Moreover, EPZ-5676 effectively disrupts histone methylation and suppresses the expression of MLL-fusion target genes197. Despite entering clinical trials based on this premise, this drug was found to have limited efficacy.
PKMT inhibitors hold immense potential as therapeutic agents for cancer treatment. While their direct application faces current limitations, innovative strategies such as combination therapies and alternative approaches targeting tumor suppressive PKMTs offer promising avenues to overcome these challenges27. Above all, alternative approaches, including miRNA-based regulation, focus on restoring the expression of tumor-suppressive PKMTs, including SETD2, to counteract tumorigenesis67,198. Enhancing the stability of methyltransferases is another strategy to maintain the activity of tumor-suppressive PKMTs and influence the epigenetic landscape and gene expression in cancer cells, thereby suppressing tumorigenic processes199. Extensive research and clinical investigations are necessary to fully explore the potential of PKMT inhibitors, thereby paving the way for personalized medicine and improved patient outcomes.
Gene expression analysis of PKMTs in various cancers
To gain a better understanding of tumor-suppressive PKMTs in various types of cancer and to provide valuable insights to aid in the development of novel therapeutic approaches targeting PKMTs, we conducted gene expression analyses of three representative PKMTs with established tumor-suppressive activity: SETD2, SETD7, and EZH2. We used several publicly available databases, including The Cancer Genome Atlas (TCGA) (https://www.cancer.gov.tcga), The Human Protein Atlas (https://www.proteinatlas.org), and Kaplan‒Meier Plotter (https://kmplot.com/analysis) (Fig. 4)200,201,202,203.



a, d, g A general pancancer overview of copy number variations (CNVs) in SETD2, SETD7, and EZH2, including copy number gain and copy number loss. The X-axis represents cancer type, while the Y-axis displays the frequencies of alterations (including both amplification and deletion) as percentages. The colors blue and red indicate deletion and amplification, respectively. b, e, h RNA expression profiles of SETD2, SETD7, and EZH2 based on RNA-seq data from The Cancer Genome Atlas. c, f, i Kaplan‒Meier analysis of recurrence-free survival (RFS) or overall survival (OS) based on the expression levels of SETD2, SETD7, and EZH2 in breast, gastric, lung, and ovarian cancers. ACC adrenocortical carcinoma, BLCA bladder urothelial carcinoma, BRCA breast invasive carcinoma, CESC cervical squamous cell carcinoma and endocervical adenocarcinoma, CHOL cholangiocarcinoma, COAD colon adenocarcinoma, DLBC lymphoid neoplasm diffuse large B-cell lymphoma, ESCA esophageal carcinoma, HNSC head and neck squamous cell carcinoma, KICH kidney chromophobe, KIRC kidney renal clear cell carcinoma, LAML acute myeloid leukemia, LGG lower-grade glioma, LIHC liver hepatocellular carcinoma, LUAD lung adenocarcinoma, LUSC lung squamous cell carcinoma, MESO mesothelioma, OV ovarian serous cystadenocarcinoma, PAAD pancreatic adenocarcinoma, PCPG pheochromocytoma and paraganglioma, PRAD prostate adenocarcinoma, READ rectal adenocarcinoma, SARC sarcoma, SKCM skin cutaneous melanoma, STAD stomach adenocarcinoma, TGCT testicular germ cell tumor, UCEC uterine corpus endometrial carcinoma, UCS uterine carcinosarcoma.
The majority of TCGA cancer types showed copy number loss of SETD2. Specifically, mesothelioma (48.81%, 41/84), kidney renal clear cell carcinoma (14.92%, 77/516), cholangiocarcinoma (13.89%, 5/36), ovarian cancer (13.5%, 79/585), breast cancer (12.5%, 134/1072), uterine carcinosarcoma (10.71%, 6/56), and gastric cancer (9.03%, 39/432), among others, showed downregulation (Fig. 4a). Although the observed downregulation of SETD2 in these cancer types suggests a potential role for SETD2 as a tumor suppressor, there is no existing research on the tumor suppressor activity or underlying mechanisms of SETD2 in cholangiocarcinoma, ovarian cancer, or uterine carcinosarcoma. Thus, we propose an exploration of the molecular mechanisms of SETD2 in these tumors. Similar to CNV analysis, TCGA RNA sequencing data indicated that the median values of fragments per kilobase of transcript per million mapped reads (FPKM) for SETD2 had low specificity in 17 cancer types (Fig. 4b). In particular, SETD2 mRNA downregulation was related to worse prognosis in terms of recurrence-free survival (RFS) in breast cancer (p < 0.001, n = 3951) and overall survival (OS) in gastric cancer (p = 0.014, n = 875) and lung cancer (p < 0.001, n = 1925) (Fig. 4c).
According to CNV distribution in the TCGA database, SETD7 shows copy number loss in ovarian cancer (9.74%, 57/585), sarcoma (6.92%, 18/260), lung squamous cell carcinoma (6.77%, 34/502), adrenocortical carcinoma (6.67%, 6/90), and other cancers (Fig. 4d). However, copy number gain of SETD7 was observed in ovarian cancer (11.28%, 66/585), uterine carcinoma (10.71%, 6/56), esophageal carcinoma (7.07%, 13/184), and sarcoma (6.92%, 18/260). In TCGA RNA sequencing data, the FPKM level of SETD7 showed low specificity in almost all cancer types except prostate and breast cancer (Fig. 4e). In addition, loss of SETD7 mRNA expression led to poor survival in ovarian cancer (p = 0.034, n = 1435) and lung cancer (p < 0.001, n = 1927) cohorts (Fig. 4f).
According to the TCGA database, EZH2 showed copy number loss in esophageal carcinoma (26.09%, 48/184), head and neck squamous cell carcinoma (16.51%, 86/512), gastric cancer (15.5%, 67/432), and lymphoid neoplasm diffuse large B-cell lymphoma (12.77%, 6/47) but a copy number gain in ovarian cancer (27.86%, 163/585), skin cutaneous melanoma (8.76%, 41/468), lung adenocarcinoma (7.41%, 38/513), and lower-grade glioma (6.24%, 31/497) (Fig. 4g). The mRNA expression level of EZH2 was decreased in thyroid, liver, pancreatic, renal, and prostate cancers compared with other cancer types in the TCGA database (Fig. 4h). Furthermore, EZH2-deficient gastric cancer patients had a shorter OS time than the controls (p = 0.0015, n = 875) (Fig. 4i).
Similar to previous research, our gene expression analysis also indicated that SETD7 and EZH2 exhibit both oncogenic and tumor-suppressive properties, depending on the cancer type. Thus, the design and development of anticancer drugs targeting these proteins require a meticulous approach and thorough investigations into their precise mechanisms in each cancer subtype.
Conclusion
This literature review provides a summary of tumor-suppressive PKMTs, their substrates, and their functions in various human cancers at the molecular, cellular, and clinicopathological levels to help understand the tumor-suppressive ability of PKMTs and to provide new insights for the development of anticancer drugs targeting these proteins. There are approximately 18 tumor-suppressive PKMTs, and most of these PKMTs contain SET domains and share common features, including the ability to methylate histone proteins and subcellular localization in the nucleus. PKMTs predominantly exhibit tumor-suppressive activity by mediating the methylation of histones, α-tubulin, p53, and β-catenin. These modifications cause gene alterations in oncogenes and tumor suppressors. PKMTs may also contribute to genomic stability by regulating DNA repair processes or cell cycle arrest. Several PKMTs, including MLL3, PRDM2/RIZ, PRDM5, SMYD4, SUV4-20H2, and EEF1AKMT3, have been reported to exhibit tumor suppressor activities. However, few studies of these proteins have been performed, and their precise mechanisms and role in tumor suppression need to be further elucidated. The other group comprises PKMTs with both oncogenic and tumor-suppressive functions, including DOT1L, EZH2, MLL2, MLL4, NSD1, SETD2, SETD3, SETDB1, and PRDM16. The functions and molecular mechanisms of these proteins are relatively well documented, and they have been frequently reported as targets for cancer therapy. However, due to the pleiotropic roles of PKMTs in tumorigenesis, administering PKMT inhibitors can lead to unintended negative consequences. Therefore, it is important to carefully consider their use with regard to the cancer type and target enzyme.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Feinberg, A. P. & Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153 (2004).
Ibrahim, R. K., Bruneau, A. & Bantignies, B. Plant O-methyltransferases: molecular analysis, common signature and classification. Plant Mol. Biol. 36, 1–10 (1998).
Weisiger, R. A. & Jakoby, W. B. Thiol S-methyltransferase. Methods Enzymol. 77, 257–262 (1981).
Yoo, J. et al. Drawing a line between histone demethylase KDM5A and KDM5B: their roles in development and tumorigenesis. Exp. Mol. Med. 54, 2107–2117 (2022).
Kouzarides, T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 12, 198–209 (2002).
Hyun, K., Jeon, J., Park, K. & Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49, e324 (2017).
Kim, J., Lee, H., Yi, S. J. & Kim, K. Gene regulation by histone-modifying enzymes under hypoxic conditions: a focus on histone methylation and acetylation. Exp. Mol. Med. 54, 878–889 (2022).
Li, G. et al. Homocysteine-targeting compounds as a new treatment strategy for diabetic wounds via inhibition of the histone methyltransferase SET7/9. Exp. Mol. Med. 54, 988–998 (2022).
Peng, H. et al. Hypoxia-hindered methylation of PTGIS in endometrial stromal cells accelerates endometriosis progression by inducing CD16(-) NK-cell differentiation. Exp. Mol. Med. 54, 890–905 (2022).
Santos-Rosa, H. et al. Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 (2002).
Ng, H. H., Ciccone, D. N., Morshead, K. B., Oettinger, M. A. & Struhl, K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc. Natl Acad. Sci. USA 100, 1820–1825 (2003).
Schotta, G. et al. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251–1262 (2004).
Wiles, E. T. & Selker, E. U. H3K27 methylation: a promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 43, 31–37 (2017).
Jacob, Y. et al. ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat. Struct. Mol. Biol. 16, 763–768 (2009).
Ferrari, K. J. et al. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62 (2014).
Shoaib, M. et al. Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 12, 4800 (2021).
Carlson, S. M., Moore, K. E., Green, E. M., Martin, G. M. & Gozani, O. Proteome-wide enrichment of proteins modified by lysine methylation. Nat. Protoc. 9, 37–50 (2014).
Guo, A. et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteom. 13, 372–387 (2014).
Hornbeck, P. V. et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 (2012).
Copeland, R. A. Protein methyltransferase inhibitors as precision cancer therapeutics: a decade of discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 373, 20170080 (2018).
Hamamoto, R. & Nakamura, Y. Dysregulation of protein methyltransferases in human cancer: an emerging target class for anticancer therapy. Cancer Sci. 107, 377–384 (2016).
Schubert, H. L., Blumenthal, R. M. & Cheng, X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 (2003).
Dillon, S. C., Zhang, X., Trievel, R. C. & Cheng, X. The SET-domain protein superfamily: protein lysine methyltransferases. Genome Biol. 6, 227 (2005).
Trievel, R. C., Beach, B. M., Dirk, L. M., Houtz, R. L. & Hurley, J. H. Structure and catalytic mechanism of a SET domain protein methyltransferase. Cell 111, 91–103 (2002).
Feng, Q. et al. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 12, 1052–1058 (2002).
Weirich, S. & Jeltsch, A. Mutations in histone lysine methyltransferases and demethylases. In Encyclopedia of Cancer, 3rd ed., in reference module in biomedical sciences, 538–550 (Elsevier, 2019).
Liao, Q. et al. Novel insights into histone lysine methyltransferases in cancer therapy: From epigenetic regulation to selective drugs. J. Pharm. Anal. 13, 127–141 (2023).
Chen, Y. et al. Increased expression of SETD7 promotes cell proliferation by regulating cell cycle and indicates poor prognosis in hepatocellular carcinoma. PLOS One 11, e0154939 (2016).
Duan, B. et al. Histone-lysine N-methyltransferase SETD7 is a potential serum biomarker for colorectal cancer patients. EBioMedicine 37, 134–143 (2018).
Gu, Y. et al. SET7/9 promotes hepatocellular carcinoma progression through regulation of E2F1. Oncol. Rep. 40, 1863–1874 (2018).
Zhang, Y. et al. The transcription factor GATA1 and the histone methyltransferase SET7 interact to promote VEGF-mediated angiogenesis and tumor growth and predict clinical outcome of breast cancer. Oncotarget 7, 9859 (2016).
Oudhoff, M. J. et al. SETD7 controls intestinal regeneration and tumorigenesis by regulating Wnt/β-catenin and Hippo/YAP signaling. Dev. Cell 37, 47–57 (2016).
Song, H. et al. Isoform-specific lysine methylation of RORα2 by SETD7 is required for association of the TIP60 coactivator complex in prostate cancer progression. Int. J. Mol. Sci. 21, 1622 (2020).
Wang, C. et al. Histone methyltransferase Setd7 regulates Nrf2 signaling pathway by phenethyl isothiocyanate and ursolic acid in human prostate cancer cells. Mol. Nutr. Food Res. 62, 1700840 (2018).
Liu, Q., Geng, H., Xue, C., Beer, T. M. & Qian, D. Z. Functional regulation of hypoxia inducible factor-1α by SET9 lysine methyltransferase. Biochim. Biophys. Act. 1853, 881–891 (2015).
Guo, S. et al. EZH2 overexpression in different immunophenotypes of breast carcinoma and association with clinicopathologic features. Diagn. Pathol. 11, 41 (2016).
Bachmann, I. M. et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 24, 268–273 (2006).
Dong, M. et al. Aberrant expression of enhancer of zeste homologue 2, correlated with HIF-1alpha, refines relapse risk and predicts poor outcome for breast cancer. Oncol. Rep. 32, 1101–1107 (2014).
Chen, Z. et al. Expression of EZH2 is associated with poor outcome in colorectal cancer. Oncol. Lett. 15, 2953–2961 (2018).
Ohuchi, M. et al. Increased EZH2 expression during the adenoma-carcinoma sequence in colorectal cancer. Oncol. Lett. 16, 5275–5281 (2018).
Varambally, S. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002).
Kleer, C. G. et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl Acad. Sci. USA 100, 11606–11611 (2003).
Cao, Q. et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 27, 7274–7284 (2008).
Purkait, S. et al. EZH2 expression in gliomas: correlation with CDKN2A gene deletion/ p16 loss and MIB-1 proliferation index. Neuropathology 35, 421–431 (2015).
Velichutina, I. et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116, 5247–5255 (2010).
Chen, Q., Zheng, P. S. & Yang, W. T. EZH2-mediated repression of GSK-3beta and TP53 promotes Wnt/beta-catenin signaling-dependent cell expansion in cervical carcinoma. Oncotarget 7, 36115–36129 (2016).
Liao, Y. et al. Inhibition of EZH2 transactivation function sensitizes solid tumors to genotoxic stress. Proc. Natl Acad. Sci. USA 119, e2105898119 (2022).
Rastgoo, N., Pourabdollah, M., Abdi, J., Reece, D. & Chang, H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia 32, 2471–2482 (2018).
Comet, I., Riising, E. M., Leblanc, B. & Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 16, 803–810 (2016).
de Vries, N. A. et al. Prolonged Ezh2 depletion in glioblastoma causes a robust switch in cell fate resulting in tumor progression. Cell Rep. 10, 383–397 (2015).
Dhar, S. et al. A tumor suppressor role for EZH2 in diffuse midline glioma pathogenesis. Acta Neuropathol. Commun. 10, 47 (2022).
Huang, J. et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J. Biol. Chem. 285, 9636–9641 (2010).
Zhong, X. et al. Overexpression of G 9a and MCM 7 in oesophageal squamous cell carcinoma is associated with poor prognosis. Histopathology 66, 192–200 (2015).
Chen, M.-W. et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70, 7830–7840 (2010).
Hua, K.-T. et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 13, 1–13 (2014).
Lee, S., Kim, J., Kim, W.-H. & Lee, Y. M. Hypoxic silencing of tumor suppressor RUNX3 by histone modification in gastric cancer cells. Oncogene 28, 184–194 (2009).
Komatsu, S. et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 30, 1139–1146 (2009).
Cho, H.-S. et al. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia 14, 476–IN478 (2012).
Huang, J. et al. Repression of p53 activity by Smyd2-mediated methylation. Nature 444, 629–632 (2006).
Saddic, L. A. et al. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 285, 37733–37740 (2010).
Viotti, M. et al. SUV420H2 is an epigenetic regulator of epithelial/mesenchymal states in pancreatic cancer. J. Cell Biol. 217, 763–777 (2018).
Li, Y. et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J. Biol. Chem. 284, 34283–34295 (2009).
Qiao, Q. et al. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J. Biol. Chem. 286, 8361–8368 (2011).
Martinez-Garcia, E. et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood 117, 211–220 (2011).
Morishita, M., Mevius, D. & di Luccio, E. In vitro histone lysine methylation by NSD1, NSD2/MMSET/WHSC1 and NSD3/WHSC1L. BMC Struct. Biol. 14, 25 (2014).
Martinelli, G. et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 32, 139–148 (2018).
Xiang, W. et al. miR-106b-5p targets tumor suppressor gene SETD2 to inactive its function in clear cell renal cell carcinoma. Oncotarget 6, 4066–4079 (2015).
Hacker, K. E. et al. Structure/function analysis of recurrent mutations in SETD2 protein reveals a critical and conserved role for a SET domain residue in maintaining protein stability and histone H3 lys-36 trimethylation. J. Biol. Chem. 291, 21283–21295 (2016).
Li, J. et al. SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget 7, 50719–50734 (2016).
Chiang, Y. C. et al. SETD2 haploinsufficiency for microtubule methylation is an early driver of genomic instability in renal cell carcinoma. Cancer Res. 78, 3135–3146 (2018).
Duns, G. et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 70, 4287–4291 (2010).
Kanu, N. et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene 34, 5699–5708 (2015).
Carvalho, S. et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 3, e02482 (2014).
Li, L., Miao, W., Huang, M., Williams, P. & Wang, Y. Integrated genomic and proteomic analyses reveal novel mechanisms of the methyltransferase SETD2 in renal cell carcinoma development. Mol. Cell Proteom. 18, 437–447 (2019).
Liu, J. et al. Loss of SETD2 induces a metabolic switch in renal cell carcinoma cell lines toward enhanced oxidative phosphorylation. J. Proteome Res. 18, 331–340 (2019).
Li, J. et al. Functional studies on primary tubular epithelial cells indicate a tumor suppressor role of SETD2 in clear cell renal cell carcinoma. Neoplasia 18, 339–346 (2016).
Zhang, Y. L. et al. Setd2 deficiency impairs hematopoietic stem cell self-renewal and causes malignant transformation. Cell Res. 28, 476–490 (2018).
Bu, J. et al. SETD2-mediated crosstalk between H3K36me3 and H3K79me2 in MLL-rearranged leukemia. Leukemia 32, 890–899 (2018).
Chen, B. Y. et al. SETD2 deficiency accelerates MDS-associated leukemogenesis via S100a9 in NHD13 mice and predicts poor prognosis in MDS. Blood 135, 2271–2285 (2020).
Parker, H. et al. Genomic disruption of the histone methyltransferase SETD2 in chronic lymphocytic leukaemia. Leukemia 30, 2179–2186 (2016).
Mar, B. G. et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat. Commun. 5, 3469 (2014).
Mar, B. G. et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood 130, 2631–2641 (2017).
Dong, Y. et al. SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints. Leukemia 33, 2585–2598 (2019).
Sheng, Y. et al. Downregulation of the histone methyltransferase SETD2 promotes imatinib resistance in chronic myeloid leukaemia cells. Cell Prolif. 52, e12611 (2019).
Niu, N. et al. Loss of Setd2 promotes Kras-induced acinar-to-ductal metaplasia and epithelia-mesenchymal transition during pancreatic carcinogenesis. Gut 69, 715–726 (2020).
Niu, N. et al. Tumor cell-intrinsic SETD2 deficiency reprograms neutrophils to foster immune escape in pancreatic tumorigenesis. Adv. Sci. 10, e2202937 (2023).
Walter, D. M. et al. Systematic in vivo inactivation of chromatin-regulating enzymes identifies Setd2 as a potent tumor suppressor in lung adenocarcinoma. Cancer Res. 77, 1719–1729 (2017).
Xie, Y. et al. SETD2 regulates chromatin accessibility and transcription to suppress lung tumorigenesis. JCI Insight 8, e154120 (2023).
Kim, I. K. et al. Acquired SETD2 mutation and impaired CREB1 activation confer cisplatin resistance in metastatic non-small cell lung cancer. Oncogene 38, 180–193 (2019).
Walter, D. M. et al. Setd2 inactivation sensitizes lung adenocarcinoma to inhibitors of oxidative respiration and mTORC1 signaling. Commun. Biol. 6, 255 (2023).
Chen, Z. et al. SETD2 indicates favourable prognosis in gastric cancer and suppresses cancer cell proliferation, migration, and invasion. Biochem. Biophys. Res. Commun. 498, 579–585 (2018).
Huang, K. K. et al. SETD2 histone modifier loss in aggressive GI stromal tumours. Gut 65, 1960–1972 (2016).
Yuan, H. et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J. Clin. Invest. 127, 3375–3391 (2017).
Bushara, O. et al. Clinical and histopathologic characterization of SETD2-mutated colorectal cancer. Hum. Pathol. 131, 9–16 (2023).
Ganaie, I. A., Naqvi, S. H., Jain, S. K. & Wajid, S. Reduced expression of SETD2 and SNX9 proteins in chemically induced mammary tumorigenesis in Wistar rats: a prognostic histological and proteomic study. Protoplasma 254, 1451–1466 (2017).
Jiang, C., He, C., Wu, Z., Li, F. & Xiao, J. Histone methyltransferase SETD2 regulates osteosarcoma cell growth and chemosensitivity by suppressing Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 502, 382–388 (2018).
Bhattacharya, S., Reddy, D., Zhang, N., Li, H. & Workman, J. L. Elevated levels of the methyltransferase SETD2 causes transcription and alternative splicing changes resulting in oncogenic phenotypes. Front. Cell Dev. Biol. 10, 945668 (2022).
Kurash, J. K. et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 29, 392–400 (2008).
Donehower, L. A. et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 (1992).
Shen, C. et al. SET7/9 regulates cancer cell proliferation by influencing beta-catenin stability. FASEB J. 29, 4313–4323 (2015).
Akiyama, Y. et al. Reduced expression of SET7/9, a histone mono-methyltransferase, is associated with gastric cancer progression. Oncotarget 7, 3966–3983 (2016).
Hu, H. Y. et al. Set9, NF-kappaB, and microRNA-21 mediate berberine-induced apoptosis of human multiple myeloma cells. Acta Pharmacol. Sin. 34, 157–166 (2013).
Montenegro, M. F. et al. Tumor suppressor SET9 guides the epigenetic plasticity of breast cancer cells and serves as an early-stage biomarker for predicting metastasis. Oncogene 35, 6143–6152 (2016).
Song, Y. et al. SET7/9 inhibits oncogenic activities through regulation of Gli-1 expression in breast cancer. Tumour Biol. 37, 9311–9322 (2016).
Cao, L. et al. Downregulation of SETD7 promotes migration and invasion of lung cancer cells via JAK2/STAT3 pathway. Int. J. Mol. Med. 45, 1616–1626 (2020).
Xie, H. et al. METTL3/YTHDF2 m6A axis promotes tumorigenesis by degrading SETD7 and KLF4 mRNAs in bladder cancer. J. Cell Mol. Med. 24, 4092–4104 (2020).
Wang, Y. et al. Ezh2 Acts as a tumor suppressor in Kras-driven lung adenocarcinoma. Int. J. Biol. Sci. 13, 652–659 (2017).
Li, Z. et al. The polycomb group protein EZH2 inhibits lung cancer cell growth by repressing the transcription factor Nrf2. FEBS Lett. 588, 3000–3007 (2014).
Ernst, T. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 42, 722–726 (2010).
Bejar, R. et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 364, 2496–2506 (2011).
Guglielmelli, P. et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 118, 5227–5234 (2011).
Muto, T. et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. J. Exp. Med. 210, 2627–2639 (2013).
Sashida, G. et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat. Commun. 5, 4177 (2014).
Nangalia, J. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405 (2013).
Mullally, A. et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 17, 584–596 (2010).
Sashida, G. et al. The loss of Ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J. Exp. Med. 213, 1459–1477 (2016).
Simon, C. et al. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 26, 651–656 (2012).
Ntziachristos, P. et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18, 298–301 (2012).
Gollner, S. et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 23, 69–78 (2017).
Krill, L. et al. Overexpression of enhance of Zeste homolog 2 (EZH2) in endometrial carcinoma: An NRG oncology/Gynecologic oncology group study. Gynecol. Oncol. 156, 423–429 (2020).
Qiu, B. Q. et al. Long non-coding RNA PSMA3-AS1 promotes malignant phenotypes of esophageal cancer by modulating the miR-101/EZH2 axis as a ceRNA. Aging 12, 1843–1856 (2020).
Gan, L. et al. The polycomb group protein EZH2 induces epithelial–mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J. Hematol. Oncol. 11, 9 (2018).
Pellecchia, S. et al. The long non-coding RNA prader Willi/Angelman region RNA5 (PAR5) is downregulated in anaplastic thyroid carcinomas where it acts as a tumor suppressor by reducing EZH2 activity. Cancers 12, 235 (2020).
Fan, D. C., Zhao, Y. R., Qi, H., Hou, J. X. & Zhang, T. H. MiRNA-506 presents multiple tumor suppressor activities by targeting EZH2 in nasopharyngeal carcinoma. Auris Nasus Larynx 47, 632–642 (2020).
Yu, H. et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl Acad. Sci. USA 111, 285–290 (2014).
Sun, C. et al. EZH2 Expression is increased in BAP1-mutant renal clear cell carcinoma and is related to poor prognosis. J. Cancer 9, 3787–3796 (2018).
Zauderer, M. G. et al. EZH2 inhibitor tazemetostat in patients with relapsed or refractory, BAP1-inactivated malignant pleural mesothelioma: a multicentre, open-label, phase 2 study. Lancet Oncol. 23, 758–767 (2022).
LaFave, L. M. et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 21, 1344–1349 (2015).
Ciarapica, R. et al. Deregulated expression of miR-26a and Ezh2 in Rhabdomyosarcoma. Cell Cycle 8, 172–175 (2009).
Richter, G. H. S. et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl Acad. Sci. USA 106, 5324–5329 (2009).
McCabe, M. T. et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl Acad. Sci. USA 109, 2989–2994 (2012).
Yap, D. B. et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459 (2011).
Morin, R. D. et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185 (2010).
Ogawa, S. Genetic basis of myelodysplastic syndromes. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 96, 107–121 (2020).
Simon, J. A. & Lange, C. A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 647, 21–29 (2008).
Antonysamy, S. et al. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS One 8, e84147 (2013).
Chase, A. et al. Mutational mechanisms of EZH2 inactivation in myeloid neoplasms. Leukemia 34, 3206–3214 (2020).
Zhou, B. et al. PRDM16 sppresses MLL1r leukemia via itrinsic hstone mthyltransferase ativity. Mol. Cell 62, 222–236 (2016).
Alam, H. et al. KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell 37, 599–617.e597 (2020).
Ortega-Molina, A. et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 21, 1199–1208 (2015).
Zhang, J. et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med. 21, 1190–1198 (2015).
Dhar, S. S. et al. MLL4 is required to maintain broad H3K4me3 peaks and super-enhancers at tumor suppressor genes. Mol. Cell 70, 825–841.e826 (2018).
Du, D. et al. Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial-mesenchymal transition. EMBO Rep. 19, 135–155 (2018).
Abaev-Schneiderman, E., Admoni-Elisha, L. & Levy, D. SETD3 is a positive regulator of DNA-damage-induced apoptosis. Cell Death Dis. 10, 74 (2019).
Zhu, B. et al. The protective role of DOT1L in UV-induced melanomagenesis. Nat. Commun. 9, 259 (2018).
Berdasco, M. et al. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc. Natl Acad. Sci. USA 106, 21830–21835 (2009).
Cancer Genome Atlas Research, N. et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
Garg, M. et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood 126, 2491–2501 (2015).
Kayser, S. et al. Clinical impact of KMT2C and SPRY4 expression levels in intensively treated younger adult acute myeloid leukemia patients. Eur. J. Haematol. 99, 544–552 (2017).
Chen, C. et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25, 652–665 (2014).
Yeh, C.-H. et al. Mutation of epigenetic regulators TET2 and MLL3 in patients with HTLV-I-induced acute adult T-cell leukemia. Mol. Cancer 15, 15 (2016).
Watanabe, Y. et al. Frequent alteration of MLL3 frameshift mutations in microsatellite deficient colorectal cancer. PLoS One 6, e23320 (2011).
Rampias, T. et al. The lysine‐specific methyltransferase KMT 2C/MLL 3 regulates DNA repair components in cancer. EMBO Rep. 20, e46821 (2019).
Lee, J. et al. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl Acad. Sci. USA 106, 8513–8518 (2009).
Buyse, I. M., Shao, G. & Huang, S. The retinoblastoma protein binds to RIZ, a zinc-finger protein that shares an epitope with the adenovirus E1A protein. Proc. Natl Acad. Sci. USA 92, 4467–4471 (1995).
Steele-Perkins, G. et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 15, 2250–2262 (2001).
Piao, G. H. et al. Hyper-methylation of RIZ1 tumor suppressor gene is involved in the early tumorigenesis of hepatocellular carcinoma. Histol. Histopathol. 23, 1171–1175 (2008).
Liu, Z. Y. et al. Retinoblastoma protein-interacting zinc-finger gene 1 (RIZ1) dysregulation in human malignant meningiomas. Oncogene 32, 1216–1222 (2013).
He, L. et al. RIZ1, but not the alternative RIZ2 product of the same gene, is underexpressed in breast cancer, and forced RIZ1 expression causes G2-M cell cycle arrest and/or apoptosis. J. Cancer Res. 58, 4238–4244 (1998).
Cai, Z. et al. RIZ1 negatively regulates ubiquitin-conjugating enzyme E2C/UbcH10 via targeting c-Myc in meningioma. Am. J. Transl. Res. 9, 2645–2655 (2017).
Zhang, C. et al. RIZ1: a potential tumor suppressor in glioma. BMC Cancer 15, 990 (2015).
Wei, D. et al. The absence of PRDM2 involved the tumorigenesis of somatotroph adenomas through regulating c-Myc. J. Gene 737, 144456 (2020).
van Ree, J. H., Jeganathan, K. B., Malureanu, L. & van Deursen, J. M. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J. Cell Biol. 188, 83–100 (2010).
Tokumaru, Y. et al. Biallelic inactivation of the RIZ1 gene in human gastric cancer. Oncogene 22, 6954–6958 (2003).
Du, Y. et al. Hypermethylation in human cancers of the RIZ1 tumor suppressor gene, a member of a histone/protein methyltransferase superfamily. Cancer Res. 61, 8094–8099 (2001).
Jiang, G., Liu, L., Buyse, I. M., Simon, D. & Huang, S. Decreased RIZ1 expression but not RIZ2 in hepatoma and suppression of hepatoma tumorigenicity by RIZ1. Int. J. Cancer 83, 541–546 (1999).
Jiang, G. L. & Huang, S. Adenovirus expressing RIZ1 in tumor suppressor gene therapy of microsatellite-unstable colorectal cancers. Cancer Res. 61, 1796–1798 (2001).
Piao, Z. et al. Frequent frameshift mutations of RIZ in sporadic gastrointestinal and endometrial carcinomas with microsatellite instability. Cancer Res. 60, 4701–4704 (2000).
Yang, T. et al. RIZ1 is regulated by estrogen and suppresses tumor progression in endometrial cancer. Biochem. Biophys. Res. Commun. 489, 96–102 (2017).
Huang, S. The retinoblastoma protein-interacting zinc finger gene RIZ in 1p36-linked cancers. Front. Biosci. 4, D528–D532 (1999).
Di Zazzo, E., Porcile, C., Bartollino, S. & Moncharmont, B. Critical function of PRDM2 in the neoplastic growth of testicular germ cell tumors. Biology 5, 54 (2016).
Pastural, E. et al. RIZ1 repression is associated with insulin-like growth factor-1 signaling activation in chronic myeloid leukemia cell lines. Oncogene 26, 1586–1594 (2007).
Bond, C. E. et al. Methylation and expression of the tumour suppressor, PRDM5, in colorectal cancer and polyp subgroups. BMC Cancer 15, 20 (2015).
Watanabe, Y. et al. PRDM5 identified as a target of epigenetic silencing in colorectal and gastric cancer. Clin. Cancer Res. 13, 4786–4794 (2007).
Deng, Q. & Huang, S. PRDM5 is silenced in human cancers and has growth suppressive activities. Oncogene 23, 4903–4910 (2004).
Galli, G. G. et al. Prdm5 suppresses Apc(Min)-driven intestinal adenomas and regulates monoacylglycerol lipase expression. Oncogene 33, 3342–3350 (2014).
Shu, X.-s et al. The epigenetic modifier PRDM5 functions as a tumor suppressor through modulating WNT/β-catenin signaling and is frequently silenced in multiple tumors. PLoS One 6, e27346 (2011).
Hu, L., Zhu, Y. T., Qi, C. & Zhu, Y. J. Identification of Smyd4 as a potential tumor suppressor gene involved in breast cancer development. Cancer Res. 69, 4067–4072 (2009).
Song, J. et al. Expression patterns and the prognostic value of the SMYD family members in human breast carcinoma using integrative bioinformatics analysis. Oncol. Lett. 17, 3851–3861 (2019).
Han, S. et al. miR-1307-3p stimulates breast cancer development and progression by targeting SMYD4. J. Cancer 10, 441–448 (2019).
Yan, L. et al. Genetic alteration of histone lysine methyltransferases and their significance in renal cell carcinoma. PeerJ 7, e6396 (2019).
Noroxe, D. S. et al. Genomic profiling of newly diagnosed glioblastoma patients and its potential for clinical utility - a prospective, translational study. Mol. Oncol. 14, 2727–2743 (2020).
Shinchi, Y. et al. SUV420H2 suppresses breast cancer cell invasion through down regulation of the SH2 domain-containing focal adhesion protein tensin-3. Exp. Cell Res. 334, 90–99 (2015).
Wu, Y. et al. miR-29a contributes to breast cancer cells epithelial-mesenchymal transition, migration, and invasion via down-regulating histone H4K20 trimethylation through directly targeting SUV420H2. Cell Death Dis. 10, 176 (2019).
Pogribny, I. P. et al. Histone H3 lysine 9 and H4 lysine 20 trimethylation and the expression of Suv4-20h2 and Suv-39h1 histone methyltransferases in hepatocarcinogenesis induced by methyl deficiency in rats. Carcinogenesis 27, 1180–1186 (2006).
Van Den Broeck, A. et al. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin. Cancer Res 14, 7237–7245 (2008).
Piao, L. et al. Loss of histone H4 lysine 20 trimethylation in osteosarcoma is associated with aberrant expression ofhistone methyltransferase SUV420H2. Oncol. Lett. 20, 26 (2020).
Hong, Y. H. et al. The EEF1AKMT3/MAP2K7/TP53 axis suppresses tumor invasiveness and metastasis in gastric cancer. Cancer Lett. 544, 215803 (2022).
Knutson, S. K. et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854 (2014).
Serresi, M. et al. Ezh2 inhibition in Kras-driven lung cancer amplifies inflammation and associated vulnerabilities. J. Exp. Med. 215, 3115–3135 (2018).
Al-Ghabkari, A. & Narendran, A. Targeting EZH2-mediated methylation of histone 3 inhibits proliferation of pediatric acute monocytic leukemia cells in vitro. Cancer Biol. Ther. 22, 333–344 (2021).
Wang, Y. et al. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcaemic type. J. Pathol. 242, 371–383 (2017).
Sweis, R. F. et al. Discovery and development of potent and selective inhibitors of histone methyltransferase g9a. ACS Med. Chem. Lett. 5, 205–209 (2014).
Pappano, W. N. et al. The histone methyltransferase inhibitor A-366 uncovers a role for G9a/GLP in the epigenetics of leukemia. PLoS One 10, e0131716 (2015).
Alexandrova, E. et al. Histone methyltransferase DOT1L as a promising epigenetic target for treatment of solid tumors. Front. Genet. 13, 864612 (2022).
Kim, W., Kim, R., Park, G., Park, J. W. & Kim, J. E. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J. Biol. Chem. 287, 5588–5599 (2012).
Daigle, S. R. et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025 (2013).
Gelato, K. A., Adler, D., Ocker, M. & Haendler, B. Targeting epigenetic regulators for cancer therapy: modulation of bromodomain proteins, methyltransferases, demethylases, and microRNAs. Expert Opin. Ther. Targets 20, 783–799 (2016).
Bhattacharya, S. & Workman, J. L. Regulation of SETD2 stability is important for the fidelity of H3K36me3 deposition. Epigenetics Chromatin 13, 40 (2020).
Gyorffy, B. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731 (2010).
Gyorffy, B., Lanczky, A. & Szallasi, Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer 19, 197–208 (2012).
Gyorffy, B., Surowiak, P., Budczies, J. & Lanczky, A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 8, e82241 (2013).
Szasz, A. M. et al. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7, 49322–49333 (2016).
Knutson, S. K. et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl Acad. Sci. USA 110, 7922–7927 (2013).
Qi, W. et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl Acad. Sci. USA 109, 21360–21365 (2012).
McCabe, M. T. et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012).
Bradley, W. D. et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem. Biol. 21, 1463–1475 (2014).
Knutson, S. K. et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8, 890–896 (2012).
Campbell, J. E. et al. EPZ011989, A potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med. Chem. Lett. 6, 491–495 (2015).
Song, X. et al. Selective inhibition of EZH2 by ZLD10A blocks H3K27 methylation and kills mutant lymphoma cells proliferation. Biomed. Pharmacother. 81, 288–294 (2016).
Beguelin, W. et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692 (2013).
Xie, H. et al. Chronic myelogenous leukemia-initiating cells require polycomb group protein EZH2. Cancer Discov. 6, 1237–1247 (2016).
Verma, S. K. et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 3, 1091–1096 (2012).
Kung, P. P. et al. Correction to design and synthesis of pyridone-containing 3,4-dihydroisoquinoline-1(2H)-ones as a novel class of enhancer of Zeste Homolog 2 (EZH2) inhibitors. J. Med. Chem. 59, 11196 (2016).
Garapaty-Rao, S. et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem. Biol. 20, 1329–1339 (2013).
Vaswani, R. G. et al. Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-Cell lymphomas. J. Med. Chem. 59, 9928–9941 (2016).
Lu, B. et al. Discovery of EBI-2511: A highly potent and orally active EZH2 inhibitor for the treatment of non-Hodgkin’s lymphoma. ACS Med. Chem. Lett. 9, 98–102 (2018).
Konze, K. D. et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8, 1324–1334 (2013).
Yamagishi, M. et al. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 29, 2321–2337.e2327 (2019).
Honma, D. et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 108, 2069–2078 (2017).
Kung, P. P. et al. Optimization of orally bioavailable enhancer of Zeste Homolog 2 (EZH2) inhibitors using ligand and property-based design strategies: Identification of development candidate (R)-5,8-dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J. Med. Chem. 61, 650–665 (2018).
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant Nos. 2011-0016397, 2017R1A6A1A03015642, and 2020R1F1A1073219 to J.Y.C.), Republic of Korea.
Author information
Authors and Affiliations
Contributions
N.A. and Y.H.H. conceived and designed the study, performed data analysis and acquisition/interpretation, performed bioinformatics analysis, and drafted the manuscript. H.G.K., J.H.K., and J.Y.C. offered scientific leadership, conceived and designed the study, analyzed the data, and edited the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aziz, N., Hong, Y.H., Kim, H.G. et al. Tumor-suppressive functions of protein lysine methyltransferases. Exp Mol Med 55, 2475–2497 (2023). https://doi.org/10.1038/s12276-023-01117-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01117-7
