
Abstract
Dipeptidyl peptidase-4 (DPP-4) inhibitors are glucose-lowering drugs for type 2 diabetes mellitus (T2DM). We investigated whether evogliptin® (EVO), a DPP-4 inhibitor, could protect against diabetic cardiomyopathy (DCM) and the underlying mechanisms. Eight-week-old diabetic and obese db/db mice were administered EVO (100 mg/kg/day) daily by oral gavage for 12 weeks. db/db control mice and C57BLKS/J as wild-type (WT) mice received equal amounts of the vehicle. In addition to the hypoglycemic effect, we examined the improvement in cardiac contraction/relaxation ability, cardiac fibrosis, and myocardial hypertrophy by EVO treatment. To identify the mechanisms underlying the improvement in diabetic cardiomyopathy by EVO treatment, its effect on lipotoxicity and the mitochondrial damage caused by lipid droplet accumulation in the myocardium were analyzed. EVO lowered the blood glucose and HbA1c levels and improved insulin sensitivity but did not affect the body weight or blood lipid profile. Cardiac systolic/diastolic function, hypertrophy, and fibrosis were improved in the EVO-treated group. EVO prevented cardiac lipotoxicity by reducing the accumulation of lipid droplets in the myocardium through suppression of CD36, ACSL1, FABP3, PPARgamma, and DGAT1 and enhancement of the phosphorylation of FOXO1, indicating its inhibition. The EVO-mediated improvement in mitochondrial function and reduction in damage were achieved through activation of PGC1a/NRF1/TFAM, which activates mitochondrial biogenesis. RNA-seq results for the whole heart confirmed that EVO treatment mainly affected the differentially expressed genes (DEGs) related to lipid metabolism. Collectively, these findings demonstrate that EVO improves cardiac function by reducing lipotoxicity and mitochondrial injury and provides a potential therapeutic option for DCM.
Similar content being viewed by others


Introduction
Type 2 diabetes mellitus (T2DM), a common metabolic disease, accounts for over 90% of diabetes cases worldwide1,2. Poorly managed T2DM leads to various complications, including diabetic cardiomyopathy (DCM), a major cause of end-stage heart failure (HF), resulting in mortality and morbidity in patients with T2DM3,4. DCM is characterized by abnormal cardiac structure and function in diabetic patients without identified causes, such as coronary artery disease, uncontrolled hypertension, and significant valvular and congenital heart disease5,6. A wide variety of mechanisms have been suggested to be involved in this clinical condition in recent years, including cardiomyocyte oxidative stress, inflammation, fibrosis, various forms of cell death, mitochondrial dysfunction, and alterations in myocardial energetics7,8,9,10. However, the underlying mechanism has not yet been elucidated, and there is no specific therapy for DCM patients in clinical practice.
The main energy source of the adult heart is obtained from fatty acid (FA) oxidation, which accounts for 60–80%, with the remaining part being derived from glucose, lactate, and ketones11. However, in obese and diabetic patients, excessive accumulation of lipids and overactivation of lipid signaling pathways disturb the balance of FA uptake, metabolism, and oxidation in cardiomyocytes, leading to an increased flux of nonoxidative metabolic FAs and the accumulation of cardiotoxic fatty acid metabolites, thereby resulting in lipotoxic cardiomyopathy12,13. This condition not only changes cardiac metabolism but also causes the accumulation of damaged mitochondria and cardiac fibrosis14,15,16. Inhibiting FA utilization is a promising strategy for increasing cardiac efficiency in DCM.
Dipeptidyl peptidase-4 (DPP-4) inhibitors are an approved treatment for T2DM. Several studies have shown that they have a beneficial effect on cardiovascular diseases associated with metabolic syndrome and enhance blood pressure and vascular endothelial function17,18,19. Evogliptin® (EVO), a novel DPP-4 inhibitor, was developed by Dong-A, Republic of Korea, and approved as an oral antihyperglycemic drug for the treatment of T2DM by the Ministry of Food and Drug Safety of Korea in 2015. EVO has an effect on gluconeogenesis, hepatic steatosis, vascular inflammation, and whole-body composition20,21,22,23. However, the effect of DPP-4 inhibitors on HF is still unknown, and studies on EVO to understand its impact on diabetes and its complications, including DCM, are lacking. To explore these issues, we investigated the effects of EVO on DCM in db/db mice.
Materials and methods
Animal experiments
Eight-week-old male diabetic db/db mice (BKS.Cg-Dock7m+/+ Leprdb/J) and eight-week-old male nondiabetic db/m+ mice (C57BLKS/J) were purchased from Charles River (Japan). C57BKS/J mice were used as normal controls or wild-type (WT) mice. All animals were housed in a specific pathogen-free facility with controlled temperature (20–24 °C) and humidity (40–70%) on a 12 h light cycle with access to standard laboratory chow and tap water ad libitum. All experimental procedures were approved by the Inje Medical University Animal Care and Use Committee (approval No. 2011-049). After adaptive feeding for three days, WT and db/db mice with similar blood glucose levels and body weights were randomized into the following groups: untreated db/db control group and db/db+Evo group. Mice in the db/db+Evo group were intragastrically administered 100 mg/kg/day evogliptin (Dong-A, Korea) for 12 weeks, while WT and db/db control mice were administered equal amounts of 0.5% hydroxyethylcellulose (vehicle) via gavage. Body weight (BW) and food intake were measured weekly.
At the end of the treatment, the mice were sacrificed after performing LV catheterization under anesthesia with isoflurane (1.5% in 100% oxygen). Overnight-fasted mice were sacrificed by exsanguination under anesthesia by inhalation of 2% isoflurane in the ambient air of the room. Blood samples and heart tissue were collected for further experiments; the heart weight was also measured (Supplementary Fig. 1).
Echocardiography
The mice were kept warm on a heating pad while under anesthesia (1–2% isoflurane supplemented with 100% oxygen), and transthoracic echocardiography was performed. Images were acquired using a high-frequency ultrasound system (Vivid 7, GE Healthcare, Korea) to detect cardiac structure alterations and cardiac function in vivo using an echocardiogram with an i13L-14 MHz probe. Additionally, 2-D guided M-mode images of the LV in parasternal short-axis views at the papillary muscle level were recorded. The left ventricular internal diameter in diastole/systole (LVIDd/s), end-diastolic volume (EDV), end-systolic volume (ESV), stroke volume (SV), ejection fraction (EF), and fractional shortening (FS) were obtained in conscious experimental animals. According to Teichholz, EDV = (7.0/(2.4 + LVIDd)) × LVIDd3) and ESV = (7.0/(2.4 + LVIDs)) × LVIDs3) were used to calculate the EF as follows: EF = 100 × (EDV – ESV)/ESV. LV diastolic function was obtained using the transmitral inflow velocities recorded with pulsed-wave Doppler in the apical 4-chamber view, and the longitudinal component of myocardial contraction was assessed with tissue Doppler imaging with the sample volume placed at the base of the posterior wall in the parasternal long axis. The ratio of the peak velocity of early to late filling of mitral inflow (E/A) and of early diastolic myocardial relaxation (e’) to active atrial contraction in late diastole (a’), (e’/a’), E/e’, and deceleration time (DT) were calculated. All data and images were saved and analyzed using an EchoPAC PC (GE Healthcare). Three or more consecutive cardiac cycles were averaged for all analyses.
Fasting blood glucose and glucose tolerance test
Fasting blood glucose levels were measured in all mice after fasting for 16 h, once every two weeks during the treatment. Briefly, 2 μL of blood from the tail vein was collected via tail clipping and measured directly using a glucose meter (ACCU-CHEK; Roche, USA) with test strips. For the intraperitoneal glucose tolerance test (IPGTT), mice were fasted for 16 h prior to intraperitoneal injection of a 20% D-glucose solution. Blood glucose levels were determined before the injection (time 0) and at different time points after injection (30, 60, 90, 120, and 150 min).
Blood sample collection and biochemical analyses
At the end of the study, the mice were anesthetized by inhalation of 5% isoflurane. Fasting blood specimens were collected from the heart in commercial tubes containing K3EDTA as an anticoagulant, and tubes without anticoagulants were used to collect the serum by centrifugation for 5 min at 1000 × g. The plasma and serum were stored at −20 °C until analysis. HbA1c, insulin, fructosamine, creatine kinase-MB (CK-MB), total cholesterol (TC), triglycerides (TGs), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), total lipid, and myoglobin levels were determined.
Determination of TG levels in cardiac tissue
Heart samples were washed in phosphate-buffered saline (PBS) and homogenized in 1 mL of 5% Nonidet P 40 substitute solution (74385, Sigma‒Aldrich, Korea). The samples were heated at 80 °C for 3 min and then cooled to ~25 °C. The samples were then centrifuged for 3 min at 5000 × g. TG levels were determined using a triglyceride quantification kit (MAK266, Sigma-Aldrich, Korea) according to the manufacturer’s instructions.
Histological analyses
The mouse hearts were excised, washed with PBS, and fixed in 10% paraformaldehyde. The hearts were then sent to Histoire (Ansan, Korea) for staining. To measure the cross-sectional and total area of LV cardiomyocytes, sections were stained with hematoxylin and eosin (HE) and photographed using a NanoZoomer Digital Slide Scanner (Hamamatsu, Japan). Each section was quantitatively measured using ImageJ 1.48 software (NIH, MD). For assessment of myocardial fibrosis, sections were stained by Masson’s trichrome method and photographed using NanoZoomer Digital Slide Scanners (Hamamatsu Photonics, Hamamatsu, Japan). The relative fibrotic area (% of the total area) was measured quantitatively using ImageJ 1.48 software.
Electron microscopy analysis of in situ cardiac mitochondria
The dissected heart fragments (1 mm3) from experimental animals were fixed in 2.5% glutaraldehyde in PBS solution at 4 °C overnight and then with 1% osmium tetroxide in PBS for 2 h. The tissues were washed, dehydrated, and embedded, from which semithin sections were cut (0.5–1 µm). Ultrasectioning (60–90 nm) was performed, and the slices were double-stained with uranyl acetate and lead citrate and imaged using a JEM 1200 EX2 electron microscope (Jeol, Japan). The developed images were scanned on a flatbed scanner (Umax PowerLook 1100; Fremont, CA, USA) and analyzed using ImageJ software.
Permeabilized left ventricle tissue oxygen consumption
Oxygen consumption was determined polarographically using a fiber optic oxygen monitor (Instech, Plymouth Meeting, PA, USA). The cardiac fibers were separated in a small Petri dish filled with buffer (7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM DTT, 20 mM taurine, 5.7 mM ATP, 14.3 mM PCr, 6.56 mM MES, with 100 µL of 0.5 M EGTA; pH 7.1) on top of a frozen block under the dissecting scope. Next, ~2–3 mg (wet weight) of the fiber bundles was weighed. They were placed in saponin tubes for 30 min on a rotating tray in the refrigerator and then in washing buffer (105 mM K-MES, 30 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, 0.5 mg/mL BSA, pH 7.1) for 15 min. Next, 2.1 mL of assay buffer was added to each chamber. Oxygen consumption was measured in an air-saturated (220 nmol O2/mL) respiration medium. The state 4 respiratory rate was determined in the presence of 5 mM malate, 5 mM glutamate, and 5 mM succinate with rotenone as the respiratory substrate for complex I and complex II. The state 3 (active) respiration rate was determined in the presence of 0.1 mM ADP. Oxygen uptake was expressed as ng-at O/min mg protein. Data were acquired at a sample rate of 100 ms and analyzed using the OOIsensors program (Ocean Optic Inc., FL, USA). The respiratory control ratio (RCR) was calculated as the ratio of the state 3/state 4 oxygen consumption rates.
Western blotting analysis
Total protein was extracted from WT and db/db cardiac tissues using RIPA lysis buffer containing a protease and phosphatase inhibitor cocktail (Thermo Fisher, Waltham, MA, USA). The extracted proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. After blocking with 5% skim milk in TBS with 0.1% Tween 20, membranes were incubated with the following primary antibodies overnight at 4 °C: primary antibodies were purchased from Cell Signaling (TGF-β1, PGC1a, NRF1, NRF2, total OXPHOS, ACC, p-ACC Ser79, ACSL1, ATGL, FOXO1, p-FOXO1 Ser256, AMPK, p-AMPK Thr172, Akt, p-Akt Ser473, mTOR, p-mTOR Ser2448, PPARγ1/2, NF-κB p65, β-Actin, and GAPDH); Abcam (GLUT4, Col1a1, and TFAM); Invitrogen (FABP3, IGFBP7, and p-SREBP1 Ser338); and Santa Cruz Biotechnology (CD36, SREBP1, MFN1, OPA1, and DRP1). After washing, the membranes were incubated with the HRP–conjugated secondary antibody (Jackson ImmunoResearch, USA), diluted in 5% skim milk, and incubated for 1 h at room temperature. Finally, the membranes were washed with Tris-buffered saline (TBS) containing 0.1% Tween 20. Immunodetection was performed using an enhanced luminol-based chemiluminescent substrate (WESTSAVE Up, AbFrontier, Korea). GAPDH and β-Actin were used as loading controls. Quantification of each band was performed using ImageJ software.
Quantitative real-time PCR
Total RNA from WT and db/db mouse cardiac tissues was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA). Then, 1.5 µg of RNA was reverse-transcribed to cDNA using a Revert Aid First Strand cDNA Synthesis kit (Thermo Fisher) following the manufacturer’s protocol. The primers were obtained from Cosmo Genetech (Korea), and their sequences are listed in Supplementary Table 1. SYBR premix Ex Taq (Takara, Shiga, Japan) was used to perform real-time PCR with reactions prepared according to the manufacturer’s protocol (Bio-Rad, Hercules, CA, USA). All reactions were performed in triplicate. cDNA was amplified by 45 cycles using the following settings: 15 s at 95 °C, 30 s at 58 °C, and 30 s at 72 °C. The analysis was carried out using CFX Manager™ software (Bio-Rad, Hercules, CA, USA) and Microsoft Excel. The relative RNA levels were normalized to those of GAPDH. Primer sequences used for PCR are listed in Supplementary Table 1.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 8.0.1 software (San Diego, CA, USA). All data are expressed as the mean ± standard deviation (SD) or standard error (SE). Unpaired Student’s t test was used for comparisons between two groups, and one- or two-way analysis of variance (ANOVA) and post hoc Tukey’s test were used for multiple comparisons. Statistical significance was set at p < 0.05.
Results
Effects of EVO on body weight, blood glucose, HbA1c levels, food intake, and biochemical characteristics of diabetic mice
Body weight was measured every week, food intake was measured three times per week, and fasting blood glucose levels were determined before and after every two weeks of treatment. After 12 weeks of treatment, IPGTT was performed on the mice before sacrifice. Blood samples were collected, and biochemical features were analyzed (Supplementary Fig. 1). Comparisons of various physical and biochemical parameters among the three groups of mice are shown in Fig. 1 and Supplementary Table 2. The results showed that there was no significant difference in body weight and food intake between db/db and db/db+EVO mice (Fig. 1B, C). Eight-week-old db/db mice exhibited higher blood glucose levels than WT mice of the same age (p < 0.05; Fig. 1D). EVO effectively decreased fasting blood glucose levels in db/db mice after two weeks of treatment (Fig. 1D) and improved glucose homeostasis (IPGTT results) after 12 weeks of treatment (Fig. 1E). EVO treatment also significantly reduced HbA1c levels in db/db mice (p < 0.05; Fig. 1F). In addition, fructosamine, CK-MB, cholesterol, TG, total lipid, and myoglobin levels were slightly decreased in db/db+EVO mice compared with db/db mice (p > 0.05; Supplemental Table 2). The results demonstrated that EVO had no effect on body weight and food intake in db/db mice but significantly decreased glucose and HbA1c levels.

A Morphology of WT, db/db, and db/db+EVO mice. B Weekly body weight (n = 10/group). C Food intake and D fasting blood glucose level (n = 10). E Glucose tolerance test after 12 weeks of EVO treatment (n = 10/group). F HbA1c levels (n = 6). Data are presented as the mean ± SE; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns not significant.
EVO treatment improved systolic and diastolic function in db/db mice
Echocardiographic data demonstrated that db/db mice exhibited a significant decrease in both systolic and diastolic function compared with that of WT mice (Fig. 2A), while db/db+EVO mice showed a remarkable improvement in EF and FS after 12 weeks of treatment (Fig. 2B, C). Doppler flow analysis revealed a decrease in the E/A and e’/a’ ratios as well as an increase in the E/e’ ratio, indicating that LV diastolic function was notably impaired in db/db mice, while EVO significantly reversed these diastolic function parameters after 12 weeks of treatment (p < 0.05, p < 0.01, and p < 0.05, respectively; Fig. 2D–F). In addition, DT was significantly increased in the db/db group compared to the WT group (p < 0.01; Fig. 2G), but EVO treatment resulted in an attenuation of DT compared to the WT group. Collectively, db/db mice developed marked LV systolic and diastolic dysfunction, but these were significantly reversed by EVO treatment.

A Representative 2-D, M-Mode, and Doppler echocardiographic images of WT, db/db, and db/db+EVO mice. B Ejection fraction (EF%) and C fractional shortening (FS%). D Ratio of the velocities of early to late mitral flow (E/A). E Ratio of early diastolic myocardial relaxation to active atrial contraction in late diastole (e’/a’). F E/e’ ratio and G deceleration time (DT). Data are presented as the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10–20/group, ns not significant.
EVO treatment reduced cardiac hypertrophy and fibrosis in db/db mice
The hearts of db/db mice were clearly larger, and the heart weight to tibia length ratio was higher than those in WT mice (p < 0.0001; Fig. 3A, B). In addition, cardiomyocytes were clearly striated and regularly arrayed in WT mice, while myocardial fibers were disordered and myocyte cross-sectional area was increased in db/db mice, as shown by HE staining (p < 0.001; Fig. 3A, C). However, these features were also attenuated in the hearts of EVO-treated db/db mice (p < 0.05; Fig. 3B, C). Moreover, Masson’s staining showed that the interstitial fibrosis and accumulation of collagen fibers were increased in the hearts of db/db mice compared with those in WT mice (p < 0.001; Fig. 3A, D), and these parameters were significantly attenuated after 12 weeks of EVO treatment. To further verify these cardiac changes, we detected the protein expression of Col1a1 and TGF-β1 using western blotting analysis. The results showed that the expression of these proteins was increased in the hearts of db/db mice and was significantly attenuated by EVO treatment (p < 0.05; Fig. 3E–G). Additionally, insulin-like growth factor binding protein-7 (IGFBP7) has been defined as a biomarker of cardiac fibrosis24, and it has been reported that the expression of IGFBP7 is upregulated by TGF-β25. We found that IGFBP7 expression was elevated in db/db hearts and was significantly reduced in EVO-treated mouse hearts (p < 0.05; Fig. 3E, H). Moreover, NF-κB expression, which is downregulated by PGC1α and plays an important role in inflammation and cardiac hypertrophy, was attenuated by EVO treatment (p < 0.05; Fig. 3E, I). In summary, these results demonstrated that EVO treatment inhibited cardiac hypertrophy and fibrosis in db/db mice.

A Representative images of whole hearts and tibias from mice in different groups and hematoxylin and eosin- and Masson-stained heart sections. Magnification ×400. The scale bar = 100 µm. B The ratio of body weight/tibia length in different weeks of treatment (n = 10/group). C Myocyte cross-sectional area (µm2). D Interstitial fibrosis. E–I Protein expression and quantitative analysis of Cola1, TGF-β1, IGFBP7, and NF-κB in the hearts of mice as determined by western blotting. Data are presented as the mean ± SE. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 (n = 6/group).
EVO treatment attenuated myocardial mitochondrial injury and improved mitochondrial function in db/db mice
Transmission electron microscopy revealed clear evidence of mitochondrial injury. Representative images of the cardiac mitochondrial ultrastructure are shown in Fig. 4A. The results showed that EVO attenuated myocardial mitochondrial injury and the accumulation of lipid droplets, which contributed to decreased cardiac lipotoxicity in db/db mice (p < 0.01 and p < 0.05, respectively; Fig. 4B, C). The mitochondrial number and area were significantly decreased in diabetic cardiomyocytes compared with those in WT mice, but they were slightly increased in the db/db+EVO group (Fig. 4D and Supplementary Fig. 2c). There was no significant difference in mitochondrial size between the db/db and db/db+EVO groups (Fig. 4E). In addition, EVO increased the protein levels of complexes I, II, III, and IV in the electron transport chain of the mitochondria and enhanced the mitochondrial oxygen consumption rate in db/db mice (Fig. 4F, G, I). On the other hand, the PGC1α/NRFs/TFAM signaling pathway plays an important role in mitochondrial biogenesis26, and we next determined whether it was affected by EVO treatment. The present study showed that EVO treatment significantly prevented the impaired expression of PGC1α, NRF1, and TFAM in the hearts of db/db mice (p < 0.05; Fig. 4H, J–M). However, there was no significant difference in NRF2 expression between the db/db and db/db+EVO hearts (p > 0.05; Fig. 4L), and AMPK phosphorylation, the major upregulated factor upon PGC1α posttranslation modification, was not changed in EVO-treated hearts compared with db/db hearts (p > 0.05; Supplementary Fig. 2a, b). Additionally, we assessed mitochondrial dynamics, which is controlled mainly by the two opposing processes of fission and fusion. The results showed that the markers for fusion, namely, mitofusin 1 (MFN1) and optic atrophy protein 1 (OPA1), were slightly augmented in hearts from db/db mice treated with EVO compared with the db/db control group. In contrast, the marker for fission, that is, dynamin-related protein 1 (DRP1), was slightly reduced. However, there were no significant differences in these characteristics between the db/db and db/db+EVO hearts (p > 0.05; Supplementary Fig. 2d, e–g). In summary, EVO can prevent mitochondrial damage and enhance mitochondrial function via the PGC1α/NRF1/TFAM signaling pathway in the hearts of db/db mice.

A Representative transmission electron microscopy images of left ventricular cardiac tissues, scale bar = 1 μm. B–E Lipid droplets, percentage of damaged mitochondria, and mitochondrial area and size (n = 4/group). F Saponin-permeabilized cardiac fiber oxygen consumption rate. G, H Protein expression of OXPHOS complexes I, II, III, PGC-1α, NRF1, NRF2, TFAM, and GAPDH in the hearts of mice after 12 weeks of treatment was determined by western blotting. I–M Quantitative analysis of these proteins. Data are presented as the mean ± SE. *p < 0.05, **p < 0.01, ns not significant (n = 6/group).
EVO treatment alleviates cardiac lipotoxicity in db/db mice
Next, the lipotoxic status of the hearts was evaluated. Cardiac TG levels were significantly increased in the myocardium of db/db mice compared to WT and db/db+EVO mice (p < 0.0001 and p < 0.01, respectively; Fig. 5A). EVO was also shown to effectively reduce the accumulation of lipid droplets in db/db hearts, as shown in Fig. 4A and Fig. 4B. This means that EVO reduces lipid accumulation in T2DM hearts, leading to the amelioration of cardiac lipotoxicity. A wide variety of metabolic pathways and target proteins are involved in the regulation of cardiomyocyte lipid levels and their signaling, which have, in turn, been implicated in the development of lipotoxic cardiomyopathy12. The western blotting results showed that CD36, a major cardiac FA transporter, was significantly enhanced in the hearts of T2DM db/db mice compared with those of WT and db/db+EVO mice (p < 0.01 and p < 0.05, respectively; Fig. 5B, C). Overexpression of CD36 in the cardiomyocytes of diabetic db/db mice promotes CD36-mediated long-chain FA uptake and TG accumulation, inducing diabetes-mediated cardiac lipotoxicity and subsequent contractile dysfunction12. Additionally, EVO treatment significantly reduced ACSL1 expression (Fig. 5B, D). Moreover, we found that the expression of fatty acid binding protein 3 (FABP3), a chaperone for lipids and a biomarker for heart injury, was obviously increased in the myocardium of db/db mice compared with the db/m group, while EVO significantly reduced its expression (p < 0.01; Fig. 5B, E). Our study further showed that EVO treatment decreased PPARγ expression in db/db hearts but did not affect PPARα gene expression (Fig. 5B, F, G). In addition, the results of real-time PCR indicated a statistically significant increase in the mRNA expression of diacylglycerol O-acyltransferase 1 (DGAT1) in db/db mice (p < 0.05; Fig. 5H), but there was no difference in DGAT2 expression among the groups (p > 0.05, Fig. 5H). However, ACC phosphorylation was significantly decreased in diabetic cardiomyocytes compared with that in WT cardiomyocytes (p < 0.05; Supplementary Fig. 3a, b), while EVO slightly increased ACC phosphorylation. No significant differences in ATGL expression were observed among the groups (p > 0.05; Supplementary Fig. 3a, c). Because of the shift in cardiac substrate utilization from glucose to FAs and concomitant FA overload, which is also a consequence of cardiac lipotoxicity, we examined GLUT4 expression and found that EVO tended to increase the expression of GLUT4 (Supplementary Fig. 3a, d, e).

A Cardiac triglyceride level. B Protein expression of CD36, ACSL1, FABP3, PPARγ1/2, and GAPDH using western blotting analysis. C–F Quantitative analysis of these proteins. G, H mRNA expression of PPARα, PPARγ, DGAT1, and DGAT2 in the hearts was determined by RT‒PCR. I Protein expression of FOXO1 (total and phosphorylated). J, K Quantitative analysis of FOXO1 by western blotting and RT‒PCR. Data are presented as the mean ± SE. *p < 0.05, **p < 0.01, ****p < 0.0001, ns not significant (n = 6/group).
Because forkhead box protein O 1 (FOXO1) activation recruits the FA translocase CD36 to the plasma membrane and increases FA uptake and oxidation27, we further explored whether EVO markedly decreased the lipid content via FOXO1 activity. The results showed that EVO reduced FOXO1 gene expression and inhibited FOXO1 activation by increasing its phosphorylation (p < 0.05 and p < 0.01, respectively; Fig. 5I–K).
On the other hand, we found that EVO did not regulate insulin-mediated signaling through the SREBP-1c gene, which is involved in lipogenesis in the liver and kidney. Although SREBP-1c expression was significantly increased in db/db mice compared with db/m mice (Supplementary Fig. 4a, b, e), there was no significant difference in this signal between db/db mice and EVO-treated db/db mice (Supplementary Fig. 4c, d–f). However, it tended to be attenuated in EVO-treated db/db hearts (Supplementary Fig. 4d).
Collectively, we suggest that changes in lipid metabolism in the hearts of db/db EVO-treated mice are cardiac-specific effects of EVO rather than systemic effects.
To confirm this statement and further explore whether the improvement in lipotoxicity following EVO treatment was mainly due to the decrease in lipid influx, inhibition of cardiac lipid synthesis, or increase in efflux, RNA sequencing was conducted. A multidimensional scaling (MDS) analysis showed that the gene expression changes among the WT, db/db, and EVO treatment groups were clearly distinguished (Supplementary Fig. 5a). In addition, these groups expressed quantitative differences in gene expression associated with cardiomyopathy (Supplementary Fig. 5b). Importantly, through GO analysis, we determined that EVO significantly changed biological processes related to lipid metabolism in the hearts of db/db mice (Fig. 6A and Supplementary Fig. 5c). Furthermore, 24 genes associated with cellular lipid metabolism were enriched in the groups. It was confirmed that EVO changed the expression patterns of these genes based on the heatmap and volcano plots (Fig. 6B, C). In the EVO target protein map created through STRING protein network analysis, major genes related to lipid metabolism were located in the center of the entire network. This finding supports our suggestion that the major mechanism of the cardioprotective effect of EVO is through the regulation of lipid metabolism (Supplementary Fig. 5d).

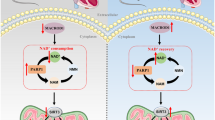
A Based on GO analysis, EVO significantly changed biological processes related to lipid metabolism in the hearts of db/db mice. B Heatmap analysis of the change patterns of 24 genes related to cellular lipid metabolism in the WT, Con, and EVO groups. C Volcano plots of the EVO vs. Con group and changes in the gene expression patterns of 24 genes related to cellular lipid metabolism. D Graphical abstract – EVO ameliorated cardiac lipotoxicity via the inhibition of overactivated lipid signaling pathways in diabetic cardiomyocytes and improved mitochondrial function via the PGC1α/NRF1/TFAM signaling pathway.
In summary, these results indicate that EVO prevented the increase in cardiac TG levels and FA transport and inhibited the overactivation of lipid signaling pathways in diabetic cardiomyocytes, thereby attenuating cardiac lipotoxicity in db/db mice (Supplementary Fig. 5d).
Discussion
In this study, we showed the cardioprotective effects of the DPP4 inhibitor EVO in diabetic db/db mice. EVO attenuated both systolic and diastolic dysfunction, cardiac hypertrophy, and fibrosis, which are the main etiologies of DCM. In addition, through additional experiments, we confirmed that EVO reduced mitochondrial dysfunction and lipotoxicity by reducing fat accumulation in the myocardium or upregulating transcription factors such as NRF1 and PGC1.
Heart failure and related morbidity and mortality are major challenges in the treatment of T2DM. Although hyperglycemia, insulin resistance, and impaired cardiac insulin metabolic signaling are associated with DCM, the pathogenesis of DCM remains complex, and there is no specific treatment to date10,28. Since the 2008 guidance document by the US Food and Drug Administration (FDA), all new glucose-lowering agents must demonstrate cardiovascular (CV) safety29. Therefore, a wide range of clinical trials have addressed the relationship between strict glycemic control and CV endpoints. Among them, sodium-glucose cotransporter type 2 (SGLT-2) and DPP-4 inhibitors are promising drugs for reducing the morbidity of CV diseases related to diabetes, as they have positive data published related to CV outcomes. In particular, DPP-4 inhibitors are not associated with hypoglycemia or weight gain and have a good safety profile; therefore, their role as a substitute for sulfonylurea is becoming important30. In addition, they can be prescribed to patients with moderate to severe chronic kidney disease.
Beyond the glucose-lowering effect, DPP-4 inhibitors positively affect vascular endpoints and other CV events31,32. Previous studies have explained the possible mechanism of the beneficial effects on body weight, blood pressure (without an increase in heart rate), postprandial lipemia, inflammatory markers, oxidative stress, and endothelial function in patients with T2DM17,33. However, cellular-level studies on the myocardium or mitochondria are lacking.
DPP-4 is widely distributed throughout most tissues of the body and acts as a plasma membrane enzyme34. It regulates postprandial glucose by rapidly inactivating GLP-1 and GIP35. Therefore, DPP4 inhibitors stimulate insulin secretion in a glucose-dependent manner and reduce glucagon secretion from the pancreas by enhancing the endogenous bioactive hormones, incretin, GLP-1, and GIP, which are responsible for the antihyperglycemic effect35,36.
Evogliptin, a new DPP-4 inhibitor developed by Dong-A company, was first approved as an oral antihyperglycemic drug for the treatment of T2DM. Several studies have determined the influence of EVO on liver gluconeogenesis, hepatic steatosis, vascular inflammation, and whole-body composition21,22,23,37. In addition, EVO directly induces fat loss through an increase in Ppargc1α in white adipose tissue in obese mice23, inhibits vascular inflammation via modulation of Sirt1/NF-κB in atherosclerosis in ApoE−/− mice21, and ameliorates fatty liver by increasing insulin sensitivity22. However, limited information is available regarding the direct effects of DPP4 inhibition with EVO on DCM. To explore these issues, the effects of EVO-induced DPP4 inhibition on the DCM model in db/db mice were investigated. We found that EVO treatment reduced blood glucose and HbA1c levels and improved glucose tolerance in db/db mice, consistent with the results of other studies37,38,39. However, in our study, EVO did not influence body weight gain, which differs from previous publications that reported that EVO decreased20 or increased body weight40. This difference may be due to the distinct research subjects, the age of the mice, or the dose of EVO. EVO treatment did not produce any significant changes in serum cholesterol, TG, HDL, LDL, and total lipid levels, which are indicators of systemic lipid metabolism in db/db mice. This finding indicates that EVO particularly impacts cardiac lipid metabolism and that these effects are not systemic.
In addition, although the early stage of DCM exhibited diastolic dysfunction, we found that EVO treatment improved both systolic and diastolic function by analyzing echocardiography data in db/db mice, including a significant increase in EF and FS, as well as the E/A and e’/a’ ratios, and a reduction in the E/e’ ratio compared with those of nontreated db/db mice.
Cardiomyopathies are characterized by cardiac hypertrophy, which can contribute to ventricular dysfunction in HF41. Myocardial fibrosis occurs as a result of the excessive accumulation of extracellular matrix proteins in the heart and type 1 collagen, mediated by TGFβ1, a well-known profibrotic factor42. This accumulation can worsen ventricular stiffness, leading to cardiac dysfunction. In the present study, EVO treatment reduced the heart weight/tibia length, myocyte cross-sectional area, interstitial fibrosis, and molecular markers of fibrosis, such as collagen 1 and TGFβ1 (Fig. 3). Recently, studies of chronic HF have shown that higher levels of IGFBP7 are associated with diabetes mellitus and obesity43. IGFBP7 has been defined as a biomarker of myocardial fibrosis and was shown to be upregulated by TGF-β25. In the present study, cardiac IGFBP7 expression was significantly decreased after EVO treatment.
Mitochondria are considered the powerhouses of cells. To maintain continuous pump function, the heart requires large amounts of high-energy phosphates and accounts for approximately 8% of the total ATP consumption of the body. Recent studies have implicated mitochondrial injury as a key factor in the pathophysiology of DCM28. Improvement in mitochondrial function is a target for potential therapeutic strategies in many diseases. Furthermore, the transcription factors NRF1 and NRF2 are master regulators of antioxidant and stress responses44,45. The PGC1a/NRFs/TFAM signaling pathway plays an important role in mitochondrial function26. The current study demonstrated that EVO treatment attenuated myocardial mitochondrial injury and enhanced the oxygen consumption rate in db/db mice, as well as elevated the expression of complexes I, II, III, and IV in the electron transport chain of mitochondria. Although there was no significant difference in NRF2 expression and AMPK phosphorylation between the db/db and db/db+EVO groups, EVO treatment significantly increased PGC1α, NRF1, and TFAM expression in the hearts of db/db+EVO mice compared to their expression in those of db/db mice. These results indicate that EVO enhanced mitochondrial function in db/db mice via PGC1α/NRF1/TFAM. In contrast to our findings, AMPK phosphorylation was increased in db/db hearts and decreased in those of sitagliptin (another DPP-4 inhibitor)-treated db/db mice at 10 weeks old, as shown in the study by M. Lenski et al.46. This difference could be due to the age of the animals and the fact that the db/db mice belonged to distinct stages of DCM development. AMPK is defined as the main upstream activator of PGC1α and plays an important role in the regulation of fatty acid oxidation and mitochondrial biogenesis47. However, the mechanism by which EVO induces the activation of AMPK or other upstream activators of PGC1α remains unknown, and further studies are needed.
Additionally, the p65 subunit of NF-κB can bind to PGC1α and is thereby associated with cardiac hypertrophy, inflammation, and metabolic disturbances in cardiac cells48,49. This study also confirmed that EVO attenuated cardiac NF-κB expression in db/db mice.
Next, we evaluated the lipotoxicity status underlying the effects of EVO in the mouse heart. In DCM, decreased glucose and increased FA utilization lead to lipid accumulation, resulting in lipotoxicity, oxidative stress, and mitochondrial dysfunction. The accumulation of lipids in the cardiac muscle in obesity and diabetes is accompanied by accelerated myocardial FA oxidation rates. Studies have shown that diet-induced obese mice and diabetic rats and insulin-resistant ob/ob and db/db mice have increased myocardial FA oxidation rates in parallel with reduced cardiac efficiency50,51. Likewise, we observed that lipid accumulation was increased in the hearts of diabetic mice, as shown by the enhanced cardiac lipid droplets and TG levels (Figs. 4A, B and 5A). Three proteins, namely, CD36, FATP4, and FABP3, play critical roles in the transmembrane trafficking of FAs in cardiomyocytes52. Among these, CD36-dependent mechanisms are important in the development of insulin resistance and, subsequently, in the pathogenesis of DCM53. FABP3 serves as an intracellular lipid chaperone that is critical for maintaining the homeostatic function of cardiac muscle54. The present study showed that CD36 and FABP3 were significantly increased in the hearts of db/db mice compared to WT mice, while EVO treatment significantly alleviated this increase after 12 weeks of treatment. EVO treatment also significantly reduced ACSL1 expression, which is considered a building block in the synthesis of TG and other complex lipids. Moreover, previous studies have demonstrated that mice with cardiac-specific overexpression of PPARα or PPARγ, the transcription factors involved in regulating lipid metabolism, exhibited a phenotype similar to that of DCM55,56. Our results showed that PPARγ mRNA expression was increased in db/db mouse hearts but was significantly decreased in db/db mice treated with EVO. In contrast, DGAT1, a downstream target of PPARs that contributes to the final step in TG synthesis, was attenuated by EVO treatment. Furthermore, PPARs are ligand-activated transcription factors associated with FA uptake and oxidation; increased PPAR expression can activate several target genes, such as FABP3, CD36, and FATP4, thereby increasing these processes57. PPARs in turn are upregulated by PGC1α. Additionally, PPARγ may be a direct target gene of SREBP-1c, a transcription factor that controls lipogenesis. Increased SREBP-1c leads to augmented PPARγ, resulting in an increase in lipogenesis. However, in the present study, EVO slightly attenuated SREBP-1c gene expression in db/db hearts, but there was no effect in the db/db liver and kidney.
A recent study suggested that impaired insulin signaling activates FOXO1 and increases PPARα expression in cardiomyocytes, causing oxidative stress and DCM58. Our results showed that EVO reduced FOXO1 gene expression and inhibited FOXO1 activation by increasing its phosphorylation. However, it did not affect PPARα mRNA expression. Previous studies indicated that CD36-mediated fatty acid uptake upregulated the activity of FOXO127.
The RNA sequencing results showed that EVO significantly altered the biological processes related to lipid metabolism in the hearts of db/db mice. Among 24 detected genes that were related to cellular lipid metabolism and enriched in groups were CD36, FABP3, DGAT1, PPARγ, and FOXO1, which are described above. These results further confirm that the improvement in lipotoxicity by EVO was mainly due to the decrease in cardiac lipid synthesis and accumulation in db/db mice.
Taken together, our findings suggest that DCM is related to lipotoxicity and that EVO ameliorates cardiac lipotoxicity via the inhibition of overactivated lipid signaling pathways in diabetic cardiomyocytes. In addition, EVO preserves mitochondrial function by increasing the activity of the PGC1α/NRF1/TFAM signaling pathway (Fig. 6D) and inhibits the development of cardiac hypertrophy and fibrosis by attenuating TGFβ/IGFBP7/Col1a1 expression. However, this potential mechanism requires further investigation.
This is the first study to demonstrate that DPP-4 inhibition by evogliptin prevents diabetic cardiomyopathy by reducing cardiac lipotoxicity, mitochondrial damage, and fibrosis in db/db mice. The present study suggests that inhibiting fatty acid utilization via DPP-4 inhibition with evogliptin is a promising strategy to elevate cardiac efficiency in DCM.
References
Galicia-Garcia, U. et al. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 21, 62–75 (2020).
Saeedi, P. et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9(th). Diabetes Res. Clin. Pract. 157, 107843 (2019).
Dal Canto, E. et al. Diabetes as a cardiovascular risk factor: an overview of global trends of macro and micro vascular complications. Eur. J. Prev. Cardiol. 26, 25–32 (2019).
Boudina, S. & Abel, E. D. Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 11, 31–39 (2010).
Paolillo, S. et al. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart. Fail. Clin. 15, 341–347 (2019).
Seferovic, P. M. et al. Type 2 diabetes mellitus and heart failure: a position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart. Fail. 20, 853–872 (2018).
Parim, B., Sathibabu Uddandrao, V. V. & Saravanan, G. Diabetic cardiomyopathy: molecular mechanisms, detrimental effects of conventional treatment, and beneficial effects of natural therapy. Heart. Fail. Rev. 24, 279–299 (2019).
Marwick, T. H., Ritchie, R., Shaw, J. E. & Kaye, D. Implications of underlying mechanisms for the recognition and management of diabetic cardiomyopathy. J. Am. Coll Cardiol. 71, 339–351 (2018).
Jia, G., Hill, M. A. & Sowers, J. R. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res. 122, 624–638 (2018).
Russo, I. & Frangogiannis, N. G. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 90, 84–93 (2016).
Goldberg, I. J., Trent, C. M. & Schulze, P. C. Lipid metabolism and toxicity in the heart. Cell Metab. 15, 805–812 (2012).
D’Souza, K., Nzirorera, C. & Kienesberger, P. C. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim. Biophys. Acta 1861, 1513–1524 (2016).
Unger, R. H. & Orci, L. Lipoapoptosis: its mechanism and its diseases. Biochim. Biophys. Acta 1585, 202–212 (2002).
Mahajan, R., Lau, D. H. & Sanders, P. Impact of obesity on cardiac metabolism, fibrosis, and function. Trends. Cardiovasc. Med. 25, 119–126 (2015).
Elezaby, A. et al. Mitochondrial remodeling in mice with cardiomyocyte-specific lipid overload. J. Mol. Cell. Cardiol. 79, 275–283 (2015).
Schrauwen, P., Schrauwen-Hinderling, V., Hoeks, J. & Hesselink, M. K. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 1801, 266–271 (2010).
Ussher, J. R. & Drucker, D. J. Cardiovascular actions of incretin-based therapies. Circ. Res. 114, 1788–1803 (2014).
Matsubara, J. et al. Dipeptidyl peptidase-4 inhibitor, sitagliptin, improves endothelial dysfunction in association with its anti-inflammatory effects in patients with coronary artery disease and uncontrolled diabetes. Circ. J. 77, 1337–1344 (2013).
Aroor, A. R. et al. Dipeptidylpeptidase inhibition is associated with improvement in blood pressure and diastolic function in insulin-resistant male Zucker obese rats. Endocrinology 154, 2501–2513 (2013).
Kim, J. H. et al. Protective effects of evogliptin on steatohepatitis in high-fat-fed mice. Int. J. Mol. Sci. 21, 6743 (2020).
Nguyen, P. A., Won, J. S., Rahman, M. K., Bae, E. J. & Cho, M. K. Modulation of Sirt1/NF-kappaB interaction of evogliptin is attributed to inhibition of vascular inflammatory response leading to attenuation of atherosclerotic plaque formation. Biochem. Pharmacol. 168, 452–464 (2019).
Kim, M. K. et al. Prevention and treatment effect of evogliptin on hepatic steatosis in high-fat-fed animal models. Arch. Pharm. Res. 40, 268–281 (2017).
Chae, Y. N. et al. Beneficial effects of evogliptin, a novel dipeptidyl peptidase 4 inhibitor, on adiposity with increased Ppargc1a in white adipose tissue in obese mice. PLoS One 10, e0144064 (2015).
Kalayci, A. et al. Echocardiographic assessment of insulin-like growth factor binding protein-7 and early identification of acute heart failure. ESC Heart Fail 7, 1664–1675 (2020).
Ko, T. et al. Cardiac fibroblasts regulate the development of heart failure via Htra3-TGF-β-IGFBP7 axis. Nat. Commun. 13, 3275 (2022).
Gureev, A. P., Shaforostova, E. A. & Popov, V. N. Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1α signaling pathways. Front. Genet. 10, 435 (2019).
Nahlé, Z. et al. CD36-dependent regulation of muscle FoxO1 and PDK4 in the PPAR delta/beta-mediated adaptation to metabolic stress. J. Biol. Chem. 283, 14317–14326 (2008).
Verma, S. K., Garikipati, V. N. S. & Kishore, R. Mitochondrial dysfunction and its impact on diabetic heart. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1098–1105 (2017).
Goldfine, A. B. Assessing the cardiovascular safety of diabetes therapies. N. Engl. J. Med. 359, 1092–1095 (2008).
American Diabetes, A. 8. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2018. Diabetes Care 41, S73–S85 (2018).
Nauck, M. A., Meier, J. J., Cavender, M. A., Abd El Aziz, M. & Drucker, D. J. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation 136, 849–870 (2017).
Mulvihill, E. E. & Drucker, D. J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 35, 992–1019 (2014).
Scheen, A. J. Cardiovascular effects of gliptins. Nat. Rev. Cardiol. 10, 73–84 (2013).
Lambeir, A. M., Durinx, C., Scharpe, S. & De Meester, I. Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 40, 209–294 (2003).
Zhong, J., Maiseyeu, A., Davis, S. N. & Rajagopalan, S. DPP4 in cardiometabolic disease: recent insights from the laboratory and clinical trials of DPP4 inhibition. Circ. Res. 116, 1491–1504 (2015).
Scheen, A. J. Cardiovascular effects of new oral glucose-lowering agents: DPP-4 and SGLT-2 inhibitors. Circ. Res. 122, 1439–1459 (2018).
Kim, T. H. et al. Hepatic role in an early glucose-lowering effect by a novel dipeptidyl peptidase 4 inhibitor, evogliptin, in a rodent model of type 2 diabetes. Eur. J. Pharmacol. 771, 65–76 (2016).
Tan, X. & Hu, J. Evogliptin: a new dipeptidyl peptidase inhibitor for the treatment of type 2 diabetes. Expert Opin. Pharmacother. 17, 1285–1293 (2016).
Jung, C. H. et al. A randomized, double-blind, placebo-controlled, phase II clinical trial to investigate the efficacy and safety of oral DA-1229 in patients with type 2 diabetes mellitus who have inadequate glycaemic control with diet and exercise. Diabetes Metab. Res. Rev. 31, 295–306 (2015).
Kim, T. H., Lee, J. H., Chae, Y. N., Jung, I. H. & Kim, M. K. Additive effects of evogliptin in combination with pioglitazone on fasting glucose control through direct and indirect hepatic effects in diabetic mice. Eur. J. Pharmacol. 830, 95–104 (2018).
Bernardo, B. C., Weeks, K. L., Pretorius, L. & McMullen, J. R. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol. Ther. 128, 191–227 (2010).
Yue, Y., Meng, K., Pu, Y. & Zhang, X. Transforming growth factor beta (TGF-beta) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 133, 124–130 (2017).
Barroso, M. C. et al. Serum insulin-like growth factor-1 and its binding protein-7: potential novel biomarkers for heart failure with preserved ejection fraction. BMC Cardiovasc. Disord. 16, 199 (2016).
He, F., Ru, X. & Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 21, 4777 (2020).
Ohtsuji, M. et al. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 283, 33554–33562 (2008).
Lenski, M., Kazakov, A., Marx, N., Böhm, M. & Laufs, U. Effects of DPP-4 inhibition on cardiac metabolism and function in mice. J. Mol. Cell. Cardiol. 51, 906–918 (2011).
Rowe, G. C., Jiang, A. & Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 107, 825–838 (2010).
Alvarez-Guardia, D. et al. The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res 87, 449–458 (2010).
Choi, B. et al. Evogliptin suppresses calcific aortic valve disease by attenuating inflammation, fibrosis, and calcification. Cells 10, 57 (2021).
Buchanan, J. et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 146, 5341–5349 (2005).
Mazumder, P. K. et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 53, 2366–2374 (2004).
Chabowski, A., Górski, J., Glatz, J. F., JJ, P. L. & Bonen, A. Protein-mediated fatty acid uptake in the heart. Curr. Cardiol. Rev. 4, 12–21 (2008).
Zhang, Y. et al. Cardiospecific CD36 suppression by lentivirus-mediated RNA interference prevents cardiac hypertrophy and systolic dysfunction in high-fat-diet induced obese mice. Cardiovasc. Diabetol. 14, 69 (2015).
Zhuang, L. et al. FABP3 deficiency exacerbates metabolic derangement in cardiac hypertrophy and heart failure via PPARalpha pathway. Front. Cardiovasc. Med. 8, 722908 (2021).
Kvandová, M., Majzúnová, M. & Dovinová, I. The role of PPARgamma in cardiovascular diseases. Physiol. Res. 65, S343–s363 (2016).
Finck, B. N. et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Invest. 109, 121–130 (2002).
Liu, F. et al. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor α. Circulation 131, 795–804 (2015).
Kyriazis, I. D. et al. KLF5 is induced by FOXO1 and causes oxidative stress and diabetic cardiomyopathy. Circ. Res. 128, 335–357 (2021).
Acknowledgements
This work was supported by the National Research Foundation of Korea and the Ministry of Education of Korea [NRF-2020R1A4A1018943 and NRF-2018R1A2A3074998].
Author information
Authors and Affiliations
Contributions
Conceptualization: T.K.P., H.K.K., and J.C.W. Methodology, investigation and data analysis: T.K.P., T.H.T.N., J.M.Y., G.S.K., and H.R.Y. Summarized the work and writing—original draft preparation: T.K.P. and H.K.K. Writing—review and editing: T.K.P., H.K., and J.C.W. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
This study was supported by a research fund from the Dong-A ST R&D Center (2019).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pham, T.K., Nguyen, T.H.T., Yi, J.M. et al. Evogliptin, a DPP-4 inhibitor, prevents diabetic cardiomyopathy by alleviating cardiac lipotoxicity in db/db mice. Exp Mol Med 55, 767–778 (2023). https://doi.org/10.1038/s12276-023-00958-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00958-6
