
Abstract
PTEN is among the most commonly lost or mutated tumor suppressor genes in human cancer. PTEN, a bona fide lipid phosphatase that antagonizes the highly oncogenic PI3K-AKT-mTOR pathway, is considered a major dose-dependent tumor suppressor. Although PTEN function can be compromised by genetic mutations in inherited syndromes and cancers, posttranslational modifications of PTEN may also play key roles in the dynamic regulation of its function. Notably, deregulated ubiquitination and deubiquitination lead to detrimental impacts on PTEN levels and subcellular partitioning, promoting tumorigenesis. While PTEN can be targeted by HECT-type E3 ubiquitin ligases for nuclear import and proteasomal degradation, studies have shown that several deubiquitinating enzymes, including HAUSP/USP7, USP10, USP11, USP13, OTUD3 and Ataxin-3, can remove ubiquitin from ubiquitinated PTEN in cancer-specific contexts and thus reverse ubiquitination-mediated PTEN regulation. Researchers continue to reveal the precise molecular mechanisms by which cancer-specific deubiquitinases of PTEN regulate its roles in the pathobiology of cancer, and new methods of pharmacologically for modulating PTEN deubiquitinases are critical areas of investigation for cancer treatment and prevention. Here, we assess the mechanisms and functions of deubiquitination as a recently appreciated mode of PTEN regulation and review the link between deubiquitinases and PTEN reactivation and its implications for therapeutic strategies.
Similar content being viewed by others


Introduction
PTEN (phosphatase and tensin homolog deleted on chromosome 10) is one of the most frequently lost or mutated tumor suppressor genes in human cancer1,2. Its encoded protein, PTEN, negatively regulates the phosphoinositide 3-kinase (PI3K)−AKT−mammalian target of rapamycin (mTOR) signaling pathway through dephosphorylation of the plasma membrane lipid phosphoinositide-3,4,5-triphosphate. As a consequence, loss of PTEN function leads to potent derepression of the PI3K-AKT pathway, which stimulates cell survival, proliferation, energy metabolism, and architecture3,4. PTEN also shows protein phosphatase, specifically dephosphorylating tyrosine-, serine- and threonine-phosphorylated polypeptides in vitro5 and several different cellular substrates, including focal adhesion kinase (FAK), cAMP-responsive element-binding protein (CREB), tyrosine kinases SRC and PTK6, and insulin receptor substrate 1 (IRS1)6,7,8,9,10. Furthermore, phosphatase-independent activities (mostly scaffolding) of PTEN regulate many processes, such as DNA replication, DNA repair, genomic stabilizing events, and cell cycle progression, have also been identified11,12,13,14, implicating the noncanonical roles of PTEN in tumorigenesis. Germline heterozygous pathogenic mutations in PTEN have been described in a variety of rare syndromes with different clinical presentations that are collectively known as PTEN hamartoma tumor syndromes (PHTSs), which exhibit features of both benign and malignant tumors15. Many modeling efforts with Pten-knockout mice have demonstrated that PTEN functions in a haplo-insufficient manner16,17,18; paradoxically, when PTEN levels are nearly completely loss, a strong cellular senescence program is triggered19,20, which is a ‘fail-safe’ brake on tumor progression21. Notably, an analysis of a series of mouse models of hypomorphic Pten has revealed the tremendous functional consequences of a subtle reduction in PTEN protein levels22,23, which can promote cancer susceptibility and favor tumor progression. Additionally, increased PTEN levels in transgenic models result in viable mice displaying a tumor-resistant, anti-Warburg metabolic state24,25. Thus, PTEN plays a critical dose-dependent role in tumor suppression, and therefore, understanding the regulatory mechanisms that fine-tune PTEN activity has become a paramount therapeutic goal (Fig. 1).

PTEN function can be compromised via genetic disruption, which results in a stepwise loss of PTEN (50% or 100%). Posttranslational modifications, including ubiquitination and deubiquitination, of PTEN can fine-tune PTEN functionality via a continuum of tumor suppression. Notably the phenotypes acquired throughout the continuum of functional PTEN loss are differentially manifested depending on tissue type.
Although PTEN function can be compromised by genetic mutations in inherited syndromes and sporadic cancers, posttranslational modifications (PTMs) of PTEN can play key roles in the dynamic regulation of its activity and function. For example, phosphorylation of PTEN affects protein stability and activity26,27. PTEN can also be SUMOylated, which increases its nuclear retention, thereby supporting its nuclear function in DNA repair mechanisms11,28. Ubiquitination requires the concerted action of activating (E1)-conjugating (E2)-ligating (E3) enzymes29. HECT-type E3 ubiquitin ligases, including neuronal precursor cell-expressed developmentally downregulated 4-1 (NEDD4-1) and WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) and 2 (WWP2), have been shown to converge at ubiquitination-mediated PTEN regulation30; specifically, PTEN monoubiquination leads to either PTEN translocation to the nucleus or exosomal transport (by NEDD4-1)31,32, while PTEN polyubiquitination suppresses its stabilization (NEDD4-1 and WWP2)33,34 or dimerization and subsequent membrane recruitment (WWP1)25,35.
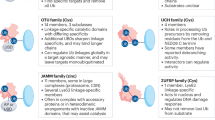
Increasing evidence has shown that deregulated deubiquitination leads to detrimental effects on PTEN levels and subcellular partitioning to promote tumorigenesis. Deubiquitinating enzymes (DUBs) are proteases that deconjugate ubiquitin from ubiquitinated substrates and thereby remodel polyubiquitin chains on target proteins to counteract the protein ubiquitination mediated by E3 ubiquitin ligases36. DUBs are categorized into two major classes, cysteine proteases and metalloproteases. The former class includes six main superfamilies36: ubiquitin-specific protease (USP), ubiquitin C-terminal hydrolase (UCH), ovarian tumor protease (OTU), Machado–Josephin domain (MJD) protease, and the recently discovered MINDY37 and ZUFSP38,39 families (Fig. 2). The JAB1/MPN/MOV34 (JAMM)-motif proteases bind to zinc and therefore are classified as metalloprotease-type DUBs40. Indeed, HAUSP/USP7, Ataxin-3, USP10, USP11, USP13, and OTUD3 have all been identified as PTEN DUBs; HAUSP specifically removes monoubiquitin from PTEN to promote its nuclear export41; Ataxin-3 restricts PTEN transcription42; and USP10, USP11, USP13 and OTUD3 increase PTEN stability in different cancer-specific contexts43,44,45. As ongoing research reveals the precise molecular mechanisms by which cancer-specific deubiquitination of PTEN regulates its roles in the pathobiology of cancer, the ability to pharmacologically modulate or otherwise counteract specific DUBs of PTEN, both selectively and in combination, is becoming a critical area of investigation for cancer prevention and treatment. In this review, we summarize the pathological and functional mechanisms of PTEN DUBs and describe how their functions dictate cancer cell biology and physiology while highlighting opportunities for therapeutic intervention.

Two classes of proteases (cysteine proteases and metalloproteases) are DUBs, with most DUBs cysteine proteases. Cysteine protease DUBs can be classified into six subfamilies based on their DUB domains: USP, UCH, OTU, MJD, MINDY, and ZUFSP. Metalloprotease DUBs include a JAMM DUB domain. USP, ubiquitin-specific protease; UCH, ubiquitin C-terminal hydrolase; OTU, ovarian tumor protease; MJD, Machado–Joseph disease protease; MINDY, motif interacting with Ub-containing novel DUB family; ZUFSP, zinc finger with UFM1-specific peptidase domain; JAMM, JAB1/MPN/Mov34 metalloenzyme.
Herpesvirus-associated ubiquitin-specific protease (HAUSP)/Ubiquitin-specific protease 7 (USP7)
HAUSP (also known as USP7) was first identified as a protein that binds herpes simplex virus E3, ubiquitin ligase ICP0, and Epstein‒Barr virus nuclear antigen 1 (EBNA1)46, indicating its relevance in key cellular processes important in viral infection. All USPs share a conserved catalytic core, while their unique substrate specificity is determined by various accessory substrate-binding domains tethered to a catalytic domain36 (Fig. 2). HAUSP contains an NH2-terminal tumor necrosis factor receptor-associated factor (TRAF)-like domain, a central catalytic core, and five ubiquitin-like (UBL) domains in the COOH terminus47. The TRAF-like domain in HAUSP can recognize the P/AxxS motifs shared by all TRAF-like domain-binding substrates, including EBNA1, the tumor suppressor p53, the ubiquitin E3 ligase MDM2 (mouse double minute 2), and the MDM2 homolog MDM448. In overexpression experiments, HAUSP has been shown to bind to, deubiquitinate, and stabilize p5349, whereas disruption of HAUSP expression in human cells and transgenic mice resulted in acquisition the opposite phenotypes, leading to stabilization and functional activation of p53 due to the destruction of MDM250,51, which suggests a dynamic role for HAUSP in the p53–MDM2 pathway. The C-terminal UBL domains can regulate the activation and specificity of HAUSP52 and function as additional platforms for substrate binding to highly basic motifs (R/KxKxxxK) within its substrates, including ICP0, UHRF1, DNMT1 and RNF16953,54,55.
HAUSP was identified as the first bona-fide PTEN deubiquitinase41 (Fig. 3) and can interact with PTEN both in vitro and in vivo. The domains of HAUSP critical for binding PTEN have not been determined, but the PTEN protein contains four P/AxxS motifs and an R/KxKxxxK motif; therefore, it will be interesting to identify the domain(s) of HAUSP that bind PTEN, the true PTEN sequence recognized by HAUSP, and the nature of their interactions. Although the E3 ligase NEDD4-1 (and/or additional E3s) monoubiquitinates PTEN at lysine residues 13 and 289 for its nuclear import32,56,57, HAUSP overexpression can lead to the deubiquitination of the monoubiquitinated PTEN protein and to subsequent nuclear exclusion of PTEN41. Notably, this phenotype is associated with more aggressive cancers, implying that when aberrantly expressed, HAUSP is an oncogene, functioning through its ability to disrupt PTEN function. Indeed, HAUSP is overexpressed and associated with unfavorable prognosis in many different types of human cancers, including brain, breast, cervical, lung, prostate, skin, stomach, and hemopoietic cancers41,54,58,59,60,61,62,63, and high HAUSP expression and PTEN nuclear exclusion are strongly and positively correlated in human cancers41,58,64,65. Intriguingly, various regulatory mechanisms in cancer influence the propensity of HAUSP to mediate PTEN deubiquitination. For example, in leukemias and prostate cancer, promyelocytic leukemia (PML) plays a critical regulatory role by inhibiting HAUSP activity through death domain-associated protein (DAXX), which in turn favors PTEN nuclear localization41. Similarly, nucleophosmin/B26 counteracts HAUSP-mediated deubiquitination and subsequent shuttling of PTEN to the cytoplasm65, supporting the notion that PTEN is delocalized in acute myeloid leukemia with mutated nucleophosmin (e.g., NPMc+). In contrast, BCR-ABL and thyroid hormone receptor-interacting protein 13 (TRIP13) enhance deubiquitination and nuclear exclusion of PTEN through activation of HAUSP in chronic myeloid leukemia and multiple myeloma, respectively66,67. These clinical and functional studies suggest that aberrant activation or overexpression of HAUSP may promote tumorigenesis, making HAUSP a target for therapeutic intervention in strategies to restore normal PTEN localization and tumor-suppressive function, as we discuss further below.

While HAUSP/USP7 induces deubiquitination and subsequent nuclear exclusion of monoubiquitinated PTEN in the nucleus, where it can control the cell cycle and genomic stability, PML-RARα, NPMc+, and BCR-ABL promote HAUSP-mediated PTEN deubiquitination in blood-borne cancers. USP11 plays a role in the maintenance of the effective levels of both nuclear and cytosolic PTEN for tumor suppression, and interestingly, its expression and activity are regulated by the PTEN/PI3K pathway. Furthermore, in the cytoplasm, USP11, USP13, and (acetylated) OTUD3 catalyze the removal of the K48-linked polyubiquitin chain on PTEN to enhance protein stability, whereas USP10 recognizes and removes the K63-linked polyubiquitin chain from PTEN, leading to PTEN recruitment to the plasma membrane. Ataxin-3 represses PTEN by inhibiting its transcription. PTEN phosphatase and tensin homolog deleted on chromosome 10, HAUSP herpesvirus-associated ubiquitin-specific protease, USP10 ubiquitin-specific protease 10, USP11 ubiquitin-specific protease 11, USP13 ubiquitin-specific protease 13, OTUD3 OTU deubiquitinase 3, PI3K phosphoinositide 3-kinase, PIP2 phosphoinositide-4,5-biphosphate, PIP3 phosphoinositide-3,4,5-triphosphate, mTOR mammalian target of rapamycin, PML promyelocytic leukemia, NPMc+ cytoplasmic nucleophosmin, TRIP13 thyroid hormone receptor-interacting protein 13.
Ubiquitin-specific protease 10 (USP10)
USP10 is a deubiquitinase involved in diverse cellular processes, including the DNA damage response, metabolic homeostasis, and ribosome recycling. Upon DNA damage, USP10 accumulates in the nucleus, where it is phosphorylated by ATM kinase, and subsequently deubiquitinates p5368. USP10 also interacts with deubiquitinates and enhances the activity of the master energy-sensor AMP-activated protein kinase-α (AMPKα)69. Furthermore, USP10 can deubiquitinase Beclin1, a key promoter of autophagy, and protect it from degradation, thus promoting autophagy70. Interestingly, Beclin1 also controls the protein stability of USP10 by regulating its deubiquitinating activity, forming a feedback loop. Similarly, USP10 prevents lysosomal degradation of 40 S subunits of ribosomes and ensures ribosome recycling associated with autophagy71. Given the importance of the energy balance and autophagy in metabolic disease, USP10 may represent a potential drug target for metabolic syndrome. Furthermore, the role of USP10 in cancer has recently been expanded to include deubiquitinase activity for PTEN. Indeed, USP10 restores the membrane localization and phosphatase activity of PTEN by reversing the tripartite motif-containing 25 (TRIM25)−mediated K63-linked polyubiquitination found in lung cancer72,73. Since USP10 is frequently downregulated in human cancers, including lung, gastric, colorectal, and small intestinal carcinomas74,75, restoration of USP10 function may represent a new therapeutic strategy for cancer prevention and treatment through PTEN reactivation.
Ubiquitin-specific protease 11 (USP11)
USP11 was originally identified as one of inherited X-linked retinal disorder genes at Xp11.2376, although a common deletion within the USP11 interval had been also found in ovarian cancer77. X-linked tumor suppressor genes are potentially significant to tumorigenesis because they can be functionally silenced by loss of heterozygosity or mutation of a single allele78, and skewed X inactivation may lead toward or against disease79. Interestingly, a recent and extensive review described USP11 as a predictive and prognostic factor in human cancers of various histologies80; indeed, USP11 is often repressed in brain, breast, skin, and prostate cancers but upregulated in colorectal and hepatocellular carcinomas80. As a deubiquitinase, USP11 interacts with multiple substrate proteins linked to cancer-related pathways. For example, USP11 recruits BRCA1 to chromatin by deubiquitinating PALB2 (partner and localizer of BRCA2) in a cell cycle-dependent manner or stabilizing MYCN in neuroblastoma81. Notably, USP11 has been found to be rapidly lost after DNA damage in a manner dependent on ATM/ATR induction. In brain tumors, USP11 deubiquitinates and stabilizes PML, and its transcription is inhibited by the Notch effector Hey182. USP11 can also regulate immune cell differentiation by deubiquitinating and stabilizing the NF-κB inhibitor IκB83. However, there was insufficient direct genetic evidence to determine the precise role of USP11 in tumorigenesis.
More recently, we found that while mice lacking Usp11 displayed increased susceptibility to PTEN-dependent tumor initiation, growth and metastasis, USP11 antagonized PI3K/AKT activity by reversing polyubiquitination and subsequently upregulating PTEN expression both in vitro and in vivo43, revealing it to be both an X-linked tumor suppressor and an important physiological PTEN deubiquitinase (Fig. 3). The downregulation of USP11 in breast, kidney, thyroid and prostate cancers is closely related to PTEN protein instability, regulating the occurrence and progression of these cancers, and is correlated with worsened prognosis43,84. USP11 also acts as an indicator of cell density, thereby controlling the physiological dose of PTEN. Furthermore, we discovered that PTEN-induced FOXO activation promotes USP11 transcription, which in turn stabilizes PTEN, suggesting that PTEN autoregulates itself through a PI3K-FOXO-USP11 feedforward loop to create a PTEN ‘integrated circuit’ that induces tumor suppression. Similarly, the activity of USP11 is regulated by phosphorylation mediated by PI3K-AKT-mTOR-S6 kinase signaling in diffuse large B-cell lymphoma85. Determining whether S6K-phosphorylated USP11 modulates PTEN levels will, however, require further investigation. Interestingly, both PTEN and USP11 participate in the homologous recombination (HR) DNA repair pathway12,86, and their deficiency results in hypersensitivity to PARP inhibition86,87. Further work is needed to precisely characterize the function/activity of the USP11-PTEN axis in DNA repair by the HR pathway to maintain genomic stability and suppress tumorigenesis.
Ubiquitin-specific protease 13 (USP13)
USP13 is localized adjacent to PIK3CA in the 3q26.3 locus, which is frequently amplified in human cancers such as brain, lung, ovarian, esophageal and cervical cancers, and high USP13 expression is correlated with poor survival outcomes88,89,90,91. In ovarian tumors, upregulation of USP13 enhances deubiquitination and stabilization of ACLY (ATP citrate lyase) and OGDH (oxoglutarate dehydrogenase), two key enzymes that drive glutaminolysis, ATP generation, and lipid synthesis in cancer metabolism89, and MCL1, a pivotal member of the antiapoptotic BCL-2 family of proteins91,92. As an oncogene, USP13 stabilizes c-Myc by antagonizing FBXL14-mediated ubiquitination to maintain glioma stem cell stemness, establish tumorigenic potential and promote cholangiocarcinoma nucleotide metabolism88,93. As previously mentioned, USP10 mediates the deubiquitination of Beclin1, and USP13 can directly regulate the deubiquitination of USP10 to promote the formation of autophagosomes70. USP13 can deubiquitinate RAP80 (receptor-associated protein 80) and promote the recruitment of the RAP80-BRCA1 complex to damage sites, fine-tuning the DNA repair system94. USP13 has also been identified as ERAD E3 ligase gp78-associated deubiquitinase for Ubl4A, a component of the ERAD chaperone complex, and thus promotes ER quality control95. Importantly, overexpression of Usp13 accelerates tumorigenesis, enhances tumor metastasis, and causes poor outcomes in transgenic mouse models of ovarian cancer96, underscoring its importance in promoting tumorigenesis in vivo. In addition to its oncogenic roles, USP13 exerts a tumor-suppressive role by deubiquitinating PTEN in different types of cancers. For example, overexpression of USP13 blocks the AKT signaling pathway and suppresses tumor cell proliferation, invasion, and glycolysis by upregulating PTEN, while USP13 levels are downregulated in breast, bladder, and oral squamous tumors, in correlation with PTEN levels45,97,98 (Fig. 3). These studies suggest that USP13 plays context-dependent oncogenic and tumor-suppressive roles and that up- or downregulation of USP13 and its target substrates/pathways can contribute to tumorigenesis.
OTU domain-containing protein 3 (OTUD3)
By regulating the deubiquitination of diverse key substrate proteins, the OTU (ovarian tumor protease) family member OTUD3 plays an important role in the processes of innate antiviral immunity, metabolism homeostasis, and tumorigenesis. OTUD3 has been identified as an acetylation-dependent deubiquitinase that restricts innate antiviral immune signaling99. Mechanistically, acetylation of the core OTU domain in OTUD3 markedly enhances deubiquitinase activity on MAVS (mitochondrial antiviral-signaling protein), thereby inhibiting the innate antiviral immune response. Upon viral infection, sirtuin 1 (SIRT1) is recruited to deacetylate OTUD3, leading to the inactivation of OTUD3, which relieves MAVS suppression. OTUD3 has also been recently implicated in the control of energy metabolism100. While OTUD3 regulates various genes involved in glucose and lipid metabolism by deubiquitinating and stabilizing peroxisome proliferator-activated receptor-delta (PPARδ), Otud3-deficient mice fed a high-fat diet developed greater obesity, dyslipidemia, and insulin resistance, suggesting that aberrant OTUD3 expression may be associated with obesity and a high risk of diabetes.
Emerging evidence has suggested cancer-associated functions of OTUD3 in multiple types of human cancer. For example, OTUD3 interacts with the ZFP36 ring finger protein through its OTU region and stabilizes it by inhibiting FBXW7-mediated ubiquitination, which in turn induces VEGF-C mRNA decay to prevent lymphatic metastasis of human esophageal cancer101. Furthermore, OTUD3 has been identified as a potent deubiquitinase for PTEN and thus a tumor suppressor in breast cancer (Fig. 3). OTUD3 (OTU region) directly interacts with PTEN (C2 domain), deubiquitinating and stabilizing the PTEN protein to suppress PI3K-AKT signaling44. OTUD3 transgenic mice exhibit higher PTEN expression and show a reduced tendency for breast cancer tumorigenesis. The reduction in OTUD3 expression, concomitant with decreased PTEN protein levels, correlates with breast cancer aggressiveness and poor prognosis. As the full activation of OTUD3 may require its acetylation99,100, it will be interesting to determine whether OTUD3 acetylation is also involved in PTEN regulation. Nevertheless, an intriguing puzzle has been suggested following a recent study of the accelerated development of lung carcinomas after deletion of Otud3 in mice102. In contrast to its level in breast cancer, OTUD3 is highly expressed in human lung cancer, and its upregulation is associated with unfavorable prognoses. Furthermore, in lung cancer, OTUD3 fails to regulate PTEN and, in contrast, maintains the stability of the oncoprotein GRP78 (glucose-regulated protein 78-kDa), showing the tumor tissue complexity of the functional role of played by a given deubiquitinase. These findings suggest that future studies should optimize the accurate stratification of deubiquitinase-targeted therapies for specific organs or tissues.
Ataxin-3 (ATXN3)
Machado–Joseph disease (MJD, also known as spinocerebellar ataxia type 3 or SCA3) is the most common dominant ataxia in the world and is caused by abnormal expansion of CAG repeats in a coding region of ATXN3, which produces an elongated polyglutamine (polyQ) tract in the Ataxin-3 protein103. Ataxin-3 contains an NH2-terminal ubiquitin-protease (Josephin) domain and COOH-terminal polyQ stretch and ubiquitin-interacting motifs103. As a deubiquitinase, Ataxin-3 plays a role in protein quality control and DNA repair by deubiquitinating several essential substrates, including the neuroprotective E3 ligases Parkin and CHIP104,105 and the DNA damage response and repair mediators p53, MDC1, RNA polymerase II, and CHK1106. A small interfering RNA (siRNA) screen for deubiquitinases revealed that three MJD subfamily members, including Ataxin-3, can inhibit PTEN expression42,107. However, Ataxin-3 regulates PTEN transcript abundance but not protein stability, suggesting that its role is independent of direct PTEN deubiquitination.
Targeting PTEN DUBs for cancer therapy
PTEN is a bona fide lipid phosphatase that opposes the activation of the highly oncogenic PI3K-AKT-mTOR pathway and is considered a major dose-dependent tumor suppressor. While PTEN itself is not considered a ‘druggable’ target, the pathological mechanisms that modulate PTEN protein levels and activity offer possible routes for cancer therapy. Furthermore, the predominant genetic change associated with loss of function is deletion of only a single gene copy of PTEN, underscoring the importance of targeting the nongenomic mechanisms of PTEN loss of function for the prevention and treatment of cancer. Along with the previously mentioned biological and clinical relevance of PTEN DUBs in tumorigenesis, PTEN DUBs may represent promising targets for therapeutic PTEN reactivation regimens in many types of cancer. Therefore, the activity of PTEN DUBs can likely be pharmacologically manipulated to fully reactivate PTEN, resulting in new and innovative approaches to the prevention and treatment of cancer (Table 1). Indeed, a small-molecule inhibitor of HAUSP/USP7, P5091, has been shown to restore the monoubiquitination and nuclear localization of endogenous PTEN and to induce cell growth arrest and apoptosis in blood-born cancers58,66. In addition to PTEN, p53 is upregulated by P5091, but its cytotoxic activity is not dependent on p53108. Other recently developed (pre)clinical HAUSP inhibitors (e.g., FT671, XL188, and GNE6640)109,110,111 will need to be used to establish a portfolio of HAUSP-PTEN axis-targeting drugs for use in future cancer therapies. Additionally, successful PTEN reactivation through disruption of the PML-DAXX-HAUSP complex by Trisenox (arsenic trioxide), which is currently used to treat patients with acute promyelocytic leukemia41, may pave the way to clinical trials for prevention and therapy for solid tumors at large.
Given the significance of USP10, USP11, USP13 and OTUD3 in PTEN stability, the development of a potent PTEN activation approach through manipulation of these DUBs may represent an attractive strategy for cancer prevention and treatment. For example, as AMPK-mediated phosphorylation of USP10 enhances USP10 activity69, treatment with metformin, which is used clinically to activate AMPK112, can lead to the upregulation of USP10 and, thus, PTEN-induced tumor suppression. In addition, resveratrol and psammaplysene A have been found to induce USP11 transcription mediated through FOXO and, in turn, appreciably elevate PTEN protein levels by increasing PTEN deubiquitination and hence PTEN stability43. Thus, new stratifications based on up- and downregulation of PTEN DUBs can ensure PTEN protein localization and activity and optimize PTEN DUB-targeted therapies, which may be specifically tailored to human cancers that do not exhibit homozygous (biallelic) loss of PTEN.
Concluding remarks
PTEN antagonizes the oncogenic PI3K-AKT-mTOR signaling pathway, which is frequently activated in cancers. PTEN deletions are often found in more aggressive tumors and are associated with worsened prognosis, increased tumor metastases, and a greater chance of recurrence after treatment. Emerging evidence has also shown that, similar to the genomic disruptions that inactivate a given PTEN allele, ‘nongenomic’ pathological mechanisms that reduce PTEN protein levels and activity are associated with cancer. As a result, identifying active deubiquitinating enzymes that directly modulate PTEN protein stability and activity for therapeutic purposes has become a high priority for cancer researchers. Indeed, new discoveries of DUBs that interact with PTEN have changed our understanding of PTEN function and regulation. HAUSP/USP7, USP10, USP11, USP13, OTUD3, and Ataxin-3 have all been recently identified as PTEN DUBs that control PTEN activity in different cancer-specific contexts. However, these DUBs play context-dependent tumor suppressor or oncogenic roles in cancer progression, and in different contexts, both their up- and downregulation can be hallmarks of tumor cells leading to malignancies; therefore, a complete understanding of how each individual DUB functionally influences tumorigenesis or tumor suppression remains unclear, and further in vivo investigation is required. Although researchers have extensively described the specificity of PTEN DUBs, as discussed herein, their ubiquitin linkage specificity (i.e., K6, K11, K27, K29, K33, K48 or K63-linked mono- and polyubiquitin chains) with respect to PTEN is still being elucidated. In addition, whether and how the complex relationship between PTEN DUBs (e.g., HAUSP−USP10113, HAUSP−USP11114 and USP10−USP1370) strengthens their activity toward PTEN requires further study. It will be interesting to evaluate the possible crosstalk between deubiquitination and other posttranslational modifications, such as acetylation115, methylation116, and SUMO ylation11, during the control of PTEN stability and activity. Given recent discoveries revealing that distinct PTEN isoforms and active PTEN dimers are related to specific PTEN functions3, it will be interesting to determine whether and how the aforementioned and several other PTEN DUBs117,118,119 impact the stability, localization and biological activity of dimeric PTEN and various PTEN isoforms. PTEN is a major tumor suppressor protein whose expression and activity often serve as the bases of diagnostic and prognostic assessment; however, no available therapy that directly targets PTEN itself is currently available. With the link of DUBs to reactivated PTEN established, the pharmacological manipulation of DUBs holds great clinical promise and suggests innovative and effective therapeutic approaches.
References
Hollander, M. C., Blumenthal, G. M. & Dennis, P. A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer. 11, 289–301 (2011).
Salmena, L., Carracedo, A. & Pandolfi, P. P. Tenets of PTEN tumor suppression. Cell 133, 403–414 (2008).
Lee, Y. R., Chen, M. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 19, 547–562 (2018).
Song, M. S., Salmena, L. & Pandolfi, P. P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 (2012).
Myers, M. P. et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl Acad. Sci. 94, 9052–9057 (1997).
Gu, T. et al. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 71, 2821–2825 (2011).
Shi, Z. et al. Ataxin-3 promotes testicular cancer cell proliferation by inhibiting anti-oncogene PTEN. Biochem. Biophys. Res. Commun. 503, 391–396 (2018).
Tamura, M. et al. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 274, 20693–20703 (1999).
Wozniak, D. J. et al. PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat. Commun. 8, 1508 (2017).
Zhang, S. et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 17, 461–469 (2011).
Bassi, C. et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 341, 395–399 (2013).
Shen, W. H. et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170 (2007).
Song, M. S. et al. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 144, 187–199 (2011).
Wang, G. et al. PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 25, 1189–1204 (2015).
Eng, C. PTEN: one gene, many syndromes. Hum. Mutat. 22, 183–198 (2003).
Di Cristofano, A., De Acetis, M., Koff, A., Cordon-Cardo, C. & Pandolfi, P. P. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet 27, 222–224 (2001).
Di Cristofano, A. et al. Impaired Fas response and autoimmunity in Pten+/- mice. Science 285, 2122–2125 (1999).
Papa, A. et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157, 595–610 (2014).
Alimonti, A. et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Invest. 120, 681–693 (2010).
Chen, Z. et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 (2005).
Berger, A. H., Knudson, A. G. & Pandolfi, P. P. A continuum model for tumour suppression. Nature 476, 163–169 (2011).
Alimonti, A. et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 42, 454–458 (2010).
Trotman, L. C. et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 1, E59 (2003).
Garcia-Cao, I. et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 149, 49–62 (2012).
Lee, Y. R. et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 364, eaau0159 (2019).
Maccario, H., Perera, N. M., Davidson, L., Downes, C. P. & Leslie, N. R. PTEN is destabilized by phosphorylation on Thr366. Biochem. J. 405, 439–444 (2007).
Vazquez, F. et al. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J. Biol. Chem. 276, 48627–48630 (2001).
Gonzalez-Santamaria, J. et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 3, e393 (2012).
Hershko, A. & Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998).
Song, M. S. & Pandolfi, P. P. The HECT family of E3 ubiquitin ligases and PTEN. Semin. Cancer Biol. 85, 43–51 (2021).
Putz, U. et al. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal. 5, ra70 (2012).
Trotman, L. C. et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 (2007).
Maddika, S. et al. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 13, 728–733 (2011).
Wang, X. et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 (2007).
Lee, Y. R. et al. WWP1 Gain-of-Function Inactivation of PTEN in Cancer Predisposition. N. Engl. J. Med. 382, 2103–2116 (2020).
Nijman, S. M. et al. A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 (2005).
Abdul Rehman, S. A. et al. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell. 63, 146–155 (2016).
Haahr, P. et al. ZUFSP Deubiquitylates K63-Linked Polyubiquitin Chains to Promote Genome Stability. Mol. Cell. 70, 165–174.e166 (2018).
Kwasna, D. et al. Discovery and Characterization of ZUFSP/ZUP1, a Distinct Deubiquitinase Class Important for Genome Stability. Mol. Cell. 70, 150–164.e156 (2018).
Clague, M. J. et al. Deubiquitylases from genes to organism. Physiol. Rev. 93, 1289–1315 (2013).
Song, M. S. et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817 (2008).
Sacco, J. J. et al. The deubiquitylase Ataxin-3 restricts PTEN transcription in lung cancer cells. Oncogene 33, 4265–4272 (2014).
Park, M. K. et al. PTEN self-regulates through USP11 via the PI3K-FOXO pathway to stabilize tumor suppression. Nat. Commun. 10, 636 (2019).
Yuan, L. et al. Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nat. Cell Biol. 17, 1169–1181 (2015).
Zhang, J. et al. Deubiquitylation and stabilization of PTEN by USP13. Nat. Cell Biol. 15, 1486–1494 (2013).
Holowaty, M. N., Sheng, Y., Nguyen, T., Arrowsmith, C. & Frappier, L. Protein interaction domains of the ubiquitin-specific protease, USP7/HAUSP. J. Biol. Chem. 278, 47753–47761 (2003).
Hu, M. et al. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111, 1041–1054 (2002).
Sarkari, F., Sheng, Y. & Frappier, L. USP7/HAUSP promotes the sequence-specific DNA binding activity of p53. PLoS One. 5, e13040 (2010).
Li, M. et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648–653 (2002).
Cummins, J. M. et al. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428 1 (2004).
Kon, N. et al. Inactivation of HAUSP in vivo modulates p53 function. Oncogene 29, 1270–1279 (2010).
Faesen, A. C. et al. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell. 44, 147–159 (2011).
An, L. et al. Dual-utility NLS drives RNF169-dependent DNA damage responses. Proc. Natl Acad. Sci. 114, E2872–E2881 (2017).
Cheng, J. et al. Molecular mechanism for USP7-mediated DNMT1 stabilization by acetylation. Nat. Commun. 6, 7023 (2015).
Zhang, Z. M. et al. An Allosteric Interaction Links USP7 to Deubiquitination and Chromatin Targeting of UHRF1. Cell Rep. 12, 1400–1406 (2015).
Howitt, J. et al. Ndfip1 regulates nuclear Pten import in vivo to promote neuronal survival following cerebral ischemia. J. Cell Biol. 196, 29–36 (2012).
Howitt, J. et al. Ndfip1 represses cell proliferation by controlling Pten localization and signaling specificity. J. Mol. Cell Biol. 7, 119–131 (2015).
Carra, G. et al. Therapeutic inhibition of USP7-PTEN network in chronic lymphocytic leukemia: a strategy to overcome TP53 mutated/deleted clones. Oncotarget 8, 35508–35522 (2017).
Gao, L. et al. Proteome Analysis of USP7 Substrates Revealed Its Role in Melanoma Through PI3K/Akt/FOXO and AMPK Pathways. Front. Oncol. 11, 650165 (2021).
Hernandez-Perez, S. et al. DUB3 and USP7 de-ubiquitinating enzymes control replication inhibitor Geminin: molecular characterization and associations with breast cancer. Oncogene 36, 4817 (2017).
Li, N., Geng, F., Liang, S. M. & Qin, X. USP7 inhibits TIMP2 by up-regulating the expression of EZH2 to activate the NF-kappaB/PD-L1 axis to promote the development of cervical cancer. Cell Signal. 96, 110351 (2022).
Wang, Z. et al. Abrogation of USP7 is an alternative strategy to downregulate PD-L1 and sensitize gastric cancer cells to T cells killing. Acta Pharm. Sin. B. 11, 694–707 (2021).
Zhao, G. Y. et al. USP7 overexpression predicts a poor prognosis in lung squamous cell carcinoma and large cell carcinoma. Tumour Biol. 36, 1721–1729 (2015).
Collaud, S. et al. Lung neuroendocrine tumors: correlation of ubiquitinylation and sumoylation with nucleo-cytosolic partitioning of PTEN. BMC Cancer. 15, 74 (2015).
Noguera, N. I. et al. Nucleophosmin/B26 regulates PTEN through interaction with HAUSP in acute myeloid leukemia. Leukemia 27, 1037–1043 (2013).
Li, C. et al. TRIP13 modulates protein deubiquitination and accelerates tumor development and progression of B cell malignancies. J. Clin. Invest. 131, e146893 (2021).
Morotti, A. et al. BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia 28, 1326–1333 (2014).
Yuan, J., Luo, K., Zhang, L., Cheville, J. C. & Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 140, 384–396 (2010).
Deng, M. et al. Deubiquitination and Activation of AMPK by USP10. Mol. Cell. 61, 614–624 (2016).
Liu, J. et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234 (2011).
Meyer, C., Garzia, A., Morozov, P., Molina, H. & Tuschl, T. The G3BP1-Family-USP10 Deubiquitinase Complex Rescues Ubiquitinated 40S Subunits of Ribosomes Stalled in Translation from Lysosomal Degradation. Mol. Cell. 77, 1193–1205.e1195 (2020).
He, Y. et al. The deubiquitinase USP10 restores PTEN activity and inhibits non-small cell lung cancer cell proliferation. J. Biol. Chem. 297, 101088 (2021).
He, Y. M. et al. TRIM25 activates AKT/mTOR by inhibiting PTEN via K63-linked polyubiquitination in non-small cell lung cancer. Acta Pharmacol. Sin. 43, 681–691 (2022).
Kim, K. et al. Prognostic significance of USP10 and p14ARF expression in patients with colorectal cancer. Pathol. Res. Pract. 216, 152988 (2020).
Song, J. S. et al. Dual loss of USP10 and p14ARF protein expression is associated with poor prognosis in patients with small intestinal adenocarcinoma. Tumour Biol. 40, 1010428318808678 (2018).
Swanson, D. A., Freund, C. L., Ploder, L., McInnes, R. R. & Valle, D. A ubiquitin C-terminal hydrolase gene on the proximal short arm of the X chromosome: implications for X-linked retinal disorders. Hum. Mol. Genet. 5, 533–538 (1996).
Yang-Feng, T. L., Li, S., Han, H. & Schwartz, P. E. Frequent loss of heterozygosity on chromosomes Xp and 13q in human ovarian cancer. Int. J. Cancer. 52, 575–580 (1992).
Spatz, A., Borg, C. & Feunteun, J. X-chromosome genetics and human cancer. Nat. Rev. Cancer. 4, 617–629 (2004).
Medema, R. H. & Burgering, B. M. The X factor: skewing X inactivation towards cancer. Cell 129, 1253–1254 (2007).
Liao, Y., Zhou, D., Wang, P., Yang, M. & Jiang, N. Ubiquitin specific peptidase 11 as a novel therapeutic target for cancer management. Cell Death Disco. 8, 292 (2022).
Herold, S. et al. Recruitment of BRCA1 limits MYCN-driven accumulation of stalled RNA polymerase. Nature 567, 545–549 (2019).
Wu, H. C. et al. USP11 regulates PML stability to control Notch-induced malignancy in brain tumours. Nat. Commun. 5, 3214 (2014).
Sun, W. et al. USP11 negatively regulates TNFalpha-induced NF-kappaB activation by targeting on IkappaBalpha. Cell Signal. 22, 386–394 (2010).
Zhang, H. et al. Long noncoding RNA lnc-DILC stabilizes PTEN and suppresses clear cell renal cell carcinoma progression. Cell Biosci. 9, 81 (2019).
Kapadia, B. et al. Fatty Acid Synthase induced S6Kinase facilitates USP11-eIF4B complex formation for sustained oncogenic translation in DLBCL. Nat. Commun. 9, 829 (2018).
Wiltshire, T. D. et al. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J. Biol. Chem. 285, 14565–14571 (2010).
Gonzalez-Billalabeitia, E. et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Disco. 4, 896–904 (2014).
Fang, X. et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J. Exp. Med. 214, 245–267 (2017).
Han, C. et al. Amplification of USP13 drives ovarian cancer metabolism. Nat. Commun. 7, 13525 (2016).
Wu, Y. et al. Amplification of USP13 drives non-small cell lung cancer progression mediated by AKT/MAPK signaling. Biomed. Pharmacother. 114, 108831 (2019).
Zhang, S. et al. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat. Commun. 9, 215 (2018).
Morgan, E. L. et al. The deubiquitinase (DUB) USP13 promotes Mcl-1 stabilisation in cervical cancer. Oncogene 40, 2112–2129 (2021).
Zhou, Q. et al. Targeting CLK3 inhibits the progression of cholangiocarcinoma by reprogramming nucleotide metabolism. J. Exp. Med. 217, e20191779 (2020).
Li, Y. et al. USP13 regulates the RAP80-BRCA1 complex dependent DNA damage response. Nat. Commun. 8, 15752 (2017).
Chu, Y. et al. The Chaperone BAG6 Regulates Cellular Homeostasis between Autophagy and Apoptosis by Holding LC3B. iScience 23, 101708 (2020).
Kwon, J. et al. USP13 promotes development and metastasis of high-grade serous ovarian carcinoma in a novel mouse model. Oncogene 41, 1974–1985 (2022).
Man, X. et al. USP13 functions as a tumor suppressor by blocking the NF-kB-mediated PTEN downregulation in human bladder cancer. J. Exp. Clin. Cancer Res. 38, 259 (2019).
Qu, Z., Zhang, R., Su, M. & Liu, W. USP13 serves as a tumor suppressor via the PTEN/AKT pathway in oral squamous cell carcinoma. Cancer Manag. Res. 11, 9175–9183 (2019).
Zhang, Z. et al. Acetylation-Dependent Deubiquitinase OTUD3 Controls MAVS Activation in Innate Antiviral Immunity. Mol. Cell. 79, 304–319.e307 (2020).
Zhou, N. et al. Deubiquitinase OTUD3 regulates metabolism homeostasis in response to nutritional stresses. Cell Metab. 34, 1023–1041.e1028 (2022).
Wang, M. et al. Nicotine-mediated OTUD3 downregulation inhibits VEGF-C mRNA decay to promote lymphatic metastasis of human esophageal cancer. Nat. Commun. 12, 7006 (2021).
Du, T. et al. The deubiquitylase OTUD3 stabilizes GRP78 and promotes lung tumorigenesis. Nat. Commun. 10, 2914 (2019).
Costa Mdo, C. & Paulson, H. L. Toward understanding Machado-Joseph disease. Prog. Neurobiol. 97, 239–257 (2012).
Durcan, T. M. & Fon, E. A. Mutant ataxin-3 promotes the autophagic degradation of parkin. Autophagy 7, 233–234 (2011).
Scaglione, K. M. et al. Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol. Cell. 43, 599–612 (2011).
Chakraborty, A. et al. Deficiency in classical nonhomologous end-joining-mediated repair of transcribed genes is linked to SCA3 pathogenesis. Proc. Natl Acad. Sci.117, 8154–8165 (2020).
Shi, Y. et al. PTEN is a protein tyrosine phosphatase for IRS1. Nat. Struct. Mol. Biol. 21, 522–527 (2014).
Chauhan, D. et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 22, 345–358 (2012).
Turnbull, A. P. et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature 550, 481–486 (2017).
Lamberto, I. et al. Structure-guided development of a potent and selective non-covalent active-site Inhibitor of USP7. Cell Chem. Biol. 24, 1490–1500 (2017).
Kategaya, L. et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 550, 534–538 (2017).
Hardie, D. G., Ross, F. A. & Hawley, S. A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 (2012).
Han, Y. & Yun, C. C. Ubiquitin-specific peptidase 7 (USP7) and USP10 mediate deubiquitination of human NHE3 regulating its expression and activity. FASEB J. 34, 16476–16488 (2020).
Maertens, G. N., El Messaoudi-Aubert, S., Elderkin, S., Hiom, K. & Peters, G. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO J. 29, 2553–2565 (2010).
Okumura, K. et al. PCAF modulates PTEN activity. J. Biol. Chem. 281, 26562–26568 (2006).
Zhang, J. et al. PTEN Methylation by NSD2 Controls Cellular Sensitivity to DNA Damage. Cancer Disco. 9, 1306–1323 (2019).
Ren, D., Sun, Y., Li, D., Wu, H. & Jin, X. USP22-mediated deubiquitination of PTEN inhibits pancreatic cancer progression by inducing p21 expression. Mol. Oncol. 16, 1200–1217 (2022).
Shen, W. M., Yin, J. N., Xu, R. J., Xu, D. F. & Zheng, S. Y. Ubiquitin specific peptidase 49 inhibits non-small cell lung cancer cell growth by suppressing PI3K/AKT signaling. Kaohsiung J. Med. Sci. 35, 401–407 (2019).
Deng, R. et al. BAP1 suppresses prostate cancer progression by deubiquitinating and stabilizing PTEN. Mol. Oncol. 15, 279–298 (2021).
Gavory, G. et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat. Chem. Biol. 14, 118–125 (2018).
Reverdy, C. et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 19, 467–477 (2012).
Burkhart, R. A. et al. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol. Cancer Res. 11, 901–911 (2013).
Du, T. et al. A small molecule inhibitor of deubiquitinase OTUD3 promotes DR5-induced apoptosis in lung cancer through ER stress modulation. Res. Square. https://www.researchsquare.com/article/rs-871606/v1 (2021).
Gertz, M. et al. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl Acad. Sci. 110, E2772–E2781 (2013).
Wang, Q., Li, L. & Ye, Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 283, 7445–7454 (2008).
Acknowledgements
We would like to thank T. Garvey for critical editing of the manuscript. The scientific illustrations were created with BioRender.com. This work was supported in part by grants from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2021R1A2C1006413, S.J.S.), National Institutes of Health (CA196740 and CA258100, M.S.S.), and the Department of Defense (W81XWH-20-1-0379, M.S.S.).
Author information
Authors and Affiliations
Contributions
The research was conceived and designed by A.C., M.K.P., S.J.S., and M.S.S. The manuscript was written by A.C., M.K.P., S.J.S. and M.S.S.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Christine, A., Park, M.K., Song, S.J. et al. The equilibrium of tumor suppression: DUBs as active regulators of PTEN. Exp Mol Med 54, 1814–1821 (2022). https://doi.org/10.1038/s12276-022-00887-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00887-w
This article is cited by
-
SQLE Knockdown inhibits bladder cancer progression by regulating the PTEN/AKT/GSK3β signaling pathway through P53
Cancer Cell International (2023)
