
Abstract
Receptor-interacting serine threonine protein kinase 1 (RIPK1) has emerged as a central molecular switch in controlling the balance between cell survival and cell death. The pro-survival role of RIPK1 in maintaining cell survival is achieved via its ability to induce NF-κB-dependent expression of anti-apoptotic genes. However, recent advances have identified the pro-death function of RIPK1: posttranslational modifications of RIPK1 in the tumor necrosis factor receptor 1 (TNFR1)-associated complex-I, in the cytosolic complex-IIb or in necrosomes regulate the cytotoxic potential of RIPK1, forming an early cell death checkpoint. Since the kinase activity of RIPK1 is indispensable in RIPK3- and MLKL-mediated necroptosis induction, while it is dispensable in apoptosis, a better understanding of this early cell death checkpoint via RIPK1 might lead to new insights into the molecular mechanisms controlling both apoptotic and necroptotic modes of cell death and help develop novel therapeutic approaches for cancer. Here, we present an emerging view of the regulatory mechanisms for RIPK1 activity, especially with respect to the early cell death checkpoint. We also discuss the impact of dysregulated RIPK1 activity in pathophysiological settings and highlight its therapeutic potential in treating human diseases.
Similar content being viewed by others

Introduction
Apoptosis is considered to be an evolutionarily conserved cell death process for normal development and homeostasis, whereas necrosis is historically considered to be an accidental, uncontrolled and passive cell death phenomenon evoked by environmental perturbations1,2. The discovery of the caspase homolog CED-3 and the Apaf-1 homolog CDD-4 in the nematode C. elegans led to the establishment of apoptosis as a regulated cell death pathway3,4. The apoptosis pathway in mammals is characterized by the activation of a series of caspase cascades that ensure cell death occurs in a controlled manner5,6. Consequently, caspase inhibition is capable of protecting cells against either extrinsic or intrinsic cytotoxic damage. However, in certain types of cells, genetic or pharmacological inhibition of caspases is unable to block cell death upon the ligation of death receptors, including the tumor necrosis factor receptor 1 (TNFR1), but rather facilitates the necrotic cell death process7,8,9. The molecular mechanism of such paradoxical death receptor-mediated necrotic cell death has long remained unknown. It is now becoming increasingly clear that receptor-interacting protein kinase 1 (RIPK1) is a key player in the control of this phenomenon, as RIPK1 is required to execute necrosis in a caspase-independent manner. Importantly, RIPK1 acts upstream of another key protein kinase, RIPK3, by forming a microfilament-like complex called the necrosome, leading to the activation of the pro-necrotic enzyme mixed lineage kinase domain-like pseudokinase (MLKL), which causes cell membrane disruption and ultimately results in programmed necrosis, which is called necroptosis, an alternative pathway for programmed cell death (PCD)10,11. Furthermore, a recent structural study indicated that activation of RIPK1 is required for the induction of both RIPK1/RIPK3 hetero- and RIPK3 homo-oligomerization in the early stages of necroptosis, which leads to an ordered, helical formation of necrosome rods12. Subsequently, such a configuration for necrosome-associated RIPK1/RIPK3 oligomers functions to facilitate MLKL oligomerization and execute necroptosis12, indicating that RIPK1 indeed functions as a signaling hub in RIPK3-dependent necroptotic cell death.
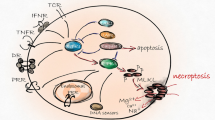
RIPK1 was initially identified as an adapter protein that interacts with the death domain (DD) of CD95 or TNFR1 via a homotypic DD-DD interaction that leads to a plasma membrane-bound death receptor (DR) signaling complex (also known as complex-I)13,14. Subsequently, complex-I internalizes and dissociates from the plasma membrane and recruits the Fas-associated death domain (FADD) and caspase-8 to form a cytoplasmic death-inducing signaling complex (DISC, also known as complex-IIa) that elicits characteristic apoptotic cell death13,14. Subsequently, another protein interaction motif in RIPK1, termed RIP homotypic interaction motif (RHIM), was identified, and this motif mediates interactions with RIPK3 to trigger necroptosis15,16. In addition, RIPK1 interacts with numerous adapter proteins and with the inhibitory κB kinase (IKK) complex, resulting in the activation of nuclear factor-κB (NF-κB)17,18,19. This process then drives the transcriptional induction of prosurvival genes such as Bcl2, A20 and cFLIP by an NF-κB-dependent process that protects against apoptosis, and this is referred to as the NF-κB-dependent late cell death checkpoint (also called the transcription-dependent cell death checkpoint20,21 (Fig. 1)). Previous studies have demonstrated that NF-κB-dependent transcription of pro-survival genes regulates apoptotic signals in multiple ways. For instance, cFLIP, a catalytically inactive caspase-8 homolog, interacts with procaspase-8 and prevents the processing of caspase-8 by counteracting the cytotoxic activity of complex-IIa22,23. Thus, it is believed that NF-κB-dependent transcription of c-FLIP expression keeps the sublethal activation of DISC in check, thus preventing DR-mediated apoptosis. On the other hand, other NF-κB-inducible genes, such as Bcl2 and Bcl-XL, also function as late cell death checkpoints by regulating the mitochondria-mediated apoptotic pathway24. Thus, RIPK1 has long been studied as a key molecule in determining cell fate in the field of NF-κB signaling. However, recent studies have reported that the posttranslational modifications (PTMs) of RIPK1 repress the cytotoxic potential of RIPK1 in complex-I and thereby protect cells from cell death by counteracting the assembly of complex-IIb (RIPK1-FADD-caspase-8), which is independent of the transcriptional activation of NF-κB and is referred to as the RIPK1-dependent early cell death checkpoint25,26,27 (Fig. 1). Given that the cytotoxic potential of RIPK1 is essential for engaging the necroptotic cell death machinery via RIPK328, insights into this regulation have not only led to an improved understanding of a fundamental signaling node in necroptosis but also offered an opportunity to develop new therapeutic approaches in necroptosis-associated human diseases29,30. Since transcription-dependent cell death regulation by NF-κB activation has been discussed extensively elsewhere29,31, in this review, we mainly focus on recent developments in the study of the early cell death checkpoint where RIPK1 acts to regulate two PCD pathways in the forms of apoptosis and necroptosis.

Ligation of TNFR1 triggers the formation of the primary TNFR1 complex, referred to as “complex-I,” by the recruitment of TRADD and RIPK1, which in turn recruits several E3 ubiquitin ligases. The early cell death checkpoint is initiated by the conjugation of K63 polyUb chains or M1-linked linear Ub chains on RIPK1 by TRAF2/5, cIAP1/2 and MIB2 or LUBAC. The ubiquitinated RIPK1 within complex-I keeps RIPK1 in a survival mode and prevents the formation of a secondary cytoplasmic complex, referred to as “complex-IIb,” in a gene transcription-independent manner. This process also allows docking of TAK1 with TAB2 or TAB3 as well as of the IKK complex (IKK-α/-β/NEMO) to activate NF-κB, which functions as a late cell death checkpoint by the transcriptional induction of survival genes, including Bcl2, A20 and c-FLIP. Deubiquitination of RIPK1 by either A20, CYLD and Cezanne or the inhibition of E3 ubiquitin ligases promotes the sequential formation of the pro-apoptotic complex-IIb consisting of RIPK1, FADD and caspase-8 to mediate RIPK1-dependent apoptosis (RDA). Alternatively, in the absence of apoptosis, RIPK1 interacts with RIPK3 and MLKL to form necrosomes, which elicits downstream events for necroptosis. Under necroptotic conditions, several E3 Ub ligases, such as PELI1, cIAP1/2 and LUBAC, are also proposed to target cytosolic RIPK1 within complex-IIb or necrosomes for ubiquitination. Abbreviations: TNFR1, tumor necrosis factor receptor 1; TRADD, TNFR1-associated death domain protein; RIPK1/3, receptor-interacting serine threonine protein kinase 1/3; TRAF2/5, TNF receptor-associated Factor 2/5; cIAP1/2, cellular inhibitor of apoptosis 1/2; MIB2, Mind Bomb-2; LUBAC, linear ubiquitin chain assembly complex; TAK1, transforming growth factor β activated kinase 1; TAB2, TAK1-binding protein 2, IKK Inhibitory kappa B kinase, NF-κB nuclear factor-kappa B, Bcl2 B-cell lymphoma 2, c-FLIP cellular FLICE-inhibitory protein, CYLD cylindromatosis lysine 63 deubiquitinase, FADD Fas-associated death domain, MLKL mixed lineage kinase domain-like pseudokinase, PELI1, Pellino 1.
Regulation of the cytotoxic potential of RIPK1 by ubiquitination in complex-I
RIPK1 is well known to play a key role in TNF signaling. Within seconds of TNF exposure, RIPK1 is recruited into TNFR1 and associates with an adapter protein, the TNFR1-associated death domain protein (TRADD), to form complex-I31. TRADD serves as an adapter for the formation of the network of polyubiquitin chains mediated by E3 ubiquitin (Ub) ligases that closely regulate the dynamic assembly of complex-I with transforming growth factor β activated kinase (TAK1) and NF-κB upstream kinase, called the IκB kinase (IKK) cascade. Although the ubiquitination of RIPK1 acts as a platform for the late cell death checkpoint by activating the NF-κB transcriptional response, it also functions to suppress the translocation of RIPK1 into cytoplasmic DISC (also known as complex-IIb), which restricts PCD by inactivating cytotoxic RIPK129,31. It has been reported that RIPK1 undergoes an extensive ubiquitination process with multiple types of polyubiquitin linkages, including K63, K48, K11 and linear ubiquitination in complex-I, implying that a number of E3 ubiquitin ligases and ubiquitin cofactors are involved in this process (Table 1)32,33,34,35.
TNF receptor-associated Factor 2 (TRAF2) is one of the first RING domain-containing E3 Ub ligases discovered in TNF signaling that mediates K63-linked ubiquitination of RIPK136. The E3 Ub ligase activity of TRAF2 seems to be required for RIPK1 ubiquitination, as it has been observed that TNF-induced RIPK1 ubiquitination is restored by the reconstitution of wild-type TRAF2 but not by the RING domain mutant TRAF2 in TRAF2 knockout cells37. However, in vitro structural analysis showed that the RING domain of TRAF2 did not possess K63-linked Ub ligase activity38,39. Thus, it is still controversial whether TRAF2 directly ubiquitinates RIPK1 or works together with other E3 Ub ligases. At this point, one interesting observation is that murine embryonic fibroblasts (MEFs) derived from TRAF2−/− mice exhibited intact levels of RIPK1 ubiquitination and NF-κB activation in response to TNF40. Subsequent genetic experiments showed that TNF-induced RIPK1 ubiquitination and NF-κB activation were markedly attenuated in TRAF2 and TRAF5 double-knockout cells41,42,43, indicating that TRAF2 and TRAF5 have overlapping functions during RIPK1 ubiquitination. Thus, the exact functions of TRAF5 in mediating RIPK1 ubiquitination need to be further examined.
The cellular inhibitors of apoptosis 1 and 2 (cIAP1 and cIAP2, cIAP1/2) are also RING domain-containing E3 Ub ligases that belong to the inhibitor of apoptosis protein (IAP) family. Both of them are recruited into complex-I and play an essential role in modulating the K63-linked ubiquitination of RIPK144,45,46. However, redundancy at this point cannot be excluded because cells bearing the cIAP1/2-interacting motif mutant of TRAF2 show reduced RIPK1 ubiquitination upon TNFR1 ligation39. Furthermore, RIPK1 ubiquitination is dependent on cIAP1/2 and TRAF2 interactions, but the RING domain of TRAF2 is dispensable38,47,48, demonstrating that TRAF2 may function to recruit cIAP1/2 in complex-I. Similarly, while TRAF2 is not always required for RIPK1 ubiquitination, data from in vitro and genetic mouse models indicate that cIAP1/2 is almost always required for RIPK1 ubiquitination and that this modification acts to prevent TNF-induced cell death49. Thus, genetic or pharmacological ablation of cIAP1/2 is sufficient to impair RIPK1 ubiquitination and dramatically increase the cytotoxic potential of RIPK1 via complex-IIb formation44,45,46. Consistent with the notion that cIAP1/2-mediated RIPK1 ubiquitination inhibits cell death by preventing complex-I from transitioning to complex-II, the deficiency of cIAP1 and cIAP2 leads to embryonic lethality that could be retarded by a single allele loss of RIPK149.
In addition to TRAF2 and cIAP1/2, the linear ubiquitin chain assembly complex (LUBAC, consisting of the catalytic subunit HOIP, HOIL-1 and the regulatory subunit SHARPIN) was identified as an additional E3 ubiquitin ligase in complex-I35,50,51. In contrast to K63-linked ubiquitination by TRAF2 and cIAP1/2, LUBAC exclusively catalyzes the M1-linked linear ubiquitination of complex-I components, including TNFR1 and RIPK152,53,54. This type of ubiquitination has a cell death suppression function similar to the K63-linked ubiquitination of RIPK1, as described above, based on observations that the suppression of HOIP or SHARPIN expression enhances complex-IIb formation and caspase-8-mediated cell death upon TNFR1 ligation in most cellular systems55,56,57. Mechanistically, LUBAC-mediated ubiquitination may delay the flux of RIPK1 to complex-IIb by tethering RIPK1 to complex-I and thus negatively regulating both apoptosis and necroptosis58,59. Consistent with these in vitro observations, loss of LUBAC components also results in severe inflammation and early embryonic lethality in genetic mouse models55,60. These results thus indicate that the M1-linked ubiquitin chain formed by LUBAC is indispensable in protecting RIPK1-mediated cell death, which functions as an important regulator of the early cell death checkpoint. Interestingly, it has been reported that the recruitment of LUBC into complex-I requires TRAF2 and cIAP1, but not RIPK1, whereas knockdown of HOIL-1 and HOIP reduces RIPK1 ubiquitination35,51. Thus, it is still unclear whether RIPK1 is a direct substrate for the E3 ubiquitin ligase activity of LUBAC, and the precise mechanisms and ubiquitination sites on RIPK1 for LUBAC remain to be further elucidated. In this regard, it has been reported that ABIN-1 (A20-binding inhibitor of NF-κB activation 1), a ubiquitin-binding protein, can interact with both M1-linked and K63-linked ubiquitin chains61. Further studies have revealed that the recruitment of ABIN-1 into complex-I by association with LUBAC and cIAP1/2 leads to the suppression of RIPK1-dependent apoptosis and necroptosis62,63. It is thus hypothesized that ABIN-1 functions as a critical link between M1/K63-linked ubiquitination to control the cytotoxic activity of RIPK1 by LUBAC.
Mind Bomb-2 (MIB2) is another E3 Ub ligase that has been reported to regulate the cytotoxic potential of RIPK164. In this study, MIB2 was capable of inducing the K63-linked ubiquitination of RIPK1 at different sites, including K115 and K377, and such ubiquitination repressed the kinase activity of RIPK164. Consequently, depletion of MIB2 sensitized cells to RIP1-dependent cell death via increased complex-II formation64. Thus, these data support a role for MIB2 as an additional early cell death checkpoint regulator. However, this study also questioned the functional relevance of K63-linked ubiquitination of RIPK1 in NF-κB signaling based on observations; depletion of MIB2 has little effect on NF-kB activation after TNF stimulation64.
Previously, it was reported that internalization of the TNFR1 signaling complex via endocytic trafficking plays an important role in complex-IIb formation in nonphagocytic cells65,66,67,68. In addition, the ubiquitin interacting motif (UIM) is found in proteins either involved in ubiquitination or in those known to interact with ubiquitin-like modifiers that function to regulate TNFR1 internalization and its cellular trafficking69,70. Recently, it has been reported that ankyrin repeat domain 13a (ANKRD13a), which carries multiple copies of UIM, interacts with ubiquitinated RIPK1 upon TNFR1 ligation, which sets a higher signal threshold for the cytotoxic potential of RIPK1: loss of AKRD13a can switch TNF signaling from survival to death by promoting the formation of complex-IIb71. However, because ANKRD13a is not an E3 Ub ligase itself, it is unclear how ANKRD13 negatively regulates the cytotoxic potential of RIPK1. Intriguingly, epsin proteins (epsin 1 or epsin 2, a family of ubiquitin-binding endocytic adapters) were found to be adapter proteins that interact with the components of complex-I, including RIPK1, HOIL-1 and HOIP, in a UIM-dependent manner72. These findings thus indicate that UIM-containing adapter proteins such as ANKRD13a and epsin 1/2 are involved in TNF signaling by keeping the specific endocytic assembly in check to inhibit the RIPK1 transition to complex-IIb, thereby reducing RIPK1 cytotoxicity. Further structural analyses via spatial compartmentalization and via integrated networks of these UIM-containing proteins in complex-IIb are necessary to elucidate the precise mechanisms by which UIM-containing adapter proteins limit the cytotoxic potential of RIPK1.
Regulation of the cytotoxic potential of RIPK1 by deubiquitination in complex-I
In addition to investigations on RIPK1 ubiquitination-mediated E3 Ub ligases, as discussed earlier, substantial progress has been made in understanding the role of RIPK1 deubiquitination regulated by multiple deubiquitinating enzymes (DUBs), including A20, CYLD and Cezanne.
The zinc finger protein A20 (also known as TNFAIP3) plays an essential role in limiting inflammatory responses by the negative feedback regulation of NF-κB signaling73. As one of the early NF-κB response genes, the expression of A20 is induced by TNF and recruited into TNFR1 complex-I and acts as a dual ubiquitin editing enzyme on RIPK173. Once A20 is recruited into complex-I, the N-terminal ovarian tumor (OTU) domain functions as a DUB that removes K63-linked polyUb chains from RIPK1 to prevent excessive NF-κB activation74,75. Interestingly, the C-terminal domain of A20, composed of seven zinc fingers, has been shown to function as an E3 Ub ligase that mediates K48-linked ubiquitination of RIPK1, thereby targeting its proteasomal degradation74, suggesting that A20 restricts the accessibility of IKKs into complex-I via its dual Ub editing activity of RIPK1. Accordingly, A20-deficient cells exhibit severe defects in the termination of TNF-induced NF-κB activation via aberrant ubiquitination of RIPK1 in complex-I76. Given the ability of A20 to terminate NF-κB activity, A20 is expected to possess a pro-cell death function. However, a growing body of evidence indicates that A20 protects most cells from death receptor-mediated cell death77,78. In fact, A20-deficient mice show premature death phenotypes due to severe inflammation76,79. Based on these observations, it seems that A20 likely exerts its anti-cell death function independently of its DUB activity. However, it has been reported that A20 is recruited into complex-I via LUBAC-mediated linear ubiquitination80. Even though A20 is unable to function as a DUB to cleave M1-linked linear ubiquitination, A20-deficient cells were found to have markedly decreased M1 ubiquitination of RIPK181, consistent with the results that A20 stabilizes the M1-linked ubiquitin chains in complex-I82. The cumulative evidence suggests that A20 plays a role in regulating the early cell death checkpoint during RIPK1-mediated cell death.
CYLD was initially identified as a tumor suppressor mutated in cylindromatosis characterized by a predisposition toward forming benign tumors of skin appendages83,84. Further in vitro studies revealed that CYLD functions as a DUB by removing K63 ubiquitin chains from various target proteins, including RIPK1, TRAF2, and NEMO/IKK-γ85,86,87,88. Consistent with the well-established DUB function, CYLD promotes formation of the cytosolic RIPK1 signaling complex, including either complex-II or necrosome, which elicits the RIPK1-dependent dual modes of PCD81,89,90,91. Importantly, genetic deletion of CYLD in mice results in the accumulation of ubiquitinated RIPK1, which is associated with the attenuation of the early wave of germ cell apoptosis92, suggesting that the DUB function of CYLD preferentially targets RIPK1 rather than TRAF2 or NEMO/IKK-γ to promote RIPK1-dependent cell death. While A20 hydrolyzes K63 and K48, but not M1 ubiquitination linkages, CYLD hydrolyzes K63 and M1 ubiquitination linkages induced by cIAP1/2 and LUBAC, respectively81. The removal of LUBAC-mediated M1 ubiquitination linkages by CYLD further modifies the overall ubiquitination status of complex-I to facilitate RIPK1-driven cytotoxicity81,89. Consistent with in vitro observations, reduced levels of CYLD expression have been reported in various human cancer tissues93,94. Accordingly, genetic studies have shown that CYLD is frequently inactivated by somatic mutations in many cancers, including hepatocellular carcinoma, multiple myeloma, and head and neck cancer95,96,97. In this regard, it is likely that s CYLD mutation or deficiency in some cancers decreases its deubiquitination activity against K63-linked or M1-linked Ub chains, leading to insufficient activation of RIPK1-mediated cell death.
In addition, it has been reported that Cezanne, a member of the A20 family of deubiquitinating cysteine proteases, also functions as a DUB on RIPK198. Cezanne can be induced following TNF exposure and is recruited into complex-I, where it cleaves polyubiquitin chains from RIPK199,100, suggesting that both Cezanne and A20 are endogenous negative regulators of the NF-κB signaling pathway by a negative feedback loop to prevent excessive activation at the level of complex-I. This deubiquitinating property of Cezanne against RIPK1 supports its role in regulating RIPK1-mediated cell death. However, since Cezanne and A20 possess overlapping biochemical properties, further characterization of divergent specificities toward other TNFR1 signaling components is required to provide an explanation for their redundant functions.
Regulation of the cytotoxic potential of RIPK1 by autophosphorylation
The kinase activity of RIPK1 is required for the apoptosis induced by RIPK1 deubiquitination. Loss of the E3 Ub ligases cIAP1 and cIAP2 in TNF-treated cells is sufficient to induce the formation of death-inducing complex-IIb containing RIPK1, FADD and caspase-8 to drive RIPK1 activation101,102,103. Such results suggest that the ubiquitination and phosphorylation processes of RIPK1 are closely interlinked. Furthermore, it has also been reported that genotoxic stress-mediated depletion of cIAP1 and cIAP2 causes spontaneous association of RIPK1/FADD/caspase-8 in the cytosol even in the absence of TNFR1 ligation104,105. This TNFR1-independent RIPK1-containing cytosolic platform has been referred to as the Ripoptosome, and it relies on RIPK1 kinase activity that can cause either apoptosis or necroptosis, depending on the cellular context106. However, how deubiquitination of RIPK1 affects its kinase activity in complex-IIb or Ripoptosome remains largely unresolved. Ectopic expression of RIPK1 is able to induce the dimerization and/or oligomerization of RIPK1 via its death domain107. Furthermore, autophosphorylation at S161 and S166 of RIPK1 induces conformational changes that allow RIPK1 to interact with death effectors such as FADD and RIPK3, thereby facilitating the formation of complex-IIb or necrosomes for the induction of apoptosis or necroptosis, respectively108,109,110. Such observations suggest that the transition of RIPK1 into cytosolic complex-IIb as a result of ubiquitination may result in its oligomerization in either complex-II or necrosomes, leading to its autophosphorylation at serines 161 and 166. Notably, reconstitution of the RIPK1-S161A mutant in RIPK1-deficient cells did not affect TNF-induced necroptosis111. Importantly, recent studies using mice containing an S166A RIPK1 knock-in allele showed that S166 phosphorylation plays a critical role in triggering RIPK1 activation but is insufficient to trigger RIPK1-mediated cell death108. Thus, it is possible that RIPK1 S166 phosphorylation permits the autophosphorylation of additional sites on RIPK1 to drive RIPK1-mediated PCD. These findings provide experimental evidence that RIPK1 autophosphorylation at S166 can serve as a biomarker of RIPK1 kinase activity.
Reactive oxygen species (ROS) have long been considered a driving force for RIPK1-dependent necroptosis, but a mechanistic understanding has been lacking9,112,113. Recent studies have revealed that the enhanced levels of ROS induced by TNF or some chemotherapeutic agents act as a positive feedback loop to enhance necrosome formation via RIPK1 autophosphorylation at Ser161114,115,116. Mechanistically, ROS are capable of promoting RIPK1 autophosphorylation at Ser161, which promotes RIPK1 oligomerization and RIPK1/RIPK3 interactions116. This suggests that ROS induce a positive feedback loop during necrosome formation by targeting RIPK1. However, ROS production during necroptosis also occurs downstream of RIPK19; thus, the signaling pathways governing the crosstalk between ROS and RIPK1 in the course of necroptosis require further investigation.
Regulation of the cytotoxic potential of RIPK1 by transphosphorylation
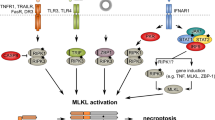
As described above, the autophosphorylation of RIPK1 at Ser 161 and S166 in complex-II functions as a positive regulator of the kinase activity of RIPK1. In contrast, recent studies have revealed that phosphorylation of RIPK1 by several kinases in complex-I acts as a fine-tuning negative mechanism to repress RIPK1 activation26,27,117 (Fig. 2).

Upon TNFR1 ligation, RIPK1 is recruited to membrane-associated complex-I and is ubiquitinated and then functions as a scaffold to interact with IKK and its related kinases that play essential roles in NF-κB and p38 MAPK activation. In addition to the role of the upstream kinases for NF-κB signaling, TAK1 and IKKs directly phosphorylate the ubiquitinated RIPK1 bound to complex-I, which suppresses the cytotoxic potential of RIPK1 via an unknown mechanism, possibly by preventing the translocation of RIPK1 into complex-IIb. In addition to TAK1 and IKKs, TBK1/IKKε and MK2 also inhibit the full activation of RIPK1 via direct phosphorylation of RIPK1 that is bound to complex-I and of the cytosolic pool of RIPK1, respectively. If the phosphorylation-mediated early cell death checkpoint is disrupted by inactivation of these kinases, RIPK1 is autophosphorylated via an allosteric effect and switches to an active death-inducing mode where it subsequently phosphorylates necrosome-associated RIPK3 to initiate necroptosis. The key kinases that phosphorylate RIPK1 at distinct sites are indicated. A broken arrow indicates indirect phosphorylation sites on RIPK1 for TBK1/IKKε. Abbreviations: TNFR1 tumor necrosis factor receptor 1, RIPK1/3 receptor-interacting serine threonine protein kinase 1/3, MAPK mitogen-activated protein kinase, TAK1 transforming growth factor β activated kinase 1, IKK inhibitory kappa B kinase, TBK1/IKKε TANK-binding kinase 1/IKK-epsilon, MK2 MAPK-activated protein kinase 2.
Upon TNFR1 ligation, TAK1 and IKKs (IKK-α/-β/-γ) are recruited into complex-I by interacting with ubiquitinated RIPK1, which functions as an upstream kinase for NF-κB-dependent prosurvival functions118. While activation of TAK1 and/or IKKs is dependent on their ability to interact with ubiquitinated RIPK1, they also act at the early cell death checkpoint to repress the cytotoxic potential of RIPK1 through phosphorylation25,119. Researchers at the Bertrand group made the unexpected observation that inhibition of IKKα or IKKβ activity induces RIPK1-dependent apoptosis or necroptosis by promoting the formation of complex-IIb or necrosomes in an NF-κB-independent manner120. In line with these findings, both in vitro and in vivo studies showed that inhibiting IKKβ activation results in excessive RIPK1 phosphorylation and RIPK1-mediated necroptosis27,121. Subsequent work has revealed that IKKα and/or IKKβ directly phosphorylate RIPK1 at Ser25 in complex-I, leading to suppression of RIPK1 kinase activity and its autophosphorylation122. Thus, IKKα/IKKβ appears to function as an early checkpoint to shut down the pro-death activity of RIPK1. However, reconstitution of a phospho-deficient alanine mutant (S25A) of RIPK1 in RIPK1-deficient cells is not sufficient to completely prevent RIPK1-mediated PCD upon TNFR1 ligation122. This finding indicates that, in addition to Ser25, IKKα/IKKβ might phosphorylate other sites at RIPK1. While proteomic studies using mass spectrometric analysis have revealed the existence of many additional residues of RIPK1 that are phosphorylated, such as Ser25, Ser166, S331 and Ser416120,122, the functional consequence of these phosphorylation events has not been examined. Furthermore, it is also unclear whether the phosphorylation of RIPK1 by IKKα/IKKβ directly affects the recruitment of RIPK1 into complex-IIb or prevents the dissociation of RIPK1 from complex-I. Apart from IKKα/IKKβ, the noncanonical IKK-related kinases TANK-binding kinase 1 (TBK1) and IKKε have recently been reported to phosphorylate RIPK1 in complex-I, and this phosphorylation prevents RIPK1-dependent PCD123,124. Similar to IKKα/IKKβ, the recruitment of these kinases into complex-I depends on RIPK1 and its K63-linked and linear ubiquitination by cIAP1/2 and LUBAC124. However, in contrast to IKKα/IKKβ, TBK1 and IKKε do not function as upstream kinases of the canonical NF-κB activation pathway125, indicating that they solely limit RIPK1 cytotoxicity by preventing the formation of complex-IIb or necrosomes without affecting the transcription-dependent late cell death checkpoint. Accordingly, TBK1 is able to directly phosphorylate human RIPK1 at T189 (equivalent to T190 mouse RIPK1) to regulate the cytotoxic potential of RIPK1123,124. Since inhibition of either IKKα/IKKβ or TBK1/IKKε is sufficient to trigger RIPK1-dependent PCD upon TNFR1 ligation, further work is required to elucidate whether their respective phosphorylation sites in RIPK1 have any redundancy in regulating RIPK1 cytotoxicity.
In addition to IKKs, activation of TAK1 also functions as an important negative regulator of RIPK1 cytotoxicity through phosphorylation. Genetic deletion of the pharmacological inhibition of TAK1 elicits RIPK1-mediated PCD without affecting RIPK1 ubiquitination in complex-I, in which TAK1 acts to repress RIPK1 autophosphorylation, independent of IKK-mediated NF-κB activation120,126. In line with this, a TAK1 deficiency not only sensitized cells to TNF-induced PCD but also enhanced the interaction between RIPK1 and RIPK3 under the conditions of a caspase-8 blockade116,117. Thus, activation of RIPK1 cytotoxicity by manipulation of its upstream kinase activities, such as those of TAK1 and IKKs, may provide new therapeutic options for TNF-mediated diseases. In fact, small molecular inhibitors of TAK1 or IKKβ are capable of inducing RDA in a TNF-dependent manner in some cancer and autoimmune disease models by dysregulating RIPK1 phosphorylation127,128. Recently, it was reported that S320 in human RIPK1 (equivalent to S321 in mouse RIPK1) is phosphorylated by TAK1 and thus attenuates RIPK1 cytotoxicity129. Accordingly, loss of S320 phosphorylation of RIPK1 drives RIPK1-mediated PCD by facilitating the formation of complex-II and necroptosis under NF-κB defective conditions129. Importantly, in cIAP1/2 double knockout cells, RIPK1-S320 phosphorylation following TNF treatment was almost completely negated129, suggesting that the K63-linked ubiquitination of RIPK1 mediated by cIAP1/2 may promote recruitment of TAK1 into complex-I. Further work is needed to accurately define the role of specific types of ubiquitin conjugation events in regulating RIPK1 phosphorylation mediated by TAK1.
MAPK-activated protein kinase 2 (MK2), a kinase downstream of p38 MAPK, was initially identified as a downstream effector of TNFR1 since it is essential for TNF biosynthesis via posttranscriptional regulation130,131. Unexpectedly, when studying the therapeutic potential of the cIAP1/2 inhibitor (smac mimetics; SM) in a leukemia model, it was noticed that MK2 inhibitors sensitized SM-induced cytotoxicity in a RIPK1-dependent manner132. Furthermore, it was observed that MK2 knockout mice showed enhanced cytotoxic sensitivity to TNF administration133. These perplexing observations led to the idea that the p38-MK2 axis might have an additional role in regulating cell death signaling downstream of TNFR1. In fact, this notion is supported by subsequent observations that MK2 phosphorylates human RIPK1 at Ser 320 and S335 (equivalent to S321 and S335 in mouse RIPK1) to inhibit RIPK1 cytotoxicity in TNFR1 signaling34,134,135. Thus, it appears that MK2 plays a role in limiting TNF cytotoxicity in the timely and coordinated TNFR1 complex, independent of the generation of ligands, including TNF. Further studies of the in vivo physiological significance of this dual action of MK2 could provide new perspectives for understanding TNF-driven cell death and inflammation. Since the phosphorylation of RIPK1 at different serine residues by IKKs and MK2 simultaneously provides a prosurvival signal by limiting RIPK1 cytotoxicity, the functional implication of multiple RIPK1 phosphorylation events by its upstream kinases remains to be further elucidated. In this regard, MK2 inhibition selectively enhances RIPK1-mediated cell death during TNF signaling when IKKs or cIAP1/2 is inhibited or depleted34,134. Mechanistically, RIPK1 phosphorylation by MK2 negatively regulates complex-IIb-mediated cell death by suppressing RIPK1 autophosphorylation34,134. More importantly, the current body of knowledge seems to suggest that IKKs and MK2 may target spatially different pools of RIPK1 for phosphorylation. IKKs are recruited into complex-I and phosphorylate plasma membrane-bound RIPK1 in a ubiquitination-dependent manner25,119. In contrast, MK2 mainly phosphorylates the cytosolic pool of RIPK1, which might be subsequently recruited into complex-I34. Thus, it is possible that IKK and MK2 provide separate early cell death checkpoints for RIPK1 activation by phosphorylating spatially distinct RIPK1 pools to limit the full activation of the cytotoxic potential of RIPK1136,137. However, given that complex-IIb, which contains RIPK1, is sequentially dissociated from complex-I26,138, the primary source of RIPK1 bound to death-inducing complex-IIb is still a longstanding issue to clarify.
Regulation of the cytotoxic potential of complex-IIb/necrosome-associated RIPK1
Given that deubiquitination of RIPK1 is required for its transition from complex-I to complex-IIb29,31, it is believed that the RIPK1 tethered within complex-I is more heavily modified with diverse polyubiquitin chains than the RIPK1 tethered within complex-IIb or necrosomes. However, several pieces of evidence have suggested that necrosome-associated RIPK1 also undergoes ubiquitination during necroptosis. For instance, although CYLD functions as a DUB for RIPK1 in complex-I81, ubiquitin-like modifications of RIPK1 were observed in detergent-insoluble cytosolic fractions11, and a CYLD deficiency led to hyperubiquitinated RIPK1 in the necrosome, which impaired the formation of the RIPK1-RIPK3 complex89. These observations raise the possibility that the ubiquitination of RIPK1 in spatially distinct necrosomes may directly control its cytotoxicity. Accordingly, it has recently been reported that necrosome-associated RIPK1 undergoes K63- and M1-linked polyubiquitination by cIAP1/2 and LUBAC, respectively52,139, but the functional link between these E3 Ub ligases and RIPK1 cytotoxicity during the necroptosis signaling pathway is less understood. Recent studies have proposed a possible role for specific ubiquitination sites of RIPK1 at K115 that function as positive regulators for necroptosis by facilitating RIPK1-RIPK3 interactions139. Interestingly, mutation of this ubiquitination site on RIPK1 (K115) suppressed RIPK1 autophosphorylation and necroptosis without affecting the inherent RIPK1 kinase activity139, indicating that it may also play an additional role in determining cell death by modifying the ubiquitination status of necrosome-associated RIPK1. Nevertheless, since RIPK1 ubiquitination occurs in the necrosome complex independent of RIPK3 and MLKL139, the question remains whether the ubiquitinated RIPK1 at K115 found in necrosomes was translocated from complex-IIb. A member of the Pellino family, Pellino 1 (PELI1), an E3 Ub ligase, may provide the possibility for ubiquitination of RIPK1 in complex-IIb140,141,142. PELI1 is not involved in RIPK1 ubiquitination during TNF-induced apoptosis, but it modulates K63-linked ubiquitination of RIPK1 on K115 bound to complex-IIb in a kinase-dependent manner that promotes the interaction of RIPK1 and RIPK3 to form necrosomes140. Thus, PELI1 can function as a pronecroptotic E3 Ub ligase that targets kinase-active RIPK1 in complex-IIb. Paradoxically, PELI1 was reported to exhibit a protective effect in the context of necroptosis by regulating the protein level of RIPK3143,144. PELI1 directly catalyzes K48-linked ubiquitination of RIPK3 for proteasomal degradation and thus selectively restricts RIPK3-mediated necroptosis144. Thus, this finding for PELI1 in the context of necroptosis execution is still under debate140,143,144. Since RIPK3 kinase activity is required for PELI1 recruitment145, it is possible that PELI1 differentially targets RIPK1 or RIPK3 depending on the status of RIPK3 activation. Further in vitro biochemical and in vivo animal experiments are required to confirm the pathophysiological pathway by which PELI1 regulates PCD in complex-IIb or necrosomes by RIPK1 or RIPK3.
Clinical relevance of RIPK1 cytotoxicity in inflammation and cancer immunotherapy
Unlike apoptosis, necroptosis is defined as an inflammatory form of cell death induced by the release of damage-associated molecular patterns (DAMPs) and aberrant inflammatory cytokines in dying cells146. Given the pivotal role of the kinase activity of RIPK1 in controlling necroptosis, RIPK1-mediated cell death is involved in a variety of pathophysiological settings, including inflammation and viral and bacterial infections147. Thus, regulation of the early cell death checkpoint that safeguards the cytotoxic potential of RIPK1 is of clinical relevance, potentially offering new therapeutic opportunities for chronic inflammatory diseases, such as rheumatoid arthritis, psoriasis and septic shock145,148,149. Consistent with this notion, GSK2982772, a highly specific RIPK1 inhibitor, has been developed150 and has successfully completed phase II of human clinical trials for the treatment of ulcerative colitis (NCT02903966), psoriasis (NCT02776033) and rheumatoid arthritis (NCT02858492), with an excellent safety profile151,152. Moreover, families of allosteric RIPK1 inhibitors, DNL758/788 (NCT03757325) and R552 (NCT03757351), are currently undergoing phase Ia/Ib or IIa clinical trials for various neuro-inflammatory diseases, including cutaneous lupus erythematosus, multiple sclerosis, Alzheimer’s disease and amyotrophic lateral sclerosis153.
Since unresolved inflammation is necessary for cancer progression154,155, it is believed that an aberrant innate and adaptive immune response contributes to tumorigenesis by inducing immune suppression and favoring aggressive clones for recurrence and/or metastasis156. While inhibition of RIPK1 activation has therapeutic potential for treating the abovementioned inflammatory or autoimmune diseases, the inflammatory nature of RIPK1-mediated cell death promotes immunogenicity and macrophage-mediated adaptive immune tolerance in some cancers157. Consistent with this notion, patients with severe immune deficiency, inflammatory bowel disease and arthritis have biallelic mutations of RIPK1158,159. Mechanistically, activation of RIPK1 and NF-κB signaling in dying cells stimulates immune responses, which subsequently drive dendritic cell (DC) maturation for CD8+ T-cell cross-priming160,161,162. Consequently, ectopic injection of RIPK1-derived necroptotic cells into the tumor microenvironment induces a systemic immune response driven by DCs and CD8+ T cells that promotes antitumor immunity and increased tumor antigen loading163. Thus, manipulating RIPK1 checkpoints to boost its cytotoxicity in cancer cells might be an attractive therapeutic avenue in which immunogenic cell death is a desired outcome.
Given that RIPK1 ubiquitination can safeguard the killing potential of RIPK1, small molecular mimetics of smac/DIABLO (smac-mimetics; SMs) have been developed to disrupt RIPK1 ubiquitination by antagonizing cIAP1/2164,165,166. At present, these SMs have shown promising therapeutic potential for cancer167,168. In addition to killing cancer cells, a number of studies have also reported the effects of SMs on both the innate and adaptive immune systems during cancer169. In some instances, SM administration not only induces a reversion of tumor-associated macrophages from a protumorous M2 phenotype to a proinflammatory M1-like phenotype170 but also sensitizes resistant cancer cells to inactivate by an inflammatory burst of TNF and IFN-γ171,172,173. Accordingly, several SMs, such as LCL161 and Debio1143, are undergoing initial clinical trials (http://clinicaltrials.gov). One of the most clinically advanced SMs (birinapant) is currently undergoing phase I/II clinical trials for a number of cancers, including myeloid leukemia and various solid tumors174,175,176. However, despite the promising preclinical results, the clinical trials have largely failed due to their poor efficacy when used as individual agents168,177. Thus, the challenge for future immunotherapy lies in research efforts directed toward the use of SMs as part of combination strategies to overcome resistance. In this regard, the recent discoveries of RIPK1-mediated early cell death checkpoints are of particular interest, especially with respect to combination strategies that incorporate SMs. In fact, activating the RIPK1-mediated cell death checkpoint by clinical inhibitors IKK or MK2 enhances the therapeutic efficacy of birinapant by triggering immunogenic cell death and antigen-cross priming CD8+ T cells in primary acute myeloid leukemia132,178,179. While many studies have demonstrated that necroptosis-mediated inflammatory cell death can promote antitumor immune responses163,180,181, it has also been reported that RIPK1 activity regulates tumor immunity by reprogramming tumor-associated macrophages in some cancers, independent of RIPK3157,182. At this point, there is still a limited understanding of the specific involvement of RIPK1 in the tumor immune microenvironment. Nevertheless, a better understanding of the mechanism that regulates the cytotoxic activity of RIPK1 in immune cells may offer new therapeutic options for chronic inflammatory diseases and cancer.
Conclusions and perspectives
While RIPK1 was originally characterized as a modulator of NF-κB activation through its interaction with IKKs, it is now clear that RIPK1 functions as a crucial early cell death checkpoint for determining the fate of cells, and this function is mainly regulated via its PTMs, including ubiquitination and phosphorylation. Considering the necessity of the kinase activity of RIPK1 for driving the necroptotic mode of cell death, the RIPK1-mediated cell death checkpoint could be targeted therapeutically to modulate human inflammatory diseases and cancer. In this sense, it will be important to identify pharmacological agents that directly control RIPK1 cytotoxicity to overcome some of the challenges associated with conventional immunotherapies. However, to date, the exact molecular mechanisms regulating RIPK1 ubiquitination or phosphorylation and its cytotoxic potential remain to be fully elucidated. Furthermore, it is still not clear whether ubiquitinated RIPK1 in necrosomes originates from the plasma membrane-bound complex-I or from the cytosolic pool. Thus, the compositions of the RIPK1-containing complex in complex-IIb and necrosomes and their subcellular localizations require further investigation. Studies addressing this issue would help our understanding of the molecular mechanisms that control the formation of death-inducing signaling complexes or the cytotoxic potential of RIPK1. Finally, the extra layer of complexity involved in regulating RIPK1 activity presents challenges, especially for the development of therapeutic agents that target the RIPK1-mediated cell death checkpoint for clinical applications in human diseases in the necroptosis era.
References
Fink, S. L. & Cookson, B. T. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916 (2005).
Kroemer, G., Dallaporta, B. & Resche-Rigon, M. E. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60, 619–642 (1998).
Ellis, H. M. & Horvitz, H. R. Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829 (1986).
Hengartner, M. O. Programmed cell death in the nematode C. elegans. Recent Prog. Horm. Res. 54, 213–222 (1999).
Earnshaw, W. C., Martins, L. M. & Kaufmann, S. H. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68, 383–424 (1999).
Dixit, V. M. Role of ICE-proteases in apoptosis. Adv. Exp. Med. Biol. 406, 113–117 (1996).
Kawahara, A., Ohsawa, Y., Matsumura, H., Uchiyama, Y. & Nagata, S. Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 143, 1353–1360 (1998).
Vanden, B. T. et al. More than one way to die: methods to determine TNF-induced apoptosis and necrosis. Methods Mol. Med 98, 101–126 (2004).
Lin, Y. et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 279, 10822–10828 (2004).
Vandenabeele, P., Galluzzi, L., Vanden, B. T. & Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 (2010).
Cho, Y. S. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Chen, X. et al. Mosaic composition of RIP1–RIP3 signalling hub and its role in regulating cell death. Nat. Cell Biol. 24, 471–482 (2022).
Stanger, B. Z., Leder, P., Lee, T. H., Kim, E. & Seed, B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81, 513–523 (1995).
Hsu, H., Xiong, J. & Goeddel, D. V. The TNF receptor 1–associated protein TRADD signals cell death and NF-κB activation. Cell 81, 495–504 (1995).
Sun, X. et al. RIP3, a novel apoptosis-inducing kinase. J. Biol. Chem. 274, 16871–16875 (1999).
Newton, K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 25, 347–353 (2015).
O’Donnell, M. A. & Ting, A. T. NF-κB and ubiquitination: partners in disarming RIPK1-mediated cell death. Immunol. Res. 54, 214–226 (2012).
Won, M., Byun, H. S., Park, K. A. & Hur, G. M. Post-translational control of NF-κB signaling by ubiquitination. Arch. Pharmacal Res. 39, 1075–1084 (2016).
Declercq, W., Vanden, B. T. & Vandenabeele, P. RIP kinases at the crossroads of cell death and survival. Cell 138, 229–232 (2009).
Hinz, M. & Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 15, 46–61 (2014).
Chen, Z. J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 7, 758–765 (2005).
Tsuchiya, Y., Nakabayashi, O. & Nakano, H. FLIP the switch: regulation of apoptosis and necroptosis by cFLIP. Int. J. Mol. Sci. 16, 30321–30341 (2015).
Hughes, M. A. et al. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol. Cell 61, 834–849 (2016).
Karin, M. & Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 3, 221–227 (2002).
Annibaldi, A. & Meier, P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol. Med. 24, 49–65 (2018).
Ting, A. T. & Bertrand, M. J. M. More to life than NF-κB in TNFR1 signaling. Trends Immunol. 37, 535–545 (2016).
Kang, K., Lee, S. R., Piao, X. & Hur, G. M. Post-translational modification of the death receptor complex as a potential therapeutic target in cancer. Arch. Pharmacal Res. 42, 76–87 (2019).
Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014).
Newton, K. Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harb. Perspect. Biol. 12, a036368 (2020).
Xu, Q., Choksi, S. & Liu, Z. Switching from TNF-induced inflammation to death signaling. Mol. Cell. Oncol. 5, e1392402 (2017).
Chen, G. & Goeddel, D. V. TNF-R1 signaling: a beautiful pathway. Science 296, 1634–1635 (2002).
Dynek, J. N. et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29, 4198–4209 (2010).
Peltzer, N., Darding, M. & Walczak, H. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol. 26, 445–461 (2016).
Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247 (2017).
Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844 (2009).
Hsu, H., Shu, H. B., Pan, M. G. & Goeddel, D. V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84, 299–308 (1996).
Zhang, L., Blackwell, K., Shi, Z. & Habelhah, H. The RING domain of TRAF2 plays an essential role in the inhibition of TNFα-induced cell death but not in the activation of NF-κB. J. Mol. Biol. 396, 528–539 (2010).
Yin, Q., Lamothe, B., Darnay, B. G. & Wu, H. Structural basis for the lack of E2 interaction in the RING domain of TRAF2. Biochemistry 48, 10558–10567 (2009).
Vince, J. E. et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-kappab and to prevent tnf-induced apoptosis. J. Biol. Chem. 284, 35906–35915 (2009).
Yeh, W. C. et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7, 715–725 (1997).
Tada, K. et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J. Biol. Chem. 276, 36530–36534 (2001).
Pobezinskaya, Y. L. et al. The function of TRADD in signaling through tumor necrosis factor receptor 1 and TRIF-dependent Toll-like receptors. Nat. Immunol. 9, 1047–1054 (2008).
Zhang, L. et al. TRAF2 suppresses basal IKK activity in resting cells and TNFα can activate IKK in TRAF2 and TRAF5 double knockout cells. J. Mol. Biol. 389, 495–510 (2009).
Mahoney, D. J. et al. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. Proc. Natl Acad. Sci. USA 105, 11778–11783 (2008).
Bertrand, M. J. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
Varfolomeev, E. et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFα)-induced NF-κB activation. J. Biol. Chem. 283, 24295–24299 (2008).
Zheng, C., Kabaleeswaran, V., Wang, Y., Cheng, G. & Wu, H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol. Cell 38, 101–113 (2010).
Rothe, M., Pan, M. G., Henzel, W. J., Ayres, T. M. & Goeddel, D. V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 83, 1243–1252 (1995).
Moulin, M. et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 31, 1679–1691 (2012).
Emmerich, C. H. et al. The linear ubiquitin chain assembly complex forms part of the TNF-R1 signalling complex and is required for effective TNF-induced gene induction and prevents TNF-induced apoptosis. Adv. Exp. Med. Biol. 691, 115–126 (2011).
Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
de Almagro, M. C., Goncharov, T., Newton, K. & Vucic, D. Cellular IAP proteins and LUBAC differentially regulate necrosome-associated RIP1 ubiquitination. Cell Death Dis. 6, e1800 (2015).
Rieser, E., Cordier, S. M. & Walczak, H. Linear ubiquitination: a newly discovered regulator of cell signalling. Trends Biochem. Sci. 38, 94–102 (2013).
Tokunaga, F. & Iwai, K. LUBAC, a novel ubiquitin ligase for linear ubiquitination, is crucial for inflammation and immune responses. Microbes Infect. 14, 563–572 (2012).
Peltzer, N. et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 9, 153–165 (2014).
Ikeda, F. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 471, 637–341 (2011).
Tokunaga, F. et al. SHARPIN is a component of the NF-kappaBactivating linear ubiquitin chain assembly complex. Nature 471, 633–636 (2011).
Emmerich, C. H. et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl Acad. Sci. USA 110, 15247–15252 (2013).
Fiil, B. K. et al. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol. Cell 50, 818–830 (2013).
Peltzer, N. et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 557, 112–127 (2018).
Nanda, S. K. et al. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J. Exp. Med 208, 1215–1228 (2011).
Dziedzic, S. A. et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat. Cell Biol. 20, 58–68 (2018).
Cai, J. et al. ABIN-1 is a key regulator in RIPK1-dependent apoptosis (RDA) and necroptosis, and ABIN-1 deficiency potentiates necroptosis-based cancer therapy in colorectal cancer. Cell Death Dis. 12, 140 (2021).
Feltham, R. et al. Mind bomb regulates cell death during TNF signaling by suppressing RIPK1’s cytotoxic potential. Cell Rep. 23, 470–484 (2018).
Schneider-Brachert, W. et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21, 415–428 (2004).
Mosselmans, R., Hepburn, A., Dumont, J. E., Fiers, W. & Galand, P. Endocytic pathway of recombinant murine tumor necrosis factor in L-929 cells. J. Immunol. 141, 3096–3100 (1988).
Schutze, S. et al. Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J. Biol. Chem. 274, 10203–10212 (1999).
Tchikov, V., Bertsch, U., Fritsch, J., Edelmann, B. & Schütze, S. Subcellular compartmentalization of TNF receptor-1 and CD95 signaling pathways. Eur. J. Cell Biol. 90, 467–475 (2010).
Polo, S. et al. A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature 416, 451–455 (2002).
Hofmann, K. & Falquet, L. A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem. Sci. 26, 347–350 (2001).
Won, M. et al. ANKRD13a controls early cell-death checkpoint by interacting with RIP1 independent of NF-kappaB. Cell Death Differ. 29, 1152–1163 (2021).
Song, K. et al. Epsins 1 and 2 promote NEMO linear ubiquitination via LUBAC to drive breast cancer development. J. Clin. Invest 131, e129374 (2021).
Coornaert, B., Carpentier, I. & Beyaert, R. A20: central gatekeeper in inflammation and immunity. J. Biol. Chem. 284, 8217–8221 (2009).
Wertz, I. E. et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430, 694–699 (2004).
Newton, K. et al. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134, 668–678 (2008).
Lee, E. G. et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289, 2350–2354 (2000).
Bellail, A. C., Olson, J. J., Yang, X., Chen, Z. J. & Hao, C. A20 ubiquitin ligase-mediated polyubiquitination of RIP1 inhibits caspase-8 cleavage and TRAIL-induced apoptosis in glioblastoma. Cancer Disco. 2, 140–155 (2012).
Daniel, S. et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood 104, 2376–2384 (2004).
Lu, T. T. et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity 38, 896–905 (2013).
Verhelst, K. et al. A20 inhibits LUBAC-mediated NF-κB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J. 31, 3845–3855 (2012).
Draber, P. et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 13, 2258–2272 (2015).
Polykratis, A. et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 21, 731–742 (2019).
Bignell, G. R. et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 25, 160–165 (2000).
Poblete, G. P. et al. Phenotype diversity in familial cylindromatosis: a frameshift mutation in the tumor suppressor gene CYLD underlies different tumors of skin appendages. J. Invest. Dermatol. 119, 527–531 (2002).
Trompouki, E. et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 424, 739–796 (2003).
Sun, S. C. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 8, 501–511 (2008).
Brummelkamp, T. R., Nijman, S. M., Dirac, A. M. & Bernards, R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature 424, 797–801 (2003).
Kovalenko, A. et al. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 424, 801–805 (2003).
Moquin, D. M., McQuade, T. & Chan, F. M. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8, e76841 (2013).
Legarda, D. et al. CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell Rep. 15, 2449–2461 (2016).
Ganjam, G. K. et al. Cylindromatosis mediates neuronal cell death in vitro and in vivo. Cell Death Differ. 25, 1394–1407 (2018).
Wright, A. et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev. Cell 13, 705–716 (2007).
Alameda, J. P. et al. CYLD regulates keratinocyte differentiation and skin cancer progression in humans. Cell Death Dis. 2, e208 (2011).
Hellerbrand, C. et al. Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis 28, 21–27 (2007).
Wang, L. et al. CYLD deficiency enhances metabolic reprogramming and tumor progression in nasopharyngeal carcinoma via PFKFB3. Cancer Lett. 532, 215586 (2022).
Massoumi, R. et al. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J. Exp. Med. 206, 221–232 (2009).
Shinriki, S. et al. Loss of CYLD promotes cell invasion via ALK5 stabilization in oral squamous cell carcinoma. J. Pathol. 244, 367–379 (2018).
Enesa, K. et al. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J. Biol. Chem. 283, 7036–7045 (2008).
Luong, L. A. et al. Cezanne regulates inflammatory responses to hypoxia in endothelial cells by targeting TRAF6 for deubiquitination. Circ. Res. 112, 1583–1591 (2013).
Enesa, K. et al. Hydrogen peroxide prolongs nuclear localization of NF-kappaB in activated cells by suppressing negative regulatory mechanisms. J. Biol. Chem. 283, 18582–18590 (2008).
Geserick, P. et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187, 1037–1054 (2009).
Hitomi, J. et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 (2008).
Wang, L., Du, F. & Wang, X. TNF-α induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
Feoktistova, M. et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011).
Tenev, T. et al. The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448 (2011).
Bertrand, M. J. & Vandenabeele, P. The Ripoptosome: death decision in the cytosol. Mol. Cell 43, 323–325 (2011).
Meng, H. et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc. Natl Acad. Sci. USA 115, E2001–E2009 (2018).
Laurien, L. et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nat. Commun. 11, 1747 (2020).
Wegner, K. W., Saleh, D. & Degterev, A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol. Sci. 38, 202–225 (2017).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
McQuade, T., Cho, Y. & Chan, F. K. Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 456, 409–415 (2013).
Goossens, V., Grooten, J., De Vos, K. & Fiers, W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc. Natl Acad. Sci. USA 92, 8115–8119 (1995).
Roca, F. J. & Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153, 521–534 (2013).
Sun, W. et al. Inhibition of lung cancer by 2-Methoxy-6-Acetyl-7-Methyljuglone through induction of necroptosis by targeting receptor-interacting protein 1. Antioxid. Redox Signal. 31, 93–108 (2019).
Schenk, B. & Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 34, 5796–5806 (2015).
Zhang, Y. et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329 (2017).
Delanghe, T., Dondelinger, Y. & Bertrand, M. J. M. RIPK1 kinase-dependent death: a symphony of phosphorylation events. Trends Cell Biol. 30, 189–200 (2020).
Hacker, H. & Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13 (2006).
Dondelinger, Y., Vandenabeele, P. & Bertrand, M. J. Regulation of RIPK1’s cell death function by phosphorylation. Cell Cycle 15, 5–6 (2016).
Dondelinger, Y. et al. NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015).
Piao, X. et al. 8-Geranylumbelliferone isolated from Paramignya trimera triggers RIPK1/RIPK3-dependent programmed cell death upon TNFR1 ligation. Biochem. Pharmacol. 192, 114733 (2021).
Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 10, 1729 (2019).
Lafont, E. et al. TBK1 and IKKepsilon prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018).
Xu, D. et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491 e1419 (2018).
Chau, T. L. et al. Are the IKKs and IKK-related kinases TBK1 and IKK-epsilon similarly activated? Trends Biochem. Sci. 33, 171–180 (2008).
Dondelinger, Y. et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20, 1381–1392 (2013).
Totzke, J. et al. Takinib, a selective TAK1 inhibitor, broadens the therapeutic efficacy of TNF-α inhibition for cancer and autoimmune disease. Cell Chem. Biol. 24, 1029–1039 (2017).
Kang, K. et al. 3-O-acetylrubianol C (3AR-C) induces RIPK1-dependent programmed cell death by selective inhibition of IKKβ. FASEB J. 34, 4369–4383 (2020).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Janes, K. A., Reinhardt, H. C. & Yaffe, M. B. Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell 135, 343–354 (2008).
Gaestel, M., Kotlyarov, A. & Kracht, M. Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discov. 8, 480–499 (2009).
Lalaoui, N. et al. Targeting p38 or MK2 enhances the anti-leukemic activity of Smac-mimetics. Cancer Cell 30, 499–500 (2016).
Vandendriessche, B. et al. MAPK-activated protein kinase 2-deficiency causes hyperacute tumor necrosis factor-induced inflammatory shock. BMC Physiol. 14, 5 (2014).
Jaco, I. et al. MK2 phosphorylates RIPK1 to prevent TNF-i nduced cell death. Mol. Cell 66, 698–710 e695 (2017).
Menon, M. B. et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259 (2017).
Oberst, A. MK2 balances inflammation and cell death. Nat. Cell Biol. 19, 1150–1152 (2017).
Dondelinger, Y., Delanghe, T. & Bertrand, M. M. J. MK2 puts an additional brake on RIPK1 cytotoxic potential. Cell Death Differ. 25, 457–459 (2018).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
de Almagro, M. C. et al. Coordinated ubiquitination and phosphorylation of RIP1 regulates necroptotic cell death. Cell Death Differ. 24, 26–37 (2017).
Wang, H. et al. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proc. Natl Acad. Sci. USA. 114, 11944–11949 (2017).
Moynagh, P. N. The roles of Pellino E3 ubiquitin ligases in immunity. Nat. Rev. Immunol. 14, 122–131 (2014).
Weinlich, R. & Green, D. R. The two faces of receptor interacting protein kinase-1. Mol. Cell 56, 469–480 (2014).
Seo, J. et al. CHIP controls necroptosis through ubiquitylation- and lysosomedependent degradation of RIPK3. Nat. Cell Biol. 18, 291–302 (2016).
Choi, S. W. et al. PELI1 selectively targets kinase-active RIP3 for ubiquitylation dependent proteasomal degradation. Mol. Cell 70, 920–935 (2018).
He, S. & Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 19, 912–922 (2018).
Nakano, H., Murai, S. & Moriwaki, K. Regulation of the release of damage-associated molecular patterns from necroptotic cells. Biochem. J. 479, 677–685 (2022).
Liu, Z. & Chan, F. K. Regulatory mechanisms of RIPK1 in cell death and inflammation. Semin. Cell Dev. Biol. 109, 70–75 (2021).
Witt, A. & Vucic, D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 24, 1160–1171 (2017).
Newton, K. et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576 (2016).
Harris, P. A. et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261 (2017).
Weisel, K. et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res. Ther. 23, 85 (2021).
Mifflin, L., Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 19, 553–571 (2020).
Li, S., Qu, L., Wang, X. & Kong, L. Novel insights into RIPK1 as a promising target for future Alzheimer’s disease treatment. Pharmacol. Ther. 231, 107979 (2022).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Balkwill, F. & Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 357, 539–545 (2001).
Palucka, A. K. & Coussens, L. M. The basis of oncoimmunology. Cell 164, 1233–1247 (2016).
Wang, W. et al. RIP1 kinase drives macrophage mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 34, 757–774 (2018).
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 116, 970–975 (2019).
Cuchet-Lourenço, D. et al. RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018).
Yatim, N., Cullen, S. & Albert, M. L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 17, 262–275 (2017).
Yatim, N. et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 350, 328–334 (2015).
Aaes, T. L. et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 15, 274–287 (2016).
Snyder, A. G. et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 4, eaaw2004 (2019).
Sharma, S. K., Straub, C. & Zawel, L. Development of peptidomimetics targeting IAPs. Int. J. Pept. Res. Ther. 12, 21–32 (2006).
Kipp, R. A. et al. Molecular targeting of inhibitor of apoptosis proteins based on small molecule mimics of natural binding partners. Biochemistry 41, 7344–7349 (2002).
Derrick, J. C. & Sergio, H. Smac mimetics as new cancer therapeutics. Anti-Cancer Drugs 20, 646–658 (2009).
Rathore, R., McCallum, J. E., Varghese, E., Florea, A. M. & Busselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 22, 898–919 (2017).
Fulda, S. & Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 11, 109–124 (2012).
Cetraro, P., Plaza-Diaz, J., MacKenzie, A. & Abadía-Molina, F. A review of the current impact of inhibitors of apoptosis proteins and their repression in cancer. Cancers (Basel) 14, 1671 (2022).
Lecis, D. et al. Smac mimetics induce inflammation and necrotic tumour cell death by modulating macrophage activity. Cell Death Dis. 4, e920 (2013).
Tanzer, M. C. et al. Combination of IAP antagonist and IFNgamma activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 24, 481–491 (2017).
Rettinger, E. et al. SMAC mimetic BV6 enables sensitization of resistant tumor cells but also affects Cytokine-Induced Killer (CIK) cells: a potential challenge for combination therapy. Front Pediatr. 2, 75 (2014).
Bake, V. R., Eckhardt, S., Belz, I., Fulda, K. & Synergistic, S. interaction of Smac mimetic and IFNalpha to trigger apoptosis in acute myeloid leukemia cells. Cancer Lett. 355, 224–231 (2014).
Condon, S. M. et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J. Med. Chem. 57, 3666–3677 (2014).
Cong, H. et al. Inhibitor of apoptosis protein (IAP) antagonists in anticancer agent discovery: current status and perspectives. J. Med. Chem. 62, 5750–5772 (2019).
Infante, J. R. et al. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 32, 3103–3110 (2014).
Noonan, A. M. et al. Pharmacodynamic markers and clinical results from the phase 2 study of the SMAC mimetic birinapant in women with relapsed platinum-resistant or -refractory epithelial ovarian cancer. Cancer 122, 588–597 (2016).
Brumatti, G. et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 8, 339–369 (2016).
Martínez-Limón, A., Joaquin, M., Caballero, M., Posas, F. & de, N. E. The p38 pathway: from biology to cancer therapy. Int. J. Mol. Sci. 21, 1913 (2020).
Lou, J. et al. Caspase-independent regulated necrosis pathways as potential targets in cancer management. Front. Oncol. 10, 616952 (2021).
Krysko, O. et al. Necroptotic cell death in anti-cancer therapy. Immunol. Rev. 280, 207–219 (2017).
Kang, T. B., Jeong, J. S., Yang, S. H., Kovalenko, A. & Wallach, D. Caspase-8 deficiency in mouse embryos triggers chronic RIPK1-dependent activation of inflammatory genes, independently of RIPK3. Cell Death Differ. 25, 1107–1117 (2018).
Funding
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2020R1A2C2005317; No. 2017R1A5A2015385), by the BK21 FOUR Program by Chungnam National University Research Grant 2021 and by the Macau Science and Technology Development Fund (FDCT0078/2020/A2 and 0031/2021/A1).
Author information
Authors and Affiliations
Contributions
G.M.H. conceived and designed the review. E.J., K.A.P., H-M. S., and G.M.H. wrote the manuscript. E. J and K.A.P. prepared the figures and table. H-M.S. and G.M.H. reviewed and edited the manuscript. All authors read and agreed to the final version of manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ju, E., Park, K.A., Shen, HM. et al. The resurrection of RIP kinase 1 as an early cell death checkpoint regulator—a potential target for therapy in the necroptosis era. Exp Mol Med 54, 1401–1411 (2022). https://doi.org/10.1038/s12276-022-00847-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00847-4
This article is cited by
-
Cellular heterogeneity in TNF/TNFR1 signalling: live cell imaging of cell fate decisions in single cells
Cell Death & Disease (2024)
-
Mediators of necroptosis: from cell death to metabolic regulation
EMBO Molecular Medicine (2024)
-
RIP3 in Necroptosis: Underlying Contributions to Traumatic Brain Injury
Neurochemical Research (2024)
-
Mitochondria-associated programmed cell death as a therapeutic target for age-related disease
Experimental & Molecular Medicine (2023)
