
Abstract
Isotope tracer infusion studies employing lactate, glucose, glycerol, and fatty acid isotope tracers were central to the deduction and demonstration of the Lactate Shuttle at the whole-body level. In concert with the ability to perform tissue metabolite concentration measurements, as well as determinations of unidirectional and net metabolite exchanges by means of arterial–venous difference (a-v) and blood flow measurements across tissue beds including skeletal muscle, the heart and the brain, lactate shuttling within organs and tissues was made evident. From an extensive body of work on men and women, resting or exercising, before or after endurance training, at sea level or high altitude, we now know that Organ–Organ, Cell–Cell, and Intracellular Lactate Shuttles operate continuously. By means of lactate shuttling, fuel-energy substrates can be exchanged between producer (driver) cells, such as those in skeletal muscle, and consumer (recipient) cells, such as those in the brain, heart, muscle, liver and kidneys. Within tissues, lactate can be exchanged between white and red fibers within a muscle bed and between astrocytes and neurons in the brain. Within cells, lactate can be exchanged between the cytosol and mitochondria and between the cytosol and peroxisomes. Lactate shuttling between driver and recipient cells depends on concentration gradients created by the mitochondrial respiratory apparatus in recipient cells for oxidative disposal of lactate.
Similar content being viewed by others


Introduction
The results of isotope tracer studies at the whole-body and tissue levels have clarified longstanding issues concerning metabolic regulation. In particular, the role of the mitochondrial reticulum1,2 in lactate disposal has been identified. The reticulum lowers cellular [lactate] by oxidative disposal, thus acting to develop the concentration gradients necessary for lactate fluxes across organ, tissue, and cellular compartments. Similar to other solutes, gases, and entities in physics and physiology, lactate moves from areas of high to areas of low concentrations. Hence, lactate-producing cells, organs, and tissues with high lactate levels drive carbon flux to disposal (recipient) sites with low lactate levels. In this scenario, the mitochondrial reticulum, in which lactate is disposed of via oxidation, creates the driving force for shuttling lactate around the body and within organs, tissues and cells3,4,5. Key features of the process involve the mitochondrial (m) Lactate Oxidation Complex (mLOC), which contains mitochondrial lactate dehydrogenase (mLDH), a monocarboxylate (lactate) transporter (mMCT), a scaffolding protein (CD147) and cytochrome oxidase (COx). However, within the context of this special issue on the use of isotopic tracers to study metabolism, the results of lactate tracer studies directly led to the discovery of the mLOC for lactate shuttling to occur in vivo. We begin with a discussion of mLDH.
Similar to the concept of an mLOC, discussions on the role of mLDH in intermediary metabolism have endured a long and winding history3,5,6, perhaps a path not unusual in the history of science3,7,8. Because of the fundamental importance of mLDH in physiology and metabolism, revisiting the history of discoveries is warranted at the outset of this paper. The presence of mLDH is essential to explain the role of lactate in affecting lactate flux and ultimately the regulation of energy substrate partitioning in vivo3,5,9.
Background knowledge
Today, we know that some types of facultative cells can be cultured with lactate as a preferred fuel because their mitochondrial reticulae consume and oxidize lactate directly without conversion to pyruvate in the cytosol. For instance, mitochondria of yeast (Saccharomyces cerevisiae) contain Flavocytochrome b2, a lactate-cytochrome c oxidoreductase10 that couples lactate dehydrogenation to the reduction of cytochrome c11. In fact, the association between cytochrome b2 and LDH in yeast can be traced to the 1940s12. A similar phenomenon occurs in mammalian muscle13,14,15,16,17,18,19, in persons resting at the sea level, and even in men exercising at sea level or at an altitude of 4300 m20,21. Technological limitations, such as the availability of isotope tracer methodology, the ability to catheterize veins and arteries for continual (a-v) measurements, the ability to determine tissue and systemic blood flow rates, and mass spectrometry for turnover analyses, made such data unobtainable in the 1920s. Consequently, longstanding ideas on the role of lactate in physiology originated on the basis of incomplete data on isolated, nonperfused, nonoxygenated amphibian muscles made to contract continually in ways not intended by nature22,23,24,25,26,27. More recently, with the advent of isotope tracers and nuclear magnetic resonance (NMR) spectroscopy, we now know that contractions stimulate glycolysis in muscle independent of O2 availability28,29,30,31,32,33 and that lactate is oxidized in working skeletal31,34,35 and cardiac33,36,37,38,39 muscles.
Mitochondrial lactate oxidation (the mind’s eye view of Polyphemus)
Contemporary textbooks of biochemistry and physiology abound the 19th-century concepts that metabolism is either “anaerobic” (without O2) or “aerobic” (with O2)40. Glycolytic flux from glucose and glycogen is typically depicted in textbooks as progressing to pyruvate and then to the tricarboxylic acid (TCA) cycle. However, if oxygen is absent, textbooks assert that glycolysis progresses to lactate41,42,43. This is a convenient motif, typically copied by one textbook author from another and then through serial editions of texts. Amazingly, some textbook authors who advanced the idea of lactate production due to oxygen limitation were biochemists who worked with cells in high-glucose-containing culture media under fully aerobic conditions of one atmosphere pressure in which the partial pressure of oxygen was at least 50% greater than in the arterial blood of individuals at sea-level altitudes. Because the maintenance of cells in such preparations required daily changing of the incubation media to maintain high [glucose], low [lactate], and physiological pH, the observations should have informed the investigators that lactate was produced under fully aerobic conditions. Contemporaneously, physiologists measured lactate [L] to pyruvate [P] concentration ratios (L/P) of 10 in muscles and blood in resting mammals, including humans, and further observed the L/P to rise more than 100 during submaximal exercise44. However, did any of the textbook authors ever look to determine whether isolated mitochondria oxidize lactate? For many textbook authors41,42,43, the answer is “no.” However, for a few others, the answer is “yes”45,46. Moreover, some investigators also looked for the presence of a mitochondrial monocarboxylate (lactate) transporter (mMCT) and a lactate dehydrogenase (mLDH) enzyme. Unfortunately, while some attempts failed47,48,49, fortunately, other attempts to observe mitochondrial lactate oxidation and the presence of mLDH and mMCT were successful13,50,51,52,53,54,55. Importantly, lactate oxidation in human muscle mitochondrial preparations was observed56. Regrettably, for a time, positive results were overlooked or castigated as being “controversial” and dismissed for failing to fit with established paradigms of metabolic regulation. However, now that the MitoCarta (https://www.broadinstitute.org/mitocarta/mitocarta30-inventory-mammalian-mitochondrial-proteins-and-pathways)57 and MitoMiner (https://mitominer.mrc-mbu.cam.ac.uk/release-4.0/begin.do) databases have been published, mLDH is a recognized constituent of the mitochondrial proteome. Acknowledging that lactate, not pyruvate, is the main gluconeogenic precursor and carbohydrate energy source shared at the organismal and tissue levels upsets archaic concepts of the organization of intermediary metabolism. Nevertheless, despite the evidence, in the manner of Polyphemus, some58,59 have ignored the literature and databases on mLDH and mMCT and their roles in mitochondrial lactate oxidation60.
The lactate:pyruvate affair
Regrettably and inexplicably, thus far, as the use of isotope tracers to study lactate metabolism is concerned, the literature has been muddled, creating a “Lactate:Pyruvate Affair” that needed to be corrected60. Specifically, there has been controversy over whether lactate tracers measure lactate (L) or pyruvate (P) turnover. Despite a series of analytical errors, the use of inappropriate tissue and animal models, a failure to consider L and P pool sizes in modeling results, inappropriate tracer and blood sampling sites, a failure to anticipate roles of heart and lung parenchyma on L ⇔ P interactions, and a failure to appreciate results from magnetic resonance spectroscopy (MRS) and immunocytochemistry61,62, it is clear that carbon-labeled lactate tracers can be used to quantitate lactate fluxes in situ and in vivo.
The early twentieth century
In the early literature of the 20th century, there were several threads to a contemporary understanding of lactate production and oxidation during postprandial and exercise conditions. Among those threads were the findings of Fletcher and Hopkins, who showed that lactic acid disappeared when frog muscles fatigued by electrical stimulation were placed in oxygen-rich environments63. Albeit in another field of endeavor, cancer research, another thread was the finding of Warburg that lactate was produced by fully oxygenated cancer cells64. Nonetheless, while there were data from a reputable investigator (Nobel Laureate) that lactate could be produced under fully aerobic conditions and that oxidative removal of lactate could be accomplished without reconversion to glucose, lactate and lactic acid in cell metabolism were misconstrued as waste products of oxygen-limited metabolism and fatigue agents early in the 20th century65,66,67.
Most importantly, several sets of findings in the late 20th century led to the realization of oxidative lactate disposal. Among these sets of findings was evidence that isolated mitochondrial preparations could oxidize lactate in vitro13,54 and that mitochondria contained the enzymatic apparatus to oxidize lactate13,53,68. Importantly, isotope tracer studies showed oxidative lactate disposal in mammals and humans in vivo69,70,71,72,73. Evidence obtained on individuals studied in vivo is elegantly supported by the results of studies using MRS technology28,33,34,74. These important sets of findings are addressed sequentially.
Overlooked findings
The first investigator to evaluate mitochondrial lactate metabolism was Mario Umberto Dianzani (1925–2014), who, in 1951, published a paper, translation of which is “Distribution of lactic acid oxidase in liver and kidney cells of normal rats and rats with fatty degeneration of the liver”55. A translation of a section on p. 182 is “In conclusion, the [cellular] lactic acid-oxidizing systems are localized 80 to 100% in the mitochondria.” Although Dianzani had a prolific career in hepatic toxicology and only recently retired from the Department of Experimental Medicine and Oncology, University of Turin, he apparently never followed up on his finding of mitochondrial lactate oxidation, and his seminal findings went unrecognized even by workers in the field of mitochondrial energetics.
First to report the presence of mitochondrial LDH were Nobuhisa Baba and Hari M. Sharma, who used electron microscopy (EM) to study the hearts and pectoralis muscles of rats53 (Fig. 1). Decades later, their results showing mLDH were confirmed by many others, including Brandt and Kline et al.51,52,75, Brooks et al.13, Nakae et al.76, Taylor et al.77, Lemire16, Szczesna-Kaczmarek et al.18,19, Young et al.54, and Pagliarini et al.57. Subsequently, the results were extended to human skeletal muscle mitochondria by Dubouchaud et al. in the Brooks Laboratory15 (Fig. 2). Retrospectively, based on their seminal observations of mLDH using EM, Baba and Sharma were probably the first to use the term “lactate shuttle.” Regrettably, they never followed up on their original finding of mLDH, and similar to the discovery of Dianzani, the seminal findings of Baba and Sharma went unrecognized.

Reaction products of LDH seen in the mitochondria are primarily located within the inner mitochondrial membranes (C, cristae). M matrix. X 93,000. Figure 11 from Baba and Sharma53.

LDH isoenzyme patterns differ between the cytosol and mitochondria in both tissues. Note cytosolic and mitochondrial LDH isoform differences between the cytosol and mitochondria across tissues. From Brooks et al.13.
Beyond Dianzani78 and those who replicated his seminal observation that mitochondria were capable of oxidizing lactate13,51,52,54,56,79 were the efforts of those who failed in their attempts to isolate mitochondrial preparations that contained sufficient mLDH to oxidize lactate47,48,49,80,81. In addition, there were others who routinely found LDH in their mitochondrial preparations but regarded their findings as an artifact and took steps to block mLDH, thus permitting the measured rate of exogenous pyruvate oxidation to rise82. The lesson to take from history is that mLDH is necessary for lactate oxidation. When mLDH is lost in the isolation of single fibers49 or mitochondrial vesicles68 or if oxamate is used to block mLDH13, the preparation will be able to oxidize pyruvate but not lactate.
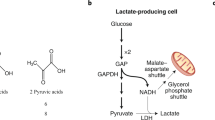
The ability of the mitochondrial reticulum to respire lactate is fundamental to the operation of lactate shuttles because lactate disposal via oxidation in the reticulum decreases the cellular [lactate], thus establishing lactate concentration gradients down which lactate fluxes. Hence, it is understandable that the rates and directions of lactate flux vary with physiological conditions (e.g., rest or exercise, early or late during exercise, endurance-trained or sedentary individuals, sea level or high altitude, carbohydrate nourished). For instance, in the postprandial state, when red skeletal muscles are glucose consumers and lactate producers4, and hence drivers of lactate shuttling, the heart, liver, and kidneys are consumers or recipients of lactate shuttle. In contrast, during exercise, white skeletal muscle and the integument are lactate producers and drivers of lactate shuttling, whereas highly oxidative (red) fiber types, cardiac tissue, liver, kidneys, and brain83,84,85,86,87 are sites of net lactate disposal. In these and other forms of shuttling, lactate fluxes from high to low concentrations with the cellular respiratory (mitochondrial) apparatus, including mLDH, functioning as removal sites3,5,88,89. These phenomena underpin the tracer-measured flux rates observed under conditions of rest and exercise35,90,91.
In a previous publication, the presence of a Postprandial Lactate Shuttle was introduced. Following a carbohydrate (CHO) meal, glycolysis, and lactate production in noncontracting red skeletal muscle92,93 raises lactate in the systemic circulation, thus providing a substrate for hepatic and renal gluconeogenesis85,94 and an energy substrate for skeletal muscle35,91,95 and the heart39,95,96, thereby forming a Postprandial Lactate Shuttle4.
Tracers and lactate turnover during rest and exercise
Pioneering work in the field of lactate kinetics in exercising mammals was performed by Florent Depocas (1923–2004) and colleagues69. Using a continuous infusion of [U-14C]lactate into dogs during rest and continuous steady-state exercise, they made several key, fundamental findings regarding lactate metabolism. These findings included (1) active lactate turnover during the resting postabsorptive condition; (2) ~1/2 of lactate formed during rest is removed through oxidation; (3) the turnover rate of lactate increases during exercise compared to rest even if there is only a minor change in blood lactate concentration; (4) the fraction of lactate disposal through oxidation increases to approximately 3/4 during exercise, and (5) a minor fraction (1/10-1/4) of lactate removed is converted to glucose via the Cori Cycle during exercise. Although the fractions are subject to species and experimental variations, the essential results have been reproduced in rats71, rabbits97, dogs69,98,99, horses100, and humans21,35,36,70,73,91,101. Depocas’ work on lactate metabolism was just one small part of his work at the Canadian NRC, where he held wide-ranging interests in the metabolic and endocrine responses of mammals to hypothermia.
Over the span of two decades, commencing in the mid-1980s, Brooks Lab Staff Research Associates Michael A. Horning, Gretchen C. Casazza and Jill A. Fattor; Brooks’ graduate students Timothy P. White, Glenn A. Gaesser, Casey M. Donovan, William C. (Bill) Stanley, Robert S. Mazzeo, Bryan C. Bergman, Benjamin F. Miller, David A. Roth, Sang-Hoon Suh, Rajaa Hussien, and Chi-An W. Emhoff; and postdoctoral fellows, visiting scholars and collaborators such as Thomas D. Fahey, Eugene E. Wolfel, Jacques Mercier, Hashimoto, Hervé Dubouchaud, Grant McClelland and Laurent A. Messonnier made concerted efforts to describe the effect of exercise intensity and training state on whole-body and working muscle lactate oxidation and gluconeogenesis from lactate in humans. The effects of exercise intensity on whole-body lactate turnover and oxidation were initially reported by Stanley70,91,95 and Mazzeo73, who showed that tracer-measured lactate turnover and oxidation scaled to the metabolic rate during exercise and that while oxidation accounted for ~50% of lactate disposal in resting men, oxidation accounted for 75–80% of lactate disposal during continuous hard exercise.
Studies of lactate metabolism in working skeletal and cardiac muscle in men during exercise were reported by Stanley et al. in 198670,91 and Edward W. Gertz et al. in 198839,96, respectively. The advantage of using tracer infusions along with simultaneous a-v difference and blood flow measurements across working muscle beds is that lactate uptake and net release, therefore, total production (= the sum of uptake and net release), can be determined simultaneously. Furthermore, another advantage of using tracer infusions along with simultaneous a-v 13CO2 and blood flow measurements across tissue muscle beds is that tissue metabolite oxidation can be measured. In addition, working muscle turnover and oxidation rates can be compared with whole-body rates. The results showed simultaneous lactate extraction (uptake) and release (production) and oxidation by working human leg muscles, which explains most whole-body lactate turnover and oxidation rates91. Importantly, due to intramuscular lactate extraction, turnover and oxidation, net chemical balance measurements underestimate true lactate production and oxidation rates for metabolites such as lactate and free fatty acids that turnover within tissue beds102. Subsequently, using variations of the same methodology, cerebral glucose, and lactate uptake and release were determined in traumatic brain injury patients and healthy controls103,104.
Another relevant but underappreciated and often unmentioned aspect of the issue of blood lactate accumulation during exercise is the assumption that the circulating lactate level rises because of net release from active muscle. Surprisingly, while net lactate release from resting muscle is common, net release from working muscle is usually transient if the power output is submaximal and held constant. As shown first by Wendell Stainsby and Hugh Welch in 1966–1967105,106 using dog muscle preparations contracting in situ, this “Stainsby Effect” of transient muscle net lactate release at exercise onset followed by a switch to net uptake from the blood by working muscle has been confirmed in exercising humans35. In this regard, noteworthy are the studies of L. Bruce Gladden on dog muscles contracting in situ107. Gladden clearly showed that lactate uptake is the concentration (substrate) and not O2-dependent, a finding that also appears to be the case in human muscle35, vide infra. Thus, it is certain that working skeletal muscle is not the sole source of blood lactate in humans during whole-body exercise. Epinephrine is more likely to signal glycolysis and lactate production in noncontracting tissues than in working muscle; in working muscle, epinephrine augments glycolysis, leading to increased lactate accumulation108,109,110.
Choice of lactate carbon tracers to determine lactate flux and disposal in vivo
As noted above, initial studies of lactate metabolism in vivo utilized a uniformly labeled tracer, i.e., U-14C]lactate69. However, with the advent of stable, nonradioactive 13C-labeled tracers, it was ethically feasible to study flux and oxidation in human subjects111. Because lactate disposal via oxidation gave rise to 13CO2, carbon-labeled lactate tracers were preferred over heavy hydrogen (i.e., deuterated, D) tracers because the oxidation product (13CO2) could be detected at the tissue level in the venous effluent of tissues such as muscle35, heart36,39, and brain103 and at the whole-body level by excretion in expired air35,73,95,112. Another advantage of the 13C-lactate tracer was that it could be used in combination with a deuterated glucose tracer, e.g., [6,6-2H]glucose, a so-called “irreversible tracer”, because the D atoms are lost to body water during glycolysis. Moreover, because a percentage (≈25%) of 13C atoms from lactate are reincorporated into glucose during the process of gluconeogenesis, the combination of [3-13C]lactate and [6,6-2H]glucose (i.e., D2-glucose) could be used to simultaneously study lactate and glucose flux rates, the lactate oxidation rate and the rate of gluconeogenesis from lactate113,114,115.
On the issue of which isotope tracer of lactate to use, the most common is [3-13C]lactate, which yields information about total lactate disposal as well as the components of oxidation and gluconeogenesis. Earlier use of uniformly labeled compounds to determine metabolite disposal rates was abandoned because of the extensive recycling and exchange of labeled lactate carbon atoms116,117. Of the alternatives, 13C from [1-13C]lactate is lost as 13CO2 in the pyruvate dehydrogenase reaction, as pyruvate is converted to acetyl-CoA upon entry into the TCA (Krebs) Cycle. This decarboxylation step shows entry into the TCA cycle, but perhaps not complete oxidation of the lactate molecule. For that, [3-13C]lactate is the preferred tracer to demonstrate lactate oxidation downstream in the TCA cycle by the actions of isocitrate and alpha-ketoglutaric dehydrogenases41,118.
Finally, with the use of carbon-labeled tracers to determine the oxidative disposal of lactate, it needs to be acknowledged that because metabolically produced 13CO2 enters the body’s bicarbonate-CO2 pool, a separate experiment under identical conditions needs to be conducted. At rest, the bicarbonate-CO2 pool turns over slowly, so for the short term (1 to several hours), an isotope dilution factor needs to be applied119. In resting humans, the correction factor for CO2 retention is large (≈50%)120. The large correction factor for estimating isotopic dilution and retention is daunting for investigators to make because it is so large. Fortunately, however, during exercise, the necessity of a bicarbonate correction factor disappears because the bicarbonate-CO2 pool turns over rapidly with no measurable 13CO2 retention or diversion of tracer to other, unmeasured metabolite pools121,122. In addition, some investigators have explored the use of so-called “acetate correction factors”123. However, the latter approach is not recommended because acetate is not bicarbonate and, most importantly, because the C1 and C2 of acetate behave differently, leaving an investigator uncertain about what the appropriate correction factor is120.
The lactate shuttle and membrane lactate transport and lactate/pyruvate exchange
With knowledge of glucose and lactate fluxes and oxidation rates in resting and exercising rats71,124, in 1984, George A. Brooks took a unique approach to explain phenomena related to lactate responses to exercise when the “Lactate Shuttle Hypothesis” was articulated125. A key element of the hypothesis was that “the shuttling of lactate through the interstitium and vasculature provides a significant carbon source for oxidation and gluconeogenesis during rest and exercise”. As such, the lactate shuttle hypothesis represents a model of how the formation and distribution of lactate represents a central means by which the coordination of intermediary metabolism in diverse tissues and different cells within tissues can be accomplished. The initial hypothesis was developed from the results of original isotope tracer studies conducted on laboratory rats in the Brooks Laboratory along with numerous other studies, many of which were cited in the previous section. Thus, the working hypothesis was developed that much of the glycolytic flux during exercise passed through lactate.
According to the Cell–Cell Lactate Shuttle hypothesis, lactate is a metabolic intermediate rather than an end product3,88,126,127,128,129. Lactate is continuously formed in and released from diverse tissues, such as skeletal muscle, skin, and erythrocytes, and also serves as an energy source in highly oxidative tissues, such as the heart, and is a gluconeogenic precursor for the liver and kidneys. Lactate exchanges among these tissues appear to occur under various conditions, ranging from postprandial rest to sustained postabsorptive exercise88,89,125.
If lactate does serve as a key metabolic intermediate that shuttles into and out of tissues at high rates, particularly during exercise, then transmembrane movement is critical. For many years, lactate was assumed to move across membranes by simple diffusion. However, by 1980, facilitated protein carrier-mediated transport of lactate across erythrocyte membranes was well documented130,131. It was not until 1990 that the characteristics of sarcolemmal membrane lactate transport were described132,133,134. The study of cell membrane lactate transport proteins took another major leap in 1994. When looking for the mevalonate (Mev) transporter gene in Chinese hamster ovary cells, Christine Kim Garcia in the Goldstein and Brown laboratories cloned and sequenced a monocarboxylate transporter that she and her colleagues termed the monocarboxylate transporter (MCT)135. In 1985, Goldstein and Brown shared the Nobel Prize for “their discoveries concerning the regulation of cholesterol metabolism”136. The newly discovered MCT was abundant in erythrocytes, the heart, and the basolateral intestinal epithelium. MCT was also detectable in oxidative muscle fiber types but not in the liver. With an interest in describing a role for MCT isoforms in the Cori Cycle, Garcia et al.137 subsequently described the isolation of a second isoform (MCT2, in effect renaming the first transporter discovered to MCT1) by screening a Syrian hamster liver library; MCT2 was initially found in the liver and testes but subsequently also in the brain138,139 and some tumor cell lines16,140,141.
Independent of Garcia et al.135,137, Price et al.142,143 identified another MCT isoform (now known as MCT4) in 1998. In terms of physiology, Halestrap and colleagues144 continued to hold traditional, Hill-Meyerhof views of an association between lactate transporter expression and oxidative metabolism even though their data showed a high correlation between MCT1 expression and mitochondrial markers144,145, which was subsequently replicated by Dubouchaud et al.15. However, with a different concept based on the knowledge that lactate was formed and oxidized continuously within muscle and heart tissue in vivo, Brooks and associates sought to identify the molecular basis of the coupling of glycolytic with oxidative pathways. In that pursuit, Brooks and colleagues first observed the presence of LDH in rat liver, cardiac and skeletal muscle mitochondria15. Subsequently, they also showed MCT1 to be in mitochondria of the same tissues50, thus allowing mitochondria to directly oxidize lactate13. With such knowledge, in 1998, Brooks extended the Cell–Cell Lactate Shuttle concept to include an intracellular component, the “Intracellular Lactate Shuttle.” As already noted, several of the observations, such as the presence of mLDH53 and mitochondrial lactate oxidation55, were similar to the vision of Polyphemus—without depth perception, swamped by the mediocrity of authority, buried and forgotten in the literature for a time but later resurrected3,4.
Compartmentation issues and controversies—the intracellular lactate shuttle
While there is growing agreement on some aspects of the Cell–Cell Lactate Shuttle, within exercise physiology and other fields, such as neurobiology139,146,147,148,149,150 and cancer research140,141,151,152, some aspects of the Intracellular Lactate Shuttle remain controversial. In essence, disagreement exists over where the first step in lactate oxidation (i.e., conversion to pyruvate) occurs. This is why a discussion over whether mitochondria contain LDH persists. If so, identifying where LDH would reside so that it is able to oxidize lactate (vide supra) is important. However, the results of light and electron microscopy studies conducted to date show that mLDH resides within the mitochondrial reticulum.
Electron microscopy
Prior to the MitoCarta and MitoMiner databases based on proteomics, those who looked for muscle mLDH located it in tissues. Studying rat heart, Baba and Sharma used methods to protect mLDH during tissue fixation for electron microscopy. Reaction products of mLDH seen in the mitochondria were “primarily located within the cristae” (Fig. 1) (their Fig. 11)53. In addition, they reported similar results for rat pectoralis Types I and II fibers. Similarly, using an antibody to LDH and gold particle secondary labeling, Brooks et al. also showed LDH located on the inner mitochondrial membranes of rat muscle, heart, and liver13. Moreover, they used agarose gel electrophoresis to separate LDH isoforms in the cytosol and mitochondria isolated from rat livers and hearts. LDH isoenzyme patterns differed between the cytosol and mitochondria in both tissues (Fig. 2).
Immunocytochemistry
In the Brooks Laboratory, Takeshi Hashimoto used confocal laser scanning microscopy, immunoprecipitation (IP), and cell fractionation techniques followed by western blotting to verify the presence of the purported Mitochondrial Lactate Oxidation Complex (mLOC) in cultured myocytes as well as in thin sections of adult rat tissues. The colocalization of MCT1, mitochondrial cytochrome oxidase (COx) and LDH in the mitochondria of adult rat skeletal muscle and astrocytes was visualized by means of combinations of primary and fluorescently labeled secondary antibodies plus MitoTracker Red and dual-wavelength scanning confocal microscopy (Fig. 3)138,153,154. Subsequently, similar techniques allowed the investigators to show the colocalization of mLOC components in cultured L6 (rat muscle-derived) cells (Fig. 4). Again, those results were confirmed by two independent techniques—IP and Western blotting of isolated cell fractions138,153,154,155. The results of IP efforts are shown in Fig. 5, and a pictorial representation of how the mLOC is organized on the inner mitochondrial membrane with projection into the intermembrane space is constructed from the results of immunocytochemistry and immunoprecipitation studies (Fig. 6).

Figures showing the cellular locations of MCT1 and MCT2 lactate transporter isoforms and the mitochondrial reticulum (cytochrome oxidase, COx) in adult rat plantaris muscle determined using confocal laser scanning microscopy (CLSM) and fluorescent probes for the respective proteins; comparisons for MCT1 in the first row (Plates A1–A3, respectively) and for MCT2 in the second row (Plates B1–B3, respectively). The localization of COx was detected in rat plantaris muscle (Plates A1 and B1). MCT1 was detected throughout the cells, including the subsarcolemmal (arrowheads) and interfibrillar (arrows) domains (Plate A2). MCT1 abundance was greatest in oxidative fibers where COx was abundant and the signal was strong. When signals from probes for MCT1 (green) and COx (red) were merged, the superimposition of the two probes was clear (yellow), a finding prominent at the interfibrillar (arrows) and sarcolemmal (arrowheads) cell domains (Plate A3). In contrast, the signal for MCT2 (Plate B2) was weak and relatively more noticeable in fibers denoted by strong staining for COx (Plates B1 and B3, broken line delineated around oxidative fibers to distinguish the faint signal for MCT2). The overlap of MCT2 and COx is insignificant, denoted by the absence of yellow in Plate B3. Scale bar = 50 μm. Sections are from the same animal. Reprinted from Hashimoto et al.154.

This complex involves the mitochondrial constituent cytochrome oxidase (COx), lactate-pyruvate transport protein (MCT1), lactate dehydrogenase (LDH), and other constituents. A Colocalization of MCT1 and the mitochondrial reticulum. MCT1 was detected at both sarcolemmal and intracellular domains (A-1). Using MitoTracker, the mitochondrial reticulum was extensively elaborated and detected at intracellular domains throughout L6 cells (A-2). When signals from probes for the lactate transporter (MCT1, green, A-1) and mitochondria (red, A-2) were merged, the superimposition of the signals (yellow) showed the colocalization of MCT1 and components of the mitochondrial reticulum, particularly at perinuclear cell domains (A-3). In Panel (B), lactate dehydrogenase (LDH) (B-1), and mitochondrial cytochrome oxidase (COx) (B-2) are imaged. The superimposition of signals for LDH (red, B-1) and COx (green, B-2) shows the colocalization of LDH in the mitochondrial reticulum (yellow) of cultured L6 rat muscle cells (D-3). Depth of field ~1 μm, scale bar = 10 μm. Reprinted from Hashimoto et al.153.

In the upper panel, representative immunoblots (IB) are shown using anti-COX, NADH-dh, LDH, or nIgG as precipitating antibodies (IPs). COX, NADH-DH, LDH, and nIgG were immunoprecipitated from mitochondrial fractions of L6 cells resuspended in a suspension medium without detergent. COX IP proteins were probed with MCT1, CD147, and LDH antibodies. MCT1, CD147, and LDH were coprecipitated with COX. NADH-dh IP pellets were probed with MCT1, COX, CD147, and LDH antibodies. Neither MCT1, CD147, nor LDH coprecipitated with NADH-DH, whereas COX coprecipitated with anti-NADH-dh. LDH IP proteins were probed with MCT1 and COX antibodies. Both MCT1 and COX coprecipitated with LDH. No protein coprecipitated with nIgG from mitochondrial fractions of L6 cells resuspended in medium without detergent (negative control). In the lower panel, the degree of coprecipitation evaluated by comparing signals in the IP and the lysate is shown. The results were categorized into four levels: +++, 80% or more precipitated; ++, ∼50% precipitated; +, 20% or less precipitated; -, no precipitation. IB immunoblot, IP immunoprecipitation. From Hashimoto et al.138.

Lactate is oxidized to pyruvate via mitochondrial LDH (mLDH) in association with COx. This endergonic lactate oxidation reaction is coupled to the exergonic redox change in COx during mitochondrial electron transport. The transport of pyruvate across the inner mitochondrial membrane is facilitated by MCT1. GP glycerol phosphate, Mal-Asp malate-aspartate, ETC electron transport chain, TCA tricarboxylic acid. Figures 4 and 5 show the results of mitochondrial respiration studies13,50,56. Modified from Hashimoto et al.138.
Both morphometry and the susceptibility to loss during mitochondrial isolation suggest that LDH resides in the intermembrane space or, similar to cytochrome c, is loosely fixed to the inner membrane. On the other hand, if it is acknowledged that cardiac and red skeletal muscle take up and oxidize lactate, while the presence of mLDH and an mLOC are denied, then opponents of the Intracellular Lactate Oxidation Complex must rely on cytosolic oxidation of lactate to pyruvate. Cytosolic oxidation of lactate is unlikely156 and contrary to the evidence available from Peter Hochachka and colleagues, who studied glycolysis in a variety of mammalian species157,158, and Bradley Zinker et al.159 in the David Wasserman laboratory, who studied glucose and lactate metabolism in the hind limb muscles of running dogs. By means of tracer glucose, they showed intramuscular lactate production from glucose to occur simultaneously with net muscle lactate uptake159. Clearly, glycolysis progressed to lactate production in the working muscles of running dogs, whereas lactate oxidation occurred in a compartment where it was oxidized, the mitochondrial reticulum.
Mitochondrial monocarboxylate uptake mMCTs, mPCs or Both?
Analogous to the controversial history of mLDH discovery was the discovery of the presence of mitochondrial monocarboxylate transporters (mMCTs)13,50,68. These were visualized in adult rat skeletal muscle154 and cultured L6 myocytes by Takeshi Hashimoto and colleagues153, Figs. 3 and 4, respectively. For a time, the isoform monocarboxylate transporter 1 (i.e., MCT1) was the only lactate/pyruvate transporter known to be expressed in the mammalian mitochondrial proteome57. Subsequently, the mitochondrial pyruvate transporter (mPC) was identified160,161. Unfortunately, at present, little effort has been expended to evaluate functional or morphometric relationships between mPCs and mMCTs, particularly in mammalian skeletal muscle. Indeed, we cannot find reports in the literature of an mPC in mammalian or human skeletal muscle. Reports of an mPC in yeast160,161 are unconvincing because of single-band Western blots that are unaccompanied by a ladder to assess the molecular weights of the putative mPC. However, with access to our own custom antibodies to MCT1, as well as a commercially available antibody to the putative mPC, we obtained images assessing the colocalization of MCT1 and mPCs in L6 cells. In those preliminary studies, colocalization analysis of mMCT1 and mPC1 in Imaris software showed an r2 of 0.8. It appears that both MCT1 and mPCs are colocalized to the mitochondria (r2 = 0.8) (Fig. 7)6. However, at the light microscopic level, it is impossible to know whether mMCTs and mPCs interact physically and functionally. Immunoprecipitation, X-ray crystallography, mass spectrometry, and protein deletion (knockout) studies are needed to definitively answer questions about mMCT and mPC colocalization and functionality and the role of mPCs in mitochondrial lactate oxidation.

Images assessing the colocalization of MCT1 and mPCs in L6 cells, which show the localization of DAPI-positive nuclei (A), MCT1 (B), mPC1 (C), and MitoTracker-positive MR (D) in L6 cells. The merged images are shown in (E). Colocalization analysis of mPC1 (C) and mitochondria D showed a Pearson correlation coefficient (r2) value of 0.8. Colocalization analysis of MCT1 (B) and mPC1 (C) showed an r2 of 0.3, largely because MCT1 occupies sarcolemmal, mitochondrial and peroxisomal compartments. A channel to represent the colocalization of MCT1 and mitochondria was created to image mMCT1; subsequent colocalization of mMCT1 with mPC1 resulted in an r2 of 0.8 (F). White dots indicate the colocalization of mMCT1 and mPC1 as observed in ImageJ software. Whole images were contrast-enhanced in (A, B, C, D, and E). Similar results were observed for mPC2. Scale bar = 20 µm. It appears that both MCT1 and the putative mPC colocalized to the mitochondria (r2 = 0.8). However, at the light microscopic level, it is impossible to know if the two proteins interact physically and functionally. Additionally, with the benefit of the Orbitrap LC/MS device, we could determine the fractional synthesis rates of mLOC and mPC proteins. From Brooks6.
Role of the mitochondrial lactate oxidation complex (mLOC) in lactate/pyruvate metabolism
Discovery of the mLOC (Fig. 5) and its role in Cell–Cell and Intracellular lactate shuttles may cause some traditional thinkers to assert something such as “the diversion of glycolytic flux from pyruvate to lactate via cytosolic LDH (cLDH) with a return to pyruvate via mLDH still means that pyruvate is the major mitochondrial energy substrate”. Such an assertion would hold at the unicellular organismic level. However, to briefly reiterate from above, lactate shuttles are based on lactate concentration differences between producer and recipient cells connected through the interstitium within a tissue bed or the vasculature at the organ system level. In the shuttling of carbohydrate carbon energy substrates, lactate concentrations are orders of magnitude greater than those of pyruvate and alanine. Hence, corporal energy substrate distribution is accomplished with lactate, not pyruvate, as the vehicle of mass-energy transfer60.
The mitochondrial lactate oxidation complex (mLOC) in cancer
As noted above, in the Brooks laboratory, Takeshi Hashimoto et al. showed the presence of the mLOC in mammalian skeletal muscle, muscle cell lines, neurons, and primary neuronal cell cultures138,153,154. In addition, his work revealed the presence of a lactate-sensitive transcription network in cultured myocytes155. Having been part of the team previously working on muscle and brain, both postmitotic tissues, Rajaa Hussien in the Brooks laboratory commenced work on breast cancer cells140 that display active mitosis. Initial efforts were on known mLOC proteins, including monocarboxylate transporters (MCT1, MCT2, and MCT4), a scaffold glycoprotein (CD147), lactate dehydrogenase (LDH isoforms A and B), and cytochrome oxidase (COx). The results were consistent with their primary hypothesis that mLOC expression and assembly are dysregulated in breast cancer140. Hussien and Brooks tested their primary hypothesis on the breast cell lines MCF-7, MDA-MB-231 and HMEC-184; the former two are transformed breast cell lines that are cancerous, while HMEC-184 is a nontransformed primary breast cell line that was used as a control. Protein expression of MCTs, CD147 and LDHs was determined by immunoblot analysis of whole-cell extracts from the three cell lines grown under standard conditions. The expression of the seven proteins in the two transformed cell lines was compared with that in the primary untransformed cell line HMEC-184. Relative to HMEC-184, in the transformed cells, some proteins (MCT2, LDH and CD147) were upregulated. Remarkably, the expression of MCT1 was low in MCF-7 cells and undetectable in MDA-MB-231 cells. A notable novel discovery from their study was the reciprocal relationship between the MCT1 and CD147 protein levels. The data from breast cancer cell lines led to the hypothesis (as yet incompletely explored) that the dysregulation of the coordinated expression of those two proteins and their respective genes contributes to the carcinogenic process in breast cancer.
To probe for the presence of mLOC proteins in cancer cells, Hussien and Brooks140 also examined the subcellular localization of MCTs and LDH isoforms in the three cell lines studied. Confocal laser scanning microscopy and related immunohistochemical techniques showed the presence of MCTs, LDH isoforms, and COx. LDH isoforms MCT2 and MCT4 were colocalized with the mitochondrial protein marker COx, but distinct from the situation in muscle, MCT1 was not mitochondrial and was localized exclusively in the plasma membrane. The localization of MCT and LDH isoforms in the two cancer cell lines (MCF-7 and MDA-MB-231) and the normal cell line (HMEC-184) was the same. MCT2, MCT4, and LDH were localized in mitochondria in addition to their localization in the plasma membrane and cytosol, whereas MCT1 was mainly localized in the plasma membrane (Fig. 8). Their data showed that in a breast cancer cell line, changes in the expression of lactate shuttle proteins occur. The reasons for these changes in mLOC protein expression, transcription, translation, and turnover in breast cancer need elucidation.

Immunohistochemical detection of LDH, MCT isoforms, and cytochrome oxidase (COx) in the control breast cell line HMEC-184, Plate A, left, and in the breast cancer cell line MCF-7, Plate B, right. LDH isoforms MCT2 and MCT4 colocalized with the mitochondrial protein marker COx (rows A, C, D) but not MCT1, which was localized mainly in the plasma membrane in the control HMEC cell line (Plate B, Row B, left). Thus, mMCT isoform expression in breast cell mitochondria differs from that in skeletal muscle, where MCT1 predominates (Figs. 4 and 5); in control and cancer breast cells, MCT-2 and -4 isoforms colocalized with the inner mitochondrial membrane component COx. The thickness of the optical sections, ~1 µm, scale bar = 1 µm. Images from Hussien and Brooks140.
Lactate signaling
The effects of increases in cellular work and subsequent lactate production on redox, reactive oxygen species (ROS), allosteric binding, and histone lactylation have been recently reviewed5. Briefly, lactate affects metabolic signaling via diverse mechanisms that include changing the L/P and NADH/NAD+ and thus cell redox; activating sirtuins via the enzyme NAM phosphoribosyl transferase (Nampt); stimulating transforming growth factor beta 2 (TGF-β2) secretion from adipose tissue; regulating gene expression by the lactylation of 28 lysine residues on histones162; and inhibiting lipolysis by activating Hydroxycarboxylic Acid Receptor-1 (HCAR-1) operating through cyclic AMP (cAMP) and cAMP response element-binding (CREB163,164. With regard to gene regulation via histone lactylation, it should be noted that it was discovered using tracer methodology ([U-13C]lactate and [U-13C]glucose) and a cancer cell line (MCF-7). As noted previously, the purported effects of lactate signaling via HCAR-1, TGF-β2, Nampt, and the lactylation of histones observed in rodent muscle and brain and cell line models await validation in humans4. The latter statement is appropriate because of the growing interest in evaluating the role of lactylation in promoting carcinogenesis in a variety of cell types, including those of the lungs165. Regrettably, thinking in the field is limited by reliance on outmoded O2 debt (i.e., O2-limited) ideas of metabolic regulation166.
Lactate and mitochondrial biogenesis
Endurance exercise has long been recognized to stimulate mitochondrial biogenesis167,168. Hence, the following question arose: ‘Could the lactatemia of exercise stimulate mitochondrial biogenesis?’ In an attempt to address the question, Hashimoto and colleagues incubated L6 cells in elevated sodium lactate added to buffered, high-glucose media and screened the cells’ genome-wide responses155. Lactate increased ROS production and upregulated 673 genes, many known to be responsive to exercise and exercise training via ROS and Ca++ stimulation. The induction of genes encoding components of the mLOC was confirmed by a polymerase chain reaction and electrophoretic mobility shift assay (PCR and EMSA, respectively). Among the many effects, lactate increased the mRNA and protein expression of MCT1 and COx. Increases in COx expression coincided with increases in peroxisome proliferator activated-receptor gamma coactivator-1 alpha (PGC1α) expression and the DNA binding activity of nuclear respiratory factor (NRF)-2. Based upon their results, Hashimoto et al. concluded that the presence of a lactate signaling cascade involves ROS production and converges on transcription factors affecting mitochondrial biogenesis.
The presence of a lactate-sensitive transcription factor network and its putative effects on muscle mitochondrial biogenesis in exercising humans have yet to be evaluated. To date, Genders et al.169 in the David Bishop Laboratory have confirmed elements of the work of Hashimoto et al.; specifically, they have advanced the field to show that low or high pH can suppress the effect of lactate on histone deacetylase (HDAC) activity and protein kinase B (aka, Ak strain transforming, Akt) signaling in L6 cells. Studies on the possibility of exercise above the lactate threshold, where there is a disproportionate increase in muscle lactate production, activating a signaling cascade leading to human skeletal muscle mitochondrial biogenesis are underway (David Bishop personal communication).
The lactate story comes full circle
Considering the above text on intact individuals in vivo, tissues, and organs in situ, and cells in vitro, as well as the results on cancer cells to date, it seems that the study of lactate metabolism has come full circle. From the perception of a lowly waste product formed as the result of a lack of oxygen, as embodied in the shuttle hypothesis, lactate is now realized to have three essential metabolic functions: an energy source, a gluconeogenic precursor, and a signaling molecule. Originally conceived of within the context of exercise physiology and supporting short- and long-term adaptive processes, some investigators propose using lactate esters and salts to sustain athletes in prolonged hard exercise bouts170 and to provide nutritive support to humans following traumatic brain injury104.
Regrettably, in clinical settings, the understanding and use of lactate in diagnosis and treatment is inadequately understood and employed. For instance, half molar lactate infusion is useful to supply nutritive support to patients with heart failure and following myocardial infarction171. Similarly, in the realm of traumatic brain injury104, a clinical trial is underway to evaluate the efficacy of lactate supplementation to support the injured brain. Nevertheless, as lamented by See and Bellomo172, in intensive care units where the blood lactate level is a biomarker for the severity of sepsis, a lack of knowledge on the sites and reasons for lactatemia hampers treatment. Fortunately, progress has been made in understanding the role of lactatemia in the etiology of diabetic ketoacidosis173. Similarly, the role of dysregulated lactate metabolism is advancing, with investigators giving thought to destroy cancer cells by blocking lactate shuttling140,141,151,174,175.
Summary
A chronology of significant events and important discoveries in the field of lactate metabolism is given in Table 1. As shown, in the latter part of the 20th century, the use of isotope tracer technologies enabled significant contributions to a revolution in understanding the diverse roles of lactate in the regulation and integration of intermediary metabolism in humans and other mammals in health and disease.
Isotope tracer technologies were central to the deduction and demonstration of the Lactate Shuttle at the whole-body level. In concert with the ability to perform tissue metabolite concentration measurements, as well as determinations of unidirectional and net metabolite exchanges by means of a-v difference and blood flow measurements across tissue beds including muscle and the brain, lactate shuttling within organs and tissues was evident. We now know that Organ–Organ, Cell–Cell, and Intracellular Lactate Shuttles operate continuously, even after eating when a Postprandial Lactate Shuttle is evident4. By means of lactate shuttling, fuel-energy substrate exchange occurs between tissues and organs such as muscle and the brain; muscle and the heart; muscle and the liver, kidneys, and gut; and muscle and other tissues. A muscle-like role of the integument is also highly suspected but unexplored. A Corporal-Microbiome Lactate Shuttle has been proposed3 but is also unexplored. Within tissues, lactate can be exchanged between white and red fibers within a muscle bed and between astrocytes and neurons in the brain. Within cells, lactate can be exchanged between the cytosol and mitochondria and between the cytosol and peroxisomes. By means of isotope tracers and related methodologies, we know that there are at least three general functions of lactate shuttling: lactate is an energy source preferred over glucose in the heart, red skeletal muscle, and brain; lactate is the major gluconeogenic precursor; and lactate is a signaling molecule. Lactate signals otherwise affect energy substrate partitioning by mass action, cell redox, the lactylation of histones, and covalent binding to HCAR-1 and signaling via CREB-dependent pathways. Discovered in the realm of exercise physiology and biochemistry, it is now possible to recognize a Postprandial Lactate Shuttle by which dietary carbohydrates are absorbed, circulated, and taken up and used or stored in diverse tissues. Isotope tracers and related technologies have made multiple fundamental discoveries possible in the field of metabolic regulation.
References
Kirkwood, S. P., Munn, E. A. & Brooks, G. A. Mitochondrial reticulum in limb skeletal muscle. Am. J. Physiol. 251, C395–C402 (1986).
Kirkwood, S. P., Packer, L. & Brooks, G. A. Effects of endurance training on a mitochondrial reticulum in limb skeletal muscle. Arch. Biochem Biophys. 255, 80–88 (1987).
Brooks, G. A. The science and translation of lactate shuttle theory. Cell Metab. 27, 757–785 (2018).
Brooks, G. A. et al. Lactate in contemporary biology: a phoenix risen. J. Physiol. 600, 1229–1251 (2022).
Brooks, G. A. Lactate as a fulcrum of metabolism. Redox Biol. 35, 101454 (2020).
Brooks, G. A. The tortuous path of lactate shuttle discovery: from cinders and boards to the lab and ICU. J. Sport Health Sci. 9, 446–460 (2020).
Passarella, S., Paventi, G. & Pizzuto, R. The mitochondrial L-lactate dehydrogenase affair. Front. Neurosci. 8, 407 (2014).
Schurr, A. Cerebral glycolysis: a century of persistent misunderstanding and misconception. Front. Neurosci. 8, 360 (2014).
Brooks, G. A. & Mercier, J. Balance of carbohydrate and lipid utilization during exercise: the "crossover" concept. J. Appl. Physiol. 76, 2253–2261 (1994).
Daff, S., Ingledew, W. J., Reid, G. A. & Chapman, S. K. New insights into the catalytic cycle of flavocytochrome b2. Biochemistry 35, 6345–6350 (1996).
Daum G, P. C., Bohni & Schatz, G. Import proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem. 252, 13028–13033 (1982).
Keilin, D. The History of Cell Respiration and Cytochrome (Cambridge U.P., 1966).
Brooks, G. A., Dubouchaud, H., Brown, M., Sicurello, J. P. & Butz, C. E. Role of mitochondrial lactate dehydrogenase and lactate oxidation in the intracellular lactate shuttle. Proc. Natl Acad. Sci. USA 96, 1129–1134 (1999).
de Bari, L., Valenti, D., Atlante, A. & Passarella, S. L-lactate generates hydrogen peroxide in purified rat liver mitochondria due to the putative L-lactate oxidase localized in the intermembrane space. FEBS Lett. 584, 2285–2290 (2010).
Dubouchaud, H., Butterfield, G. E., Wolfel, E. E., Bergman, B. C. & Brooks, G. A. Endurance training, expression, and physiology of LDH, MCT1, and MCT4 in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 278, E571–E579 (2000).
Lemire, J., Mailloux, R. J. & Appanna, V. D. Mitochondrial lactate dehydrogenase is involved in oxidative-energy metabolism in human astrocytoma cells (CCF-STTG1). PLoS ONE 3, e1550 (2008).
Passarella, S. et al. Mitochondria and L-lactate metabolism. FEBS Lett. 582, 3569–3576 (2008).
Szczesna-Kaczmarek, A. L-lactate oxidation by skeletal muscle mitochondria. Int. J. Biochem. 22, 617–620 (1990).
Szczesna-Kaczmarek, A. Regulating effect of mitochondrial lactate dehydrogenase on oxidation of cytoplasmic NADH via an "external" pathway in skeletal muscle mitochondria. Int. J. Biochem 24, 657–661 (1992).
Reeves, J. T. et al. Oxygen transport during exercise at altitude and the lactate paradox: lessons from Operation Everest II and Pikes Peak. Exerc Sport Sci. Rev. 20, 275–296 (1992).
Brooks, G. A. et al. Muscle accounts for glucose disposal but not blood lactate appearance during exercise after acclimatization to 4,300 m. J. Appl. Physiol. 72, 2435–2445 (1992).
Hill, A. The energy degraded in recovery processes of stimulated muscles. J. Phyiol. 46, 28–80 (1913).
Hill, A. V. The heat produced in contracture and muscle tone. J. Physiol. 40, 389–403 (1910).
Hill, A. V. The oxidative removal of lactic acid. J. Physiol. 48, x–xi (1914).
Meyerhof, O. Die Energieumwandlungen im Muskel I. Über die Beziehungen der Milchsaure zur Warmebildung and Arbeitsleistung des Muskels in der Anaerobiose. Pflug. Arch. fur Gesamt. Physiologie des. Menschejen und der Tiere 182, 232–283 (1920).
Meyerhof, O. Die Energieumwandlungen im Muskel II. Das Schicksal der Milchsaure in der Erholungsperiode des Muskels. Pflug. Arch. fur Gesamt. Physiologie des. Menschejen und der Tiere 182, 284–317 (1920).
Meyerhof, O. Die Energieumwandlungen im Muskel III. Kohlenhydrat- und Milchsaureumsatz im Froschmuskel. Pflug. Arch. fur Gesamt. Physiologie des. Menschejen und der Tiere 185, 11–32 (1920).
Bertocci, L. A. & Lujan, B. F. Incorporation and utilization of [3-13C]lactate and [1,2-13C]acetate by rat skeletal muscle. J. App. Physiol. 86, 2077–2089 (1999).
Conley, K. E., Kushmerick, M. J. & Jubrias, S. A. Glycolysis is independent of oxygenation state in stimulated human skeletal muscle in vivo. J. Physiol. 511, 935–945 (1998).
Connett, R. J., Gayeski, T. E. & Honig, C. R. Lactate accumulation in fully aerobic, working, dog gracilis muscle. Am. J. Physiol. 246, H120–H128 (1984).
Molé, P. A. et al. Myoglobin desaturation with exercise intensity in human gastrocnemius muscle. Am. J. Physiol. 277, R173–R180 (1999).
Richardson, R. S., Noyszewski, E. A., Leigh, J. S. & Wagner, P. D. Lactate efflux from exercising human skeletal muscle: role of intracellular PO2. J. Appl. Physiol. 85, 627–PO634 (1998).
Park, J. M. et al. Hyperpolarized 13 C NMR observation of lactate kinetics in skeletal muscle. J. Exp. Biol. 218, 3308–3318 (2015).
Bertocci, L. A., Jones, J. G., Malloy, C. R., Victor, R. G. & Thomas, G. D. Oxidation of lactate and acetate in rat skeletal muscle: analysis by 13C-nuclear magnetic resonance spectroscopy. J. Appl Physiol. 83, 32–39 (1997).
Bergman, B. C. et al. Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol. 87, 1684–1696 (1999).
Bergman, B. C., Tsvetkova, T., Lowes, B. & Wolfel, E. E. Myocardial glucose and lactate metabolism during rest and atrial pacing in humans. J. Physiol. 587, 2087–2099 (2009).
Chatham, J. C., Des Rosiers, C. & Forder, J. R. Evidence of separate pathways for lactate uptake and release by the perfused rat heart. Am. J. Physiol. 281, E794–E802 (2001).
Wisneski, J. A., Gertz, E. W., Neese, R. A. & Mayr, M. Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J. Clin. Investig. 79, 359–366 (1987).
Gertz, E. W., Wisneski, J. A., Stanley, W. C. & Neese, R. A. Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J. Clin. Investig. 82, 2017–2025 (1988).
Urry, L. A., Cain, M. L., Wasserman, S. L., Minorsky, P. V. & Orr, R. Campbell biology. In: Urry, L. A., Cain, M. L., Wasserman, S. A., Minorsky, P. V., & Orr, R. (eds) Campbell Biology, 12 edn. 164–170 (Peaarson, 2020).
Lehninger, A. L. in BIOCHEMISTRY, 1 edn, 326 (Worth, 1970).
Campbell, N. A. Biology. Benjjamin/Cummings, San Francisco (1987).
Mahler, H. R. & Cordes, E. H. BIOLOGICAL CHEMISTRY (HARPER & ROWE, 1966).
Huckabee, W. E. Relationships of pyruvate and lactate during anaerobic metabolism. II. Exercise and formation of O-debt. J. Clin. Investig. 37, 255–263 (1958).
Brooks, G. A., Fahey, T. D. & Baldwin, K. M. Exercise Physiology: Human Bioenergetics and Its Applications. John Wiley & Sons, New York. (2004).
Brooks, G. A., Fahey, T. D. & Baldwin, K. M. EXERCISE PHYSIOLOGY: Human Bioenergetics and Its Applications, V edn, vol. 1 (Kindle Direct Publishing, 2019).
Yoshida, Y. et al. Negligible direct lactate oxidation in subsarcolemmal and intermyofibrillar mitochondria obtained from red and white rat skeletal muscle. J. Physiol. 582, 1317–1335 (2007).
Sahlin, K., Fernstrom, M., Svensson, M. & Tonkonogi, M. No evidence of an intracellular lactate shuttle in rat skeletal muscle. J. Physiol. 541, 569–574 (2002).
Ponsot, E. et al. Mitochondrial tissue specificity of substrates utilization in rat cardiac and skeletal muscles. J. Cell Physiol. 203, 479–486 (2005).
Brooks, G. A., Brown, M. A., Butz, C. E., Sicurello, J. P. & Dubouchaud, H. Cardiac and skeletal muscle mitochondria have a monocarboxylate transporter MCT1. J. Appl. Physiol. 87, 1713–1718 (1999).
Kline, E. S. et al. Localization of L-lactate dehydrogenase in mitochondria. Arch. Biochem. Biophysics 246, 673–680 (1986).
Brandt, R. B., Laux, J. E., Spainhour, S. E. & Kline, E. S. Lactate dehydrogenase in rat mitochondria. Arch. Biochem. Biophysics 259, 412–422 (1987).
Baba, N. & Sharma, H. M. Histochemistry of lactic dehydrogenase in heart and pectoralis muscles of rat. J. Cell Biol. 51, 621–635 (1971).
Young, A., Oldford, C. & Mailloux, R. J. Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol. 28, 101339 (2020).
Dianzani, M. La ripartizione del sistema ossidante l'acido lactico nelle cellule del pagato e del rene di ratti normali e di ratti con degenerazione grassa del fegato. Arch. di Physiologica 50, 181–186 (1951).
Jacobs, R. A., Meinild, A. K., Nordsborg, N. B. & Lundby, C. Lactate oxidation in human skeletal muscle mitochondria. Am. J. Physiol. Endocrinol. Metab. 304, E686–E694 (2013).
Pagliarini, D. J. et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 (2008).
Glancy, B. et al. Mitochondrial lactate metabolism: history and implications for exercise and disease. J. Physiol. 599, 863–888 (2021).
Ferguson, B. S. et al. Lactate metabolism: historical context, prior misinterpretations, and current understanding. Eur. J. Appl Physiol. 118, 691–728 (2018).
Brooks, G. A. et al. The blood lactate/pyruvate equilibrium affair. Am J Physiol Endocrinol Metab. 322, E34–E43 (2022).
Wolfe, R. R., Jahoor, F. & Miyoshi, H. Evaluation of the isotopic equilibration between lactate and pyruvate. Am. J. Physiol. 254, E532–E535 (1988).
Romijn, J. A., Chinkes, D. L., Schwarz, J. M. & Wolfe, R. R. Lactate-pyruvate interconversion in blood: implications for in vivo tracer studies. Am. J. Physiol. 266, E334–E340 (1994).
Fletcher, W. M. & Hopkins, F. G. Lactic acid in amphibian muscle. J. Physiol. 35, 247–309 (1907).
Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 9, 148–163 (1925).
Hill, A. V., Long, C. N. H. & Lupton, H. Muscular exercise, lactic acid and the supply and utilisation of oxygen. Pt VII-VIII. Proc. R. Soc. B 97, 155–176 (1924).
Hill, A. V., Long, C. N. H. & Lupton, H. Muscular exercise, lactic acid and the supply and utilisation of oxygen. Pt IV-VI. Proc. R. Soc. B 97, 84–138 (1924).
Hill, A. V., Long, C. N. H. & Lupton, H. Muscular exercise, lactic acid and the supply and utilisation of oxygen. Pt I-III. Proc. R. Soc. B 96, 438–475 (1924).
Butz, C. E., McClelland, G. B. & Brooks, G. A. MCT1 confirmed in rat striated muscle mitochondria. J. Appl Physiol. (1985) 97, 1059–1066 (2004).
Depocas, F., Minaire, Y. & Chatonnet, J. Rates of formation and oxidation of lactic acid in dogs at rest and during moderate exercise. Can. J. Physiol. Pharm. 47, 603–610 (1969).
Stanley, W. C. et al. Systemic lactate kinetics during graded exercise in man. Am. J. Physiol. 249, E595–E602 (1985).
Donovan, C. M. & Brooks, G. A. Endurance training affects lactate clearance, not lactate production. Am. J. Physiol. 244, E83–E92 (1983).
Glenn, T. C. et al. Endogenous nutritive support after traumatic brain injury: peripheral lactate production for glucose supply via gluconeogenesis. J. Neurotrauma 32, 811–819 (2015).
Mazzeo, R. S., Brooks, G. A., Schoeller, D. A. & Budinger, T. F. Disposal of blood [1-13C]lactate in humans during rest and exercise. J. Appl. Physiol. 60, 232–241 (1986).
Bertocci, L. A., Haller, R. G. & Lewis, S. F. Muscle metabolism during lactate infusion in human phosphofructokinase deficiency. J. Appl Physiol. 74, 1342–1347 (1993).
Brandt, R. B., Laux, J. E., Spainhour, S. E., Bear, H. D. & Kline, E. S. Cytosolic-mitochondrial interactions (mitochondrial control of glycolysis). Prog. Clin. Biol. Res. 292, 497–506 (1989).
Nakae, Y., Stoward, P. J., Shono, M. & Matsuzaki, T. Localisation and quantification of dehydrogenase activities in single muscle fibers of mdx gastrocnemius. Histochem Cell Biol. 112, 427–436 (1999).
Taylor, S. W. et al. Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21, 281–286 (2003).
Dianzani, M. U. [Distribution of lactic acid oxidase in liver and kidney cells of normal rats and rats with fatty degeneration of the liver]. Arch. Fisiol. 50, 181–186 (1951).
De Bari, L., Atlante, A., Valenti, D. & Passarella, S. Partial reconstruction of in vitro gluconeogenesis arising from mitochondrial l-lactate uptake/metabolism and oxaloacetate export via novel L-lactate translocators. Biochem. J. 380, 231–242 (2004).
Rasmussen, H. N., van Hall, G. & Rasmussen, U. F. Lactate dehydrogenase is not a mitochondrial enzyme in human and mouse vastus lateralis muscle. J. Physiol. 541, 575–580 (2002).
Fulghum, K. L. et al. Mitochondria-associated lactate dehydrogenase is not a biologically significant contributor to bioenergetic function in murine striated muscle. Redox Biol. 24, 101177 (2019).
Chretien, D. et al. An improved spectrophotometric assay of pyruvate dehydrogenase in lactate dehydrogenase contaminated mitochondrial preparations from human skeletal muscle. Clin. Chim. Acta 240, 129–136 (1995).
Hashimoto, T. et al. Maintained exercise-enhanced brain executive function related to cerebral lactate metabolism in men. FASEB J. 32, 1417–1427 (2018).
van Hall, G. et al. Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow. Metab. 29, 1121–1129 (2009).
Gerich, J. E., Meyer, C., Woerle, H. J. & Stumvoll, M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care 24, 382–391 (2001).
Meyer, C., Dostou, J., Nadkarni, V. & Gerich, J. Effects of physiological hyperinsulinemia on systemic, renal, and hepatic substrate metabolism. Am. J. Physiol. 275, F915–F921 (1998).
Meyer, C. et al. Renal substrate exchange and gluconeogenesis in normal postabsorptive humans. Am. J. Physiol. Endocrinol. Metab. 282, E428–E434 (2002).
Brooks, G. A. Lactate production under fully aerobic conditions: the lactate shuttle during rest and exercise. Fed. Proc. 45, 2924–2929 (1986).
Brooks, G. A. Lactate shuttles in nature. Biochem Soc. Trans. 30, 258–264 (2002).
Brooks, G. A. et al. Decreased reliance on lactate during exercise after acclimatization to 4,300 m. J. Appl. Physiol. 71, 333–341 (1991).
Stanley, W. C. et al. Lactate extraction during net lactate release in legs of humans during exercise. J. Appl. Physiol. 60, 1116–1120 (1986).
James, D. E., Kraegen, E. W. & Chisholm, D. J. Effects of exercise training on in vivo insulin action in individual tissues of the rat. J. Clin. Investig. 76, 657–666 (1985).
James, D. E. et al. Intrinsic differences of insulin receptor kinase activity in red and white muscle. J. Biol. Chem. 261, 14939–14944 (1986).
Woerle, H. J. et al. Pathways for glucose disposal after meal ingestion in humans. Am. J. Physiol. Endocrinol. Metab. 284, E716–E725 (2003).
Stanley, W. C., Wisneski, J. A., Gertz, E. W., Neese, R. A. & Brooks, G. A. Glucose and lactate interrelations during moderate-intensity exercise in humans. Metab.: Clin. Exp. 37, 850–858 (1988).
Gertz, E. W. et al. Myocardial lactate metabolism: evidence of lactate release during net chemical extraction in man. Circulation 63, 1273–1279 (1981).
Drury, D. R., Wick, A. N. & Morita, T. N. Metabolism of lactic acid in extrahepatic tissues. Am. J. Physiol. 180, 345–349 (1955).
Issekutz, B. J. & Miller, H. Plasma free fatty acids during exercise and the effect of lactic acid. Proc. Soc. Exp. Biol. Med. 110, 237–239 (1962).
Issekutz, B. Jr. Effect of beta-adrenergic blockade on lactate turnover in exercising dogs. J. Appl. Physiol. 57, 1754–1759 (1984).
Weber, J. M. et al. Lactate kinetics in exercising thoroughbred horses: regulation of turnover rate in plasma. Am. J. Physiol. 253, R896–R903 (1987).
Brooks, G. A. et al. Poor relationship between arterial [lactate] and leg net release during exercise at 4,300 m altitude. Am. J. Physiol. 275, R1192–R1201 (1998).
Brooks, G. A. Role of the heart in lactate shuttling. Front Nutr. 8, 663560 (2021).
Glenn, T. C. et al. Lactate: brain fuel in human traumatic brain injury. a comparison to normal healthy control subjects. J. Neurotrauma 32, 820–832 (2015).
Brooks, G. A. & Martin, N. A. Cerebral metabolism following traumatic brain injury: new discoveries with implications for treatment. Front. Neurosci. 8, 408 (2014).
Stainsby, W. N. & Welch, H. G. Lactate metabolism of contracting dog skeletal muscle in situ. Am. J. Physiol. 211, 177–183 (1966).
Welch, H. G. & Stainsby, W. N. Oxygen debt in contracting dog skeletal muscle in situ. Respiration Physiol. 3, 229–242 (1967).
Gladden, L. B., Crawford, R. E. & Webster, M. J. Effect of lactate concentration and metabolic rate on net lactate uptake by canine skeletal muscle. Am. J. Physiol. 266, R1095–R1101 (1994).
Hamann, J. J., Kelley, K. M. & Gladden, L. B. Effect of epinephrine on net lactate uptake by contracting skeletal muscle. J. Appl. Physiol. 91, 2635–2641 (2001).
Watt, M. J., Howlett, K. F., Febbraio, M. A., Spriet, L. L. & Hargreaves, M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J. Physiol. 534, 269–278 (2001).
Kjaer, M., Farrell, P. A., Christensen, N. J. & Galbo, H. Increased epinephrine response and inaccurate glucoregulation in exercising athletes. J. Appl Physiol. 61, 1693–1700 (1986).
Mazzeo, R. S., Brooks, G. A., Budinger, T. F. & Schoeller, D. A. Pulse injection, 13 C tracer studies of lactate metabolism in humans during rest and two levels of exercise. Biomed. Mass Spectrom. 9, 310–314 (1982).
Messonnier, A. L. et al. Lactate kinetics at the lactate threshold in trained and untrained men. J. Appl. Physiol. 114, 1593–1602 (2013).
Bergman, B. C. et al. Endurance training increases gluconeogenesis during rest and exercise in men. Am. J. Physiol. Endocrinol. Metab. 278, E244–E251 (2000).
Emhoff, C. A. et al. Gluconeogenesis and hepatic glycogenolysis during exercise at the lactate threshold. J. Appl. Physiol. 114, 297–306 (2013).
Lecavalier, L., Bolli, G., Cryer, P. & Gerich, J. Contributions of gluconeogenesis and glycogenolysis during glucose counterregulation in normal humans. Am. J. Physiol. 256, E844–E851 (1989).
Brooks, G. A. & Gaesser, G. A. End points of lactate and glucose metabolism after exhausting exercise. J. Appl Physiol. Respir. Environ. Exerc Physiol. 49, 1057–1069 (1980).
Gaesser, G. A. & Brooks, G. A. Glycogen repletion following continuous and intermittent exercise to exhaustion. J. Appl Physiol. Respir. Environ. Exerc Physiol. 49, 722–728 (1980).
Brooks, G. A. & Fahey, T. D. Exercise Physiology: Human Bioenergetics and Its Applications, 1st edn. (John Wiley & Sons, Inc., 1984).
Hetenyi, G. Jr. Correction for the metabolic exchange of 14 C for 12 C atoms in the pathway of gluconeogenesis in vivo. Federation Proc. 41, 104–109 (1982).
Trimmer, J. K., Casazza, G. A., Horning, M. A. & Brooks, G. A. Recovery of (13)CO2 during rest and exercise after [1-(13)C]acetate, [2-(13)C]acetate, and NaH(13)CO3 infusions. Am. J. Physiol. 281, E683–E692 (2001).
Allsop, J. R., Wolfe, R. R. & Burke, J. F. Tracer priming the bicarbonate pool. J. Appl. Physiol. 45, 137–139 (1978).
Irving, C. S., Wong, W. W., Shulman, R. J., Smith, E. O. & Klein, P. D. [13C]bicarbonate kinetics in humans: intra- vs. interindividual variations. Am. J. Physiol. 245, R190–R202 (1983).
Schrauwen, P., Blaak, E. E., Van Aggel-Leijssen, D. P., Borghouts, L. B. & Wagenmakers, A. J. Determinants of the acetate recovery factor: implications for estimation of [13 C]substrate oxidation. Clin. Sci. 98, 587–592 (2000).
Brooks, G. A. & Donovan, C. M. Effect of endurance training on glucose kinetics during exercise. Am. J. Physiol. 244, E505–E512 (1983).
Brooks, G. A. Lactate: glycolytic end product and oxidative substrate during sustained exercise in mammals-the "lactate shuttle". In Comparative Physiology and Biochemistry—Current Topics and Trends (ed. Gilles, R.) 208–218 (Springer-Verlag, 1984).
Brooks, G. A. Glycolytic end product and oxidative substrate during sustained exercise in mammals—the "lactate shuttle". In Comparative Physiology and Biochemistry—Current Topics and Trends, Volume A, Respiration - Metabolism - Circulation, Gilles, R. (Ed.) Springer Verlag, Berlin, 208–218 (1985).
Brooks, G. A. Anaerobic threshold: review of the concept and directions for future research. Med. Sci. Sports Exerc. 17, 22–34 (1985).
Brooks, G. A. Mammalian fuel utilization during sustained exercise. Comp. Biochem. Physiol. Part B, Biochem. Mol. Biol. 120, 89–107 (1998).
Brooks, G. A. Lactate shuttle—between but not within cells? J. Physiol. 541, 333–334 (2002).
Deuticke, B. Monocarboxylate transport in erythrocytes. J. Membr. Biol. 70, 89–103 (1982).
Halestrap, A. P. Transport of pyruvate nad lactate into human erythrocytes. Evidence for the involvement of the chloride carrier and a chloride-independent carrier. Biochemical J. 156, 193–207 (1976).
Roth, D. A. & Brooks, G. A. Lactate and pyruvate transport is dominated by a pH gradient-sensitive carrier in rat skeletal muscle sarcolemmal vesicles. Arch. Biochem. Biophysics 279, 386–394 (1990).
Roth, D. A. & Brooks, G. A. Lactate transport is mediated by a membrane-bound carrier in rat skeletal muscle sarcolemmal vesicles. Arch. Biochem. Biophysics 279, 377–385 (1990).
Watt, P. W., MacLennan, P. A., Hundal, H. S., Kuret, C. M. & Rennie, M. J. L( + )-lactate transport in perfused rat skeletal muscle: kinetic characteristics and sensitivity to pH and transport inhibitors. Biochim Biophys. Acta 944, 213–222 (1988).
Garcia, C. K., Goldstein, J. L., Pathak, R. K., Anderson, R. G. & Brown, M. S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: implications for the Cori cycle. Cell 76, 865–873 (1994).
Levinovitz, A. W. & Ringertz, N. The Nobel Prize: The First 100 Years (Imperial College Press, 2001).
Garcia, C. K., Brown, M. S., Pathak, R. K. & Goldstein, J. L. cDNA cloning of MCT2, a second monocarboxylate transporter expressed in different cells than MCT1. J. Biol. Chem. 270, 1843–1849 (1995).
Hashimoto, T., Hussien, R., Cho, H. S., Kaufer, D. & Brooks, G. A. Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS ONE 3, e2915 (2008).
Pierre, K. & Pellerin, L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J. Neurochem. 94, 1–14 (2005).
Hussien, R. & Brooks, G. A. Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol. Genomics 43, 255–264 (2011).
Sonveaux, P. et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 118, 3930–3942 (2008).
Price, N. T., Jackson, V. N. & Halestrap, A. P. Cloning and sequencing of four new mammalian monocarboxylate transporter (MCT) homologues confirms the existence of a transporter family with an ancient past. Biochemical J. 329, 321–328 (1998). Pt 2.
Wilson, M. C. et al. Lactic acid efflux from white skeletal muscle is catalyzed by the monocarboxylate transporter isoform MCT3. J. Biol. Chem. 273, 15920–15926 (1998).
Bonen, A. et al. Isoform-specific regulation of the lactate transporters MCT1 and MCT4 by contractile activity. Am. J. Physiol. Endocrinol. Metab. 279, E1131–E1138 (2000).
Halestrap, A. P. The SLC16 gene family - structure, role and regulation in health and disease. Mol. Asp. Med. 34, 337–349 (2013).
Pellerin, L. & Magistretti, P. J. Sweet sixteen for ANLS. J. Cereb. Blood Flow. Metab. https://doi.org/10.1038/jcbfm.2011.149 (2011).
Schurr, A. Lactate: the ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow. Metab. 26, 142–152 (2006).
Schurr, A. Lactate: a major and crucial player in normal function of both muscle and brain. J. Physiol. 586, 2665–2666 (2008).
Schurr, A. & Gozal, E. Aerobic production and utilization of lactate satisfy increased energy demands upon neuronal activation in hippocampal slices and provide neuroprotection against oxidative stress. Front. Pharmacol. 2, 96 (2011).
Schurr, A. & Payne, R. S. Lactate, not pyruvate, is neuronal aerobic glycolysis end product: an in vitro electrophysiological study. Neuroscience 147, 613–619 (2007).
Semenza, G. L. Tumor metabolism: cancer cells give and take lactate. J. Clin. Investig. 118, 3835–3837 (2008).
San-Millan, I. & Brooks, G. A. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation ofthe Warburg Effect. Carcinogenesis. https://doi.org/10.1093/carcin/bgw127 (2016).
Hashimoto, T., Hussien, R. & Brooks, G. A. Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: evidence of a mitochondrial lactate oxidation complex. Am. J. Physiol. Endocrinol. Metab. 290, E1237–E1244 (2006).
Hashimoto, T., Masuda, S., Taguchi, S. & Brooks, G. A. Immunohistochemical analysis of MCT1, MCT2 and MCT4 expression in rat plantaris muscle. J. Physiol. 567, 121–129 (2005).
Hashimoto, T., Hussien, R., Oommen, S., Gohil, K. & Brooks, G. A. Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. FASEB J. 21, 2602–2612 (2007).
Brooks, G. A. Cell-cell and intracellular lactate shuttles. J. Physiol. 587, 5591–5600 (2009).
Hochachka, P. W. Living Without Oxygen: Closed and Open Systems in Hypoxia Tolerance (Harvard University Press, 1980).
Hochachka, P. W. Cross-species studies of glycolytic function. Adv. Exp. Med. Biol. 474, 219–229 (1999).
Zinker, B. A., Lacy, D. B., Bracy, D., Jacobs, J. & Wasserman, D. H. Regulation of glucose uptake and metabolism by working muscle. An in vivo analysis. Diabetes 42, 956–965 (1993).
Herzig, S. et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96 (2012).
Bricker, D. K. et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100 (2012).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Ahmed, K. et al. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 11, 311–319 (2010).
Bergersen, L. H. Lactate transport and signaling in the brain: potential therapeutic targets and roles in body-brain interaction. J. Cereb. Blood Flow. Metab. 35, 176–185 (2015).
Cui, Z. et al. Preliminary quantitative profile of differential protein expression between rat L6 myoblasts and myotubes by stable isotope labeling with amino acids in cell culture. Proteomics 9, 1274–1292 (2009).
Dai, X., Lv, X., Thompson, E. W. & Ostrikov, K. K. Histone lactylation: epigenetic mark of glycolytic switch. Trends Genet. https://doi.org/10.1016/j.tig.2021.09.009 (2021).
Holloszy, J. O. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 242, 2278–2282 (1967).
Davies, K. J., Packer, L. & Brooks, G. A. Biochemical adaptation of mitochondria, muscle, and whole-animal respiration to endurance training. Arch. Biochem Biophys. 209, 539–554 (1981).
Genders, A. J., Martin, S. D., McGee, S. L. & Bishop, D. J. A physiological drop in pH decreases mitochondrial respiration, and HDAC and Akt signaling, in L6 myocytes. Am. J. Physiol. Cell Physiol. 316, C404–C414 (2019).
Azevedo, J. L., Tietz, E., Two-Feathers, T., Paull, J. & Chapman, K. Lactate, fructose and glucose oxidation profiles in sports drinks and the effect on exercise performance. PLoS ONE 2, e927 (2007).
Nalos, M. et al. Half-molar sodium lactate infusion improves cardiac performance in acute heart failure: a pilot randomised controlled clinical trial. Crit. Care 18, R48 (2014).
See, E. J. & Bellomo, R. The importance of applying physiological principles of hyperlactataemia to the study of human disease. J. Physiol. 599, 1933 (2021).
Masharani, U., Strycker, L. A., Lazar, A. A., Wu, K. & Brooks, G. A. Hyperlactatemia in diabetic ketoacidosis. Diabet Med. 39, e14723 (2021).
Kennedy, K. M. & Dewhirst, M. W. Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol. 6, 127–148 (2010).
Mazan-Mamczarz, K. et al. Targeted suppression of MCT-1 attenuates the malignant phenotype through a translational mechanism. Leuk. Res. 33, 474–482 (2009).
Hill, A. V. Muscular activity and carbohydrate metabolism. Science 60, 505–514 (1924).
Hill, A. V. & Lupton, H. Muscular exercise, lactic acid and the supply and utilization of oxygen. Q. J. Med. 16, 135–171 (1923).
Krogh, A. & Lindhard, J. The relative value of fat and carbohydrate as sources of muscular energy. Biochem J. 14, 290–363 (1920).
Zuntz, V. N. Betrachtungen uber die beziehungen zwischen nahrstoffen und leistungen des korpers. in Handbuch der Biochemie des Menschen und der Tiere (ed. Oppenheimer, K.) 826–863 (1911).
Warburg, O. Über den Stoffwechsel der Tumoren. in Arbeiten aus dem Kaiser Wilhelm-Institut für Biologie (Julius Springer, 1926).
Brooks, G. A. et al. Lactate in contemporary biology: a phoenix risen(1). J. Physiol. https://doi.org/10.1113/JP280955 (2021).
Cori, C. F. & Cori, G. T. Glycogen formation in the liver from d- and l-lactic acid. J. Biol. Chem. 81, 389 (1929).
Margaria, R., Edwards, H. T. A. & Dill, D. B. The possible mechanisms of contracting and paying the oxygen debt and the rôle of lactic acid in muscular contraction. Am. J. Physiol. 106, 689–715 (1933).
Bang, O. The lactate content of the blood during and after muscular exercise in man. Skand. Arch. fur Physiologie 74, 46–82 (1936).
Krebs, H. A. & Johnson, W. A. Metabolism of ketonic acids in animal tissues. Biochem J. 31, 645–660 (1937).
Krebs, H. A., Salvin, E. & Johnson, W. A. The formation of citric and alpha-ketoglutaric acids in the mammalian body. Biochem J. 32, 113–117 (1938).
Drury, D. R. & Wick, A. N. Metabolism of lactic acid in the intact rabbit. Am. J. Physiol. 184, 304–308 (1956).
Wasserman, K. & McIlroy, M. B. Detecting the threshold of anaerobic metabolism in cardiac patients during exercise. Am. J. Cardiol. 14, 844–852 (1964).
Brooks, G. A. & Gladden, L. B. Metabolic systems: non-oxidative (glycolytic and phosphagen). in Exercise Physiology: People and Ideas (ed. Tipton, C. M.) 322–360 (American Physiological Society, 2003).
Jöbsis, F. F. & Stainsby, W. N. Oxidation of NADH during contractions of circulated mammalian skeletal muscle. Respiration Physiol. 4, 292–300 (1968).
Brooks, G. A., Brauner, K. E. & Cassens, R. G. Glycogen synthesis and metabolism of lactic acid after exercise. Am. J. Physiol. 224, 1162–1166 (1973).
Emhoff, C. A. et al. Direct and indirect lactate oxidation in trained and untrained men. J. Appl. Physiol. 115, 829–838 (2013).
Bergman, B. C. et al. Muscle net glucose uptake and glucose kinetics after endurance training in men. Am. J. Physiol. 277, E81–E92 (1999).
Hooker, A. M. & Baldwin, K. M. Substrate oxidation specificity in different types of mammalian muscle. Am. J. Physiol. 236, C66–C69 (1979).
Baldwin, K. M., Campbell, P. J. & Cooke, D. A. Glycogen, lactate, and alanine changes in muscle fiber types during graded exercise. J. Appl. Physiol. 43, 288–291 (1977).
Foster, D. W. Banting lecture 1984. From glycogen to ketones–and back. Diabetes 33, 1188–1199 (1984).
Brooks, G. A., Fahey, T. D. & Baldwin, K. M. EXERCISE PHYSIOLOGY: Human Bioenergetics and Its Applications, V edn, vol. 2 (Kindle Direct Publishing, 2019).
Schurr, A., West, C. A. & Rigor, B. M. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 240, 1326–1328 (1988).
Brooks, G. A. Current concepts in lactate exchange. Med Sci. Sports Exerc. 23, 895–906 (1991).
Schurr, A., Payne, R. S., Miller, J. J. & Rigor, B. M. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study. Brain Res. 744, 105–111 (1997).
Pellerin, L. Lactate as a pivotal element in neuron-glia metabolic cooperation. Neurochem Int. 43, 331–338 (2003).
Pilegaard, H., Terzis, G., Halestrap, A. & Juel, C. Distribution of the lactate/H + transporter isoforms MCT1 and MCT4 in human skeletal muscle. Am. J. Physiol. 276, E843–E848 (1999).
Pilegaard, H., Ordway, G. A., Saltin, B. & Neufer, P. D. Transcriptional regulation of gene expression in human skeletal muscle during recovery from exercise. Am. J. Physiol. Endocrinol. Metab. 279, E806–E814 (2000).
Bergman, B. C. et al. Evaluation of exercise and training on muscle lipid metabolism. Am. J. Physiol. 276, E106–E117 (1999).
Schurr, A., Payne, R. S., Miller, J. J., Tseng, M. T. & Rigor, B. M. Blockade of lactate transport exacerbates delayed neuronal damage in a rat model of cerebral ischemia. Brain Res 895, 268–272 (2001).
Miller, B. F. et al. Lactate and glucose interactions during rest and exercise in men: effect of exogenous lactate infusion. J. Physiol. 544, 963–975 (2002).
Miller, B. F. et al. Metabolic and cardiorespiratory responses to "the lactate clamp". Am. J. Physiol. Endocrinol. Metab. 283, E889–E898 (2002).
Valenti, D., de Bari, L., Atlante, A. & Passarella, S. L-Lactate transport into rat heart mitochondria and reconstruction of the L-lactate/pyruvate shuttle. Biochem J. 364, 101–104 (2002).
Bouzier-Sore, A. K., Voisin, P., Canioni, P., Magistretti, P. J. & Pellerin, L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J. Cereb. Blood Flow. Metab. 23, 1298–1306 (2003).
Smith, D. et al. Lactate: a preferred fuel for human brain metabolism in vivo. J. Cereb. Blood Flow. Metab. 23, 658–664 (2003).
Bartman, C. R., TeSlaa, T. & Rabinowitz, J. D. Quantitative flux analysis in mammals. Nat. Metab. https://doi.org/10.1038/s42255-021-00419-2 (2021).
Hui, S. et al. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017).
Rabinowitz, J. D. & Enerback, S. Lactate: the ugly duckling of energy metabolism. Nat. Metab. 2, 566–571 (2020).
Chen, Y. J. et al. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 12, 937–943 (2016).
Brooks, G. A. Body-mind learning: a lesson for the mind from muscle. Exerc. Sport Sci. Rev. 35, 163–165 (2007).
Poole, D. C., Rossiter, H. B., Brooks, G. A. & Gladden, L. B. The anaerobic threshold: 50+ years of controversy. J. Physiol. https://doi.org/10.1113/JP279963 (2020).
Acknowledgements
This work was supported by the National Institutes of Health grant R01 AG059715 to G.A.B. The authors thank the reviewers for providing expert commentary.
Author information
Authors and Affiliations
Contributions
GAB wrote the initial draft and edited the text depending on feedfack from the coauthors. Coauthors provided technical and conceptual feed back in editing of the final manuscript. All coauthors read and approved the final manuscript version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brooks, G.A., Curl, C.C., Leija, R.G. et al. Tracing the lactate shuttle to the mitochondrial reticulum. Exp Mol Med 54, 1332–1347 (2022). https://doi.org/10.1038/s12276-022-00802-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00802-3
This article is cited by
-
The polymorphism T1470A of the SLC16A1 gene is associated with the lactate and ventilatory thresholds but not with fat oxidation capacity in young men
European Journal of Applied Physiology (2024)
-
Wearable cellulose textile matrix self-powered biosensor sensing lactate in human sweat
Journal of Applied Electrochemistry (2024)
-
Vertical Strength Transfer Phenomenon Between Upper Body and Lower Body Exercise: Systematic Scoping Review
Sports Medicine (2024)
-
Tracing metabolic flux in vivo: motion pictures differ from snapshots
Experimental & Molecular Medicine (2022)
