
Abstract
Imbalance between fat production and consumption causes various metabolic disorders. Nonalcoholic fatty liver disease (NAFLD), one such pathology, is characterized by abnormally increased fat synthesis and subsequent fat accumulation in hepatocytes1,2. While often comorbid with obesity and insulin resistance, this disease can also be found in lean individuals, suggesting specific metabolic dysfunction2. NAFLD has become one of the most prevalent liver diseases in adults worldwide, but its incidence in both children and adolescents has also markedly increased in developed nations3,4. Progression of this disease into nonalcoholic steatohepatitis (NASH), cirrhosis, liver failure, and hepatocellular carcinoma in combination with its widespread incidence thus makes NAFLD and its related pathologies a significant public health concern. Here, we review our understanding of the roles of dietary carbohydrates (glucose, fructose, and fibers) and the gut microbiota, which provides essential carbon sources for hepatic fat synthesis during the development of NAFLD.
Similar content being viewed by others

Bridging carbohydrate metabolism and hepatic de novo lipogenesis
Carbohydrate and lipid metabolism is highly intertwined in the liver. De novo lipogenesis (DNL), a process that converts dietary carbohydrates into fat, is one such metabolic link. Dietary carbohydrates and lipids follow distinct paths from the gut lumen to other parts of the body. Due to their hydrophobicity, dietary fats, mostly composed of long-chain fatty acids, are packaged as triglycerides (TGs) into chylomicrons in intestinal enterocytes and distributed to nonhepatic tissues (e.g., adipose depots) through the lymphatic system, thereby bypassing metabolism in the liver5. On the other hand, dietary carbohydrates absorbed by the small intestine go directly to the liver. When the carbohydrate level reaches a certain threshold, diverse metabolic pathways in hepatocytes are triggered to clear away excess carbohydrates. Glycogen synthesis and DNL are such pathways. The resulting newly synthesized fatty acids are stored in hepatocytes as lipid droplets, released into the bloodstream as lipoprotein particles (e.g., very-low-density lipoproteins or VLDLs) to feed other organs, or oxidized in the liver when other energy sources are scarce (e.g., under fasting conditions). Thus, DNL is a key mechanism that contributes to the metabolic flux of dietary carbohydrates into lipids, especially in the postprandial state and under disease conditions6,7.
DNL is controlled by allosteric regulation of diverse lipogenic enzymes. Dietary carbohydrates (mainly glucose) first enter glycolysis in the cytosol and become pyruvate, the terminal product of glycolysis (Fig. 1). Pyruvate then either becomes lactate or enters the mitochondria for further oxidation in the citric acid (TCA) cycle. In high-energy states, citrate is transported back to the cytosol and converted into cytosolic acetyl-CoA by ATP citrate lyase (ACLY)8. This generation of cytosolic acetyl-CoA by ACLY is the key step in lipogenesis, as mitochondrial acetyl-CoA cannot directly participate in DNL. Notably, in obese individuals, a small fraction of short-chain fatty acids can be transported from the mitochondria to feed DNL, but the biological significance of this pathway needs more investigation9. Acetyl-CoA carboxylase (ACC) then generates malonyl-CoA from cytosolic acetyl-CoA, the commitment step in DNL. This process is allosterically promoted by cytosolic citrate. Importantly, malonyl-CoA serves as a potent allosteric inhibitor of fatty acid β-oxidation. This mechanism is thought to act as a metabolic safeguard against wasteful energy expenditure from simultaneous fatty acid synthesis and oxidation10. The final step of DNL is the conversion of malonyl-CoA into palmitate (16:0) by fatty acid synthase (FAS). Other processes, such as fatty acid elongation and desaturation, also occur after palmitate is synthesized7,10.

Dietary carbohydrates, such as glucose and fructose, are absorbed by the small intestine and delivered to the liver via the portal circulation. Alternatively, fructose or fibers reach the colon and are catabolized by the gut microbiota, producing short-chain fatty acids, including acetate. In hepatocytes, glycolysis/fructolysis provides carbon sources for ACLY-mediated generation of cytosolic acetyl-CoA from cytosolic citrate. On the other hand, acetate provides carbon for ACSS2-mediated synthesis of cytosolic acetyl-CoA, especially in low-energy environments. Acetyl-CoA can also be used for histone acetylation in the nucleus. Cytosolic acetyl-CoA is used for DNL. Metabolites of glucose or fructose also act in a signaling capacity to turn on lipogenic transcription factors, such as SREBP-1c and ChREBP, a process that is further augmented by insulin signaling.
Another crucial layer of DNL regulation occurs at the transcriptional level, which is in part hormonally controlled. Under postprandial conditions, the release of insulin stimulates the expression of lipogenic genes in the liver to promote DNL. Sterol regulatory element-binding protein (SREBP) is a well-known transcription factor that simultaneously induces many lipogenic genes. While there are several SREBP isomers, SREBP-1c is particularly insulin-sensitive compared to the other SREBP isoforms SREBP-1a and SREBP-211,12,13,14. In mice, the loss of Srebp-1c resulted in failure to induce DNL enzymes, including FAS and ACC, in response to postprandial increases in insulin levels, and this lack of response was only partially compensated by SREBP-1a/SREBP-215. Therefore, SREBP-1c is the primary isoform responsible for insulin-induced DNL in hepatocytes. The other SREBP isoforms have been linked to separate metabolic pathways; SREBP-2 is associated with cholesterol synthesis (another important feature of lipid metabolism), while SREBP-1a is mainly expressed in the spleen16,17.
In addition to insulin, full activation of the hepatic lipogenic program requires glucose18. While glucose itself has not been found to modulate transcriptional DNL activity, one glycolytic metabolite, glucose-6-phosphate (G6P), has been linked to increased FAS and ACC expression19,20. This result was further substantiated by in vitro analysis of Fas expression and glucokinase activity, the enzyme that converts glucose into G6P. Both glucose and glucokinase were found to be required to induce Fas expression in response to insulin20. Glucose-induced DNL proceeds through the upregulation of both glucokinase and SREBP-1c in response to insulin14,21. Dentin et al. further expanded on this model and implicated carbohydrate responsive element-binding protein (ChREBP) as another important transcription factor regulated by G6P to trigger DNL22. However, this link between ChREBP and dietary carbohydrates is not exclusive to glucose; hepatic ChREBP is particularly sensitive to fructose (see more details in the section below)23. Indeed, G6P may not be the only metabolite that stimulates ChREBP activity, and various other metabolites (e.g., xylulose 5-phosphate, 3-carbon glycolytic metabolites) have also been suggested to have this effect23,24,25.
Importantly, ChREBP knockdown decreased the effects of glucose on both lipogenic and glycolytic gene induction in response to insulin22,24. It is thus evident that ChREBP mediates insulin-glucose synergy26. One mechanism of action involves glucose-mediated increases in ChREBP expression, nuclear entry, and binding to the carbohydrate response element (ChoRE), which in turn induces not only lipogenic genes but also SREBP-1c27,28. ChREBP and SREBP thus rely on each other and work synergistically as follows: 1) ChREBP relies on activation by glycolytic intermediates, which, in turn, are generated by genes controlled by SREBP-1c22. 2) Meanwhile, the deletion of hepatic Chrebp reduces SREBP-1c levels29. 3) Furthermore, overexpression of hepatic Chrebp does not increase postprandial lipogenesis when Srebp-1c is lacking29.
Collectively, maximum activation of DNL requires concurrent upregulation of lipogenic and glycolytic gene expression, for which glucose and insulin are necessary to stimulate lipogenic transcription factors. In this regard, for insulin-resistant individuals with NAFLD, increases in both circulating glucose and insulin can be synergistic drivers of continuously elevated DNL, the hallmark of this disease18.
Fructose metabolism both triggers and feeds DNL
In addition to glucose, fructose is another major dietary carbohydrate in modern society that is closely linked to NAFLD/NASH development. While fructose and glucose have the same molecular formula (C6H12O6), they are metabolized by organs quite differently and have divergent effects on hepatic DNL30. For example, fructose can serve as a preferential substrate of DNL due to the faster rate of fructolysis compared to that of glycolysis31,32,33. During the primary steps of glycolysis, hexokinase and phosphofructokinase are allosterically regulated, suppressing excessive glycolysis. In contrast, ketohexokinase-c (KHK-C), the primary fructolytic enzyme, has constitutively high activity and thus bypasses these rate-limiting glycolytic steps. This results in rapid fructose catabolism, which triggers ATP depletion and carbon entry into the TCA cycle as well as DNL34. However, most in vitro hepatocyte studies used supraphysiological millimolar concentrations of fructose in the culture medium. Isotope tracing studies did not carefully differentiate between the direct and indirect contributions of fructose to the hepatic DNL pathway.
This raised the possibility that fructose-dependent DNL and NAFLD may also be linked to nonhepatic fructose catabolism. Supporting this notion, high-fructose feeding was shown to increase hepatic steatosis and inflammation in both wild-type and liver-specific Chrebp-knockout mice, which exhibit blunted hepatic fructose metabolism23. Recently, it was shown that the small intestine actively catabolizes fructose in a KHK-dependent manner, thereby reducing the amount of fructose that reaches the liver and preventing hepatic steatosis30,35. Remarkably, isotopic tracing of fructose in various genetically modified mouse models demonstrated that some fructose-derived DNL carbon sources in the liver are not derived directly from fructose itself but rather derived indirectly from fructose-derived acetate made by the gut microbiota36,37. These new data challenge the previously known role of fructose-derived direct lipogenesis in hepatocytes and emphasize the complex interaction between the intestine, liver, and gut microbiota.
What is the pathological role of fructose catabolism in the liver, then? Accumulating evidence has indicated that fructose is an especially potent inducer of hepatic DNL signaling. For example, ChREBP is more strongly responsive to fructose than glucose38. Both nuclear SREBP-1 levels and ChREBP activity were increased in the liver after fructose feeding but not glucose feeding39. Mice lacking whole-body or hepatic KHK, but not intestinal KHK, showed protection from fructose-induced hepatic steatosis40,41,42, demonstrating that hepatic fructose catabolism is a key molecular event that triggers NAFLD.
In humans, chronic high-fructose feeding in male participants increased both postprandial and steady-state serum TG levels-effects that were not seen in glucose-fed individuals43. Bolus fructose consumption also induced increased palmitate incorporation into hepatic lipoproteins as well as increased postprandial production of TGs in fructose-fed, but not glucose-fed, individuals44. KHK-C overexpression has been commonly observed in obese NASH patients45, supporting the notion that it is a proximal step of fructose metabolism needed to provide lipogenic signals to induce pathogenesis. These strong genetic mouse data in conjunction with human epidemiological and feeding studies provide promising pharmaceutical basis for the clinical use of KHK inibitors in fatty liver treatment46.
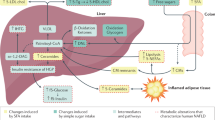
Causes and comorbidities of NAFLD and NASH
Hepatic DNL
The hallmark of NAFLD is the accumulation of lipids in the liver. What is the major source of these lipids? Early research in lean, fasted individuals suggested that DNL makes only a minimal contribution to hepatic lipid levels, as most hepatic lipids were traced back to fatty acids released from adipose tissue. However, further studies with sophisticated isotope tracing have shown that DNL plays a larger role in NAFLD patients than previously suggested47,48,49. Comparison of NAFLD patients’ livers with high versus low fat accumulation reported a 3.5-fold increase in DNL in patients with high fat accumulation but no difference in the contribution of dietary or free fatty acids to the hepatic TG pool47. Fabbrini et al. also observed that a significant portion of serum free fatty acids were not incorporated into hepatic lipid stores in obese individuals with steatosis, suggesting that these lipids did not originate from free fatty acids but rather originated from DNL49. NAFLD patients also exhibited strongly upregulated expression of DNL enzymes, such as ACC1, ACC2, and FAS50,51. Interestingly, NAFLD patients exhibited increased DNL even during fasting52, indicating the lack of metabolic flexibility in these patients as opposed to the robust suppression of DNL during fasting observed in healthy individuals44,53. This may also reflect other metabolic inflexibilities, such as insulin resistance, typically found in NAFLD patients.
While upregulated DNL is a key feature of NAFLD, it may not be the sole instigator of disease progression. Like their non-NAFLD counterparts, NAFLD patients still derive the majority of their hepatic TG stores from fatty acids released by adipocytes;52,54,55 26% of hepatic TGs were found to come from fatty acids synthesized de novo, whereas ~60% came from serum fatty acids and ~10% from diet52. Thus, in addition to upregulated DNL, other critical factors induce disease progression to NASH, as discussed below.
Hepatic inflammation and insulin resistance
Mild cases of NAFLD can be asymptomatic or benign. However, progression into NASH is characterized by inflammation, hepatocyte apoptosis, and steatosis. One longitudinal study found that individuals with NASH, but not those with only NAFLD, showed increased mortality and the increased occurrence of end-stage liver disease than the general population56,57. These results were further corroborated by Dam-Larsen et al., who concluded that a large population of individuals with simple NAFLD had a favorable long-term prognosis with no disease progression58. However, co-diagnosis with other metabolic diseases, such as type 2 diabetes, were correlated with higher rates of cirrhosis, end-stage liver disease, and death59. In a cohort of NAFLD patients without steatohepatitis, cardiovascular disease (CVD) and cancer were found to be the leading causes of mortality two decades after initial diagnosis58. It is possible that this proclivity for CVD over cirrhosis and end-stage liver disease is a result of systemic metabolic disorder and dysfunction, as NAFLD is often also co-diagnosed with insulin resistance, obesity, and hyperlipidemia60. Thus, the lethality of NAFLD may be due to hepatic manifestation of a systemic metabolic syndrome rather than the direct result of damage by steatosis.
Systemic insulin resistance is a key component in NAFLD, as patients have decreased insulin sensitivity across adipocytes, hepatocytes, and skeletal muscle61. A body of longitudinal human studies also suggests that steatosis contributes to the development of insulin resistance. Ekstedt et al. found that 78% of NAFLD patients developed impaired glucose tolerance or type 2 diabetes after their initial diagnosis57. Cohort studies of East Asian men corroborated these results and established NAFLD as a risk factor for type 2 diabetes, even in nonobese individuals62,63. Accumulated hepatic lipids, formed either by DNL or from free fatty acids, have been linked to the onset of hepatic insulin resistance. A positive correlation between high hepatic/visceral fat and hepatic insulin resistance was found in NAFLD patients64,65. In rats, 2,4-dinitrophenol (DNP), a drug that promotes β-oxidation in hepatocytes, reduced the hepatic lipid content and improved insulin sensitivity66. Conversely, systemic insulin resistance can cause hepatic lipid accumulation in NAFLD. Insulin-resistant adipocytes have higher rates of lipolysis, releasing excessive free fatty acids into the circulation that are incorporated into liver lipids. Another insulin-mediated mechanism is DNL upregulation. Glucose uptake in skeletal muscle and adipose tissue is dependent on insulin, while in hepatocytes, glucose uptake is largely insulin-independent67. Thus, this excessive influx of glucose into hepatocytes can cause steatosis via DNL68.
However, it is noteworthy that hepatic lipid accumulation does not always share a causal relationship with insulin resistance. Overexpression of diacylglycerol acyltransferase 2 (DGAT2), an important enzyme in the final production of TG synthesis, raised hepatic TG levels and caused steatosis but did not result in hepatic or systemic insulin resistance69. This finding indicates that the quality of accumulated lipids rather than their quantity matters, as further discussed below.
Lipotoxicity
As previously mentioned, not every case of NAFLD leads to chronic inflammation, hepatocyte apoptosis, and cirrhosis. As such, there must be a metabolic distinction between individuals with steatosis and those with progressive degeneration. The development of NASH and cirrhosis from NAFLD has been described by the “multiple-hit” hypothesis. The “first hit” of insulin resistance leads to lipid accumulation in the liver, which makes the organ more susceptible to the secondary “multiple hits” of oxidative stress and inflammation70. In this model, the accumulation of TGs is thought to dysregulate lipid flux throughout the liver in this “first hit”. However, Listenberger et al. suggested that TG formation may be a form of protection against lipotoxicity from free fatty acids71. The results from a similar study with obese Dgat2-knockout mice with NASH also support the findings of Listenberger et al., as a decrease in hepatic TG stores failed to dampen oxidative stress signals or NASH progression72.
Even more than the level of fatty acid influx to the liver, the nature of these fatty acids may contribute to lipotoxicity. A higher ratio of saturated fatty acids (SFAs) to monosaturated fatty acids (MUFAs) has been connected to NASH development73,74. The conversion of lipotoxic SFAs to MUFAs is mediated by stearoyl-CoA desaturase-1 (SCD1). Free fatty acid influx upregulates SCD1 expression, resulting in MUFA formation, TG storage, and ultimately, liver adaptation with no progression beyond steatosis. However, SCD1 downregulation triggers mitochondria-mediated apoptosis through death receptor signaling, ER stress, and the release of proteases in the cytoplasm from increased lysosomal permeability74. Identification of key lipotoxic lipid species (e.g., ceramides, diacylglycerol, SFAs, acylcarnitine, lipopolysaccharides) and associated metabolic and signaling pathways is thus crucial to prevent disease progression from NAFLD to NASH.
Obesity
While some NAFLD patients are lean, obesity is still prevalent in individuals with NAFLD58. In obese individuals with type 2 diabetes, regulation of adipocyte lipolysis is largely absent, which promotes the release of fatty acids into the circulation. An increase in fat deposits, particularly an increase in the mass of visceral adipose tissue, further contributes to the influx of free fatty acids into the liver60,75. Yamaguchi et al. found that the suppression of TG production failed to stop liver damage or fibrosis in obese mouse models, suggesting that increased adipocyte lipolysis and the resultant influx of fatty acids into the liver make a large contribution to NASH pathologies72. Quantitatively, the distribution of visceral fat in the body is marginal compared to the abundance of subcutaneous fat. However, large visceral deposits in obese women have been linked to suppression of the antilipolytic effect of insulin, and increased levels of free fatty acids have been found in the hepatic portal in individuals with upper body obesity75,76. Although the mechanism by which visceral adiposity selectively contributes to NAFLD remains contentious, the close proximity of visceral fat to the portal circulation may facilitate the delivery of free fatty acids and inflammatory factors (e.g., derived from immune cells in adipose depots) to the liver77.
Key DNL enzymes as potential therapeutic targets for NAFLD treatment
While there are many enzymes in the DNL pathway, only a few of them are altered in NAFLD patients and are potentially targetable for NAFLD treatment without detrimental side effects. Here, among these enzymes, we focus on two crucial lipogenic enzymes that have been extensively studied for therapeutic intervention.
ACLY
ACLY is a critical enzyme in DNL that converts cytosolic citrate into acetyl-CoA to provide carbon sources for DNL (Fig. 1). Acetyl-CoA then enters both the fatty acid synthesis and mevalonate pathways to produce fatty acids and cholesterols, respectively. ACLY dysregulation was previously implicated in diseases associated with elevated fatty acid and cholesterol synthesis, such as cancer and atherosclerosis78,79. ACLY silencing, inhibition, and genetic deletion were found to suppress cell proliferation and tumor growth in several preclinical models80,81,82,83,84,85. One recent study on atherosclerosis also found that Acly deletion in macrophages stabilized atherosclerotic plaques—a necessary preventative treatment for stroke—and attenuated the ACLY-mediated inflammatory response in the macrophages. These phenotypes were also linked to dysregulated cholesterol and fatty acid synthesis86.
Abnormally elevated expression of ACLY has been linked to NAFLD in both humans and mice. Dietary carbohydrates (both glucose and fructose) increase the mRNA expression of Acly in mice and rats7,87. In NAFLD patients, this diet-mediated ACLY induction occurs mainly through SREBP-1c51,88, but a recent mouse study showed that ChREBP is also responsible for fructose-induced expression of ACLY and other glycolytic, fructolytic, and lipogenic genes89. Furthermore, ACLY is an essential contributor to global histone acetylation. Under nutrient-rich conditions, ACLY is the primary enzyme that mediates the production of glucose-derived nuclear acetyl-CoA, the source of histone acetyl groups80. ACLY-dependent histone acetylation has also been linked to selective control of key genes involved in glucose usage in adipocytes, including phosphofructokinase (PFK) and glucose transporter 4 (GLUT4)90.
While it is evident that ACLY is transcriptionally upregulated in NAFLD, posttranslational regulation of ACLY may also be an important contributing factor to disease pathology. ACLY nuclear localization is activated by phosphorylation at residue S455 by protein kinase B (AKT)91,92. ACLY is also phosphorylated at this site by protein kinase A (PKA), which significantly increases ACLY enzyme activity (6-fold increase in Vmax compared to that of nonphosphorylated ACLY)93. Recently, BDK, a kinase of branched chain ketoacid dehydrogenase (BCKDH), was shown to phosphorylate ACLY at the same serine residue independent of AKT and is also upregulated by ChREBP94. Small molecule-mediated inhibition of BDK improved hepatic steatosis and the insulin response in insulin-resistant Zucker fatty rats. Furthermore, overexpression of BDK increased ACLY phosphorylation and hepatic DNL.
In addition to its phosphorylation, the acetylation of ACLY seems to play important roles in NAFLD. In hepatocytes, acetylation at three lysine residues has been shown to increase ACLY enzyme stability, resulting in constitutive activation of DNL95,96. Both mice and humans with NAFLD showed increased levels of not only total ACLY but also acetylated ACLY. Furthermore, while Acly knockdown suppressed steatosis in mice fed a high-fat, high-sugar diet, high levels of ACLY 3KQ (mimicking the constitutively acetylated form) induced steatosis more strongly than overexpression of ACLY wild-type96. Mechanistically, ACLY acetylation increases enzyme stability by antagonizing its ubiquitylation95,96. At least two responsible E3 ligases have been identified to negatively regulate ACLY97,98. Overexpression of one of these E3 ligases, HMG-CoA reductase degradation protein (Hrd1), decreased both acetyl-CoA and ACLY-mediated DNL in the hepatocytes of db/db and ob/ob mice97. This effect ameliorated hepatic lipid droplet accumulation and decreased hepatocyte inflammation in vivo. Hrd1 expression was also negatively correlated with NAFLD in mice, indicating that failure to activate the ubiquitin pathway and the subsequent accumulation of ACLY contribute to NAFLD development. More broadly, the proteosome-mediated pathway seems to prevent inflammation-dependent proteotoxicity in NAFLD99.
Current ACLY therapeutics
Statins, a class of HMG-CoA reductase inhibitors, have traditionally been used to attenuate atherosclerosis by reducing low-density lipoprotein (LDL) cholesterol levels. However, the emergence of small-molecule therapeutics targeting ACLY has presented new treatment options for individuals particularly sensitive to statin-related muscle symptoms or those who require a more robust reduction in LDL cholesterol. Bempedoic acid (BemA/ETC-1002), a recently FDA-approved LDL cholesterol-lowering drug, is one such therapeutic that has been singled out for ACLY inhibition; BemA was found to reduce lipogenic intermediates such as acetyl-CoA, malonyl-CoA, and HMG-CoA while subsequently increasing citrate in rat hepatocytes100. In one rodent model, BemA relieved atherosclerotic symptoms and lowered LDL cholesterol and triglyceride levels in ApoE-knockout and ApoE/AMPK β-1 double-knockout mice101. These results suggest that BemA regulates lipid metabolism and protects against LDL cholesterol and inflammation regardless of AMPK activation in the liver. As a prodrug, BemA also requires bioactivation by very long-chain acyl-CoA synthetase-1 (ACSVL1), an enzyme primarily found in the liver102. This mediation by hepatic ACSVL1 may thus circumvent muscle effects from statin usage. Current data for BemA usage in humans are primarily directed toward the treatment of atherosclerosis over NAFLD; nevertheless, phase III clinical trials have suggested positive outcomes in lowering LDL cholesterol levels when BemA is administered in conjunction with other therapeutics103.
Acetyl-CoA synthetase short-chain family member 2 (ACSS2)
ACLY activity alone clearly does not account for the cell’s entire lipogenic capacity. Cell viability was not significantly impacted in Acly-knockout mouse cell lines80. Importantly, acetate was found to be a requirement for maintaining viability in Acly-knockout cells by supporting cellular acetyl-CoA levels and DNL80. Mice heterozygous for Acly also did not show fluctuations in lipid concentration or synthesis, suggesting that the lipogenic program proceeds normally at half ACLY protein levels104. ACSS2 is one enzyme that may explain this rescue of lipogenic capacity via its involvement in an alternative acetyl-CoA-generating pathway (Fig. 1). Similar to other members of the ACS enzyme family, ACSS2 facilitates the formation of a thioester bond between acetate and CoA to form acetyl-CoA in an ATP-dependent manner.
In ACLY-knockout cell lines, the addition of exogenous acetate at physiological blood concentrations sustained acetyl-CoA levels and lipogenic metabolites via ACSS2 induction, suggesting that circulating acetate is likely adequate for compensatory lipid synthesis80,105. Similarly, ACLY silencing increased ACSS2 expression and maintained fatty acid and mevalonate synthesis pathways to preserve the ability of cancer cells to proliferate81. In mice, the loss of Acly in white adipose tissue strongly induced Acss2 expression and promoted the contribution of Acss2 to cellular acetyl-CoA pools via acetate incorporation without any discernable effect on weight80 unless mice were fed a high-carbohydrate diet, which provoked a strong dependence on ACLY in adipocytes106. In the liver, however, ACSS2 plays a major compensatory role in the context of high-fructose feeding. Hepatocyte-specific Acly-KO mice had hepatic TG levels comparable to those of their wild-type counterparts when fed a high-fructose diet37.
Despite this apparent metabolic redundancy, there is also evidence that the method of nutrient consumption—particularly, bolus versus gradual fructose feeding—results in differential ACLY and ACSS2 utilization in the liver. When fructose is fed gradually, both ACLY and ACSS2 contribute to lipogenic acetyl-CoA via citrate cleavage and acetate uptake, and suppression of both enzymes is necessary to reduce DNL37. However, bolus fructose consumption may overload the absorption capacity of the small intestine, and the resulting fructose spillover to the colon generates copious acetate by the microbiome30. This acetate significantly increases hepatic acetyl-CoA pools via ACSS237. The role of the microbiota in DNL and NAFLD will be discussed in a later section.
ACSS2 also mediates acetate usage for other cellular processes, such as energy production and gene regulation107,108. During fasting, the liver releases acetate into the circulation by keogenesis from long-chain fatty acids. Induction of ACSS2 increases the usage of serum acetate as a fuel source in organs such as the heart, skeletal muscle, and even cancers109,110,111,112,113. Furthermore, similar to ACLY, ACSS2 can translocate into the nucleus and supply acetyl-CoA for histone acetylation to regulate gene expression108,114. Takahashi et al. found that this nuclear acetate remains separate from mitochondrial acetate pools115, suggesting that most of the acetate involved in histone acetylation is produced within the nucleus. Consistently, isotope labeling of exogenous acetate is very low in acetylated histones, especially in energy-limited environments108. Nuclear ACSS2 thus not only provides acetyl-CoA for histone acetylation but also recaptures, reuses, and retains nuclear acetate.
While histone acetylation contributes to global gene regulation116, nuclear ACSS2 has been linked to specific modulation of genes involved in lysosomal biogenesis, autophagy, and hippocampal memory117,118. In one study, Acss2 silencing significantly reduced fatty acid synthesis enzymes and made mice resistant to developing hepatic steatosis upon high-fat diet feeding119. Interestingly, the authors showed that high-fat diet feeding increased nuclear ACSS2 in intestinal enterocytes, which was correlated with upregulation of lipid absorption genes119. The authors thus proposed that ACSS2 coordinates the cell’s adaptation to energy availability through control of genes in lipid metabolism. However, a mechanistic link between nuclear ACSS2 localization and the promotion of lipogenic genes has yet to be established.
Finally, ACSS1, a cousin of ACSS2, may also be relevant to DNL in NAFLD. ACSS1 is highly expressed in the liver, and its expression is induced by SREBP120. Unlike cytosolic or nuclear ACSS2, however, ACSS1 is primarily associated with mitochondria and has been linked to mitochondrial reactions such as fatty acid β-oxidation121. Nevertheless, ACSS1 has been suggested to be at least partially functionally redundant to ACSS2 and to play a background role in acetate-derived lipogenesis under hypoxic conditions113. Similarly, Gao et al. suggested that both ACSS1 and ACSS2 are necessary for the expression of lipogenic genes, as the knockdown of either ACSS1 or ACSS2 alone did not seem to inhibit histone acetylation122.
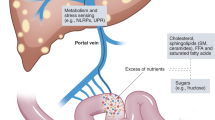
Role of the gut microbiota in NAFLD
Given that the liver is directly linked to the intestine via the portal blood and that the gut microbiome is a major source of acetate, it is not surprising that the gut microbiome plays important roles in NAFLD. Sequencing studies indicated specific changes in microbiota composition in NAFLD subjects. Healthy versus obese/NAFLD individuals showed marked population differences in members of the phyla Bacteroidetes and Firmicutes36,123. Diets supplemented with high-fructose corn syrup to induce NAFLD resulted in an increased Firmicutes:Bacteroidetes ratio124,125. However, a recent study of 10 obese patients found that fructose did not induce microbial changes126. Since this study used a solid form of fructose, it is possible that the small intestine efficiently prevented fructose spillover to the colon. Therefore, further studies with other experimental conditions (e.g., a liquid form of fructose, various doses and durations) are required. Most studies that support the causal link between the microbiota and NAFLD have reported the results of fecal transplantation from donors fed NAFLD-inducing diets127,128,129,130,131,132,133,134,135,136,137,138, which are summarized in Table 1.
The role of microbiota-derived short-chain fatty acids (SCFAs) in NAFLD
How do fructose-mediated microbial population shifts affect the onset of NAFLD? One clinical study found a link between higher SCFA concentrations and an increased Firmicutes:Bacteroidetes ratio in obese and overweight individuals138. Increases in cecal SCFA concentrations have been found in obese mice with a concurrent similar increase in the Firmicutes:Bacteroidetes ratio, supporting the clinical findings139,140. While studies often report inconsistencies in the Firmicutes to Bacteroidetes ratios in obese and NAFLD patients, increased SCFA levels are consistently reported138,141. Another potential factor is microbiome diversity. One comparative study of 154 monozygotic and dizygotic twins that were incongruous for obesity found that the twin with obesity exhibited higher Firmicutes levels and an overall decrease in gut flora diversity142. It is thus possible that this diminished microbiome diversity, rather than specific microbial populations, contributes to NAFLD pathogenesis.
One interesting development in the field is the idea that excessive microbiome-derived SCFAs feed hepatic DNL, thereby triggering NAFLD. However, different SCFAs likely have different metabolic fates, with primarily acetate feeding DNL and propionate and butyrate feeding gluconeogenesis. In one clinical study, healthy fasted individuals who received acetate and propionate did not show increases in serum free fatty acids, presumably due to hepatic glucose production rather than DNL to compensate for fasting-induced hypoglycemia143. Another study found that infusion with SCFAs lowered free fatty acids while increasing serum TG and cholesterol levels144. Isotopic tracing of cecum-infused SCFAs confirmed a significant increase in the incorporation of SCFAs into glucose, cholesterol, and lipids145. Acetate, the most abundant SCFA, is preferentially utilized by hepatocytes via ACSS2 to fuel DNL. Zhao et al. found a twofold increase in acetate after acute fructose feeding in portal blood but not systemic blood, indicating that acetate is efficiently cleared by the liver37. The authors further found that depleting the microbiome using antibiotics or silencing hepatic ACSS2 markedly blocked fructose conversion into hepatic acetyl-CoA and fatty acids. Intriguingly, fructose-induced lipogenic gene expression was intact, consistent with the notion that proximal steps of fructolysis provide a signal to transcriptionally activate the DNL program. While involved in a range of various metabolic pathways, SCFAs also have an established role as signaling molecules via G-protein coupled receptors (GPCRs)146,147. In the liver, RGS5, a protein that suppresses these GPCRs, suppresses NAFLD/NASH pathology via downregulation of lipogenic genes and inflammatory cytokines in vivo148. Thus, targeted GPCR therapeutics may represent an alternative to chronic antibiotic usage in the treatment of NAFLD/NASH.
In contrast, some studies proposed the beneficial effects of SCFAs in reducing adiposity and restoring insulin sensitivity in obese mice149,150. Additionally, in contrast to what it does in the liver, acetate in the gut-brain axis has been shown to suppress appetite by upregulating γ-aminobutyric acid (GABA) activity in the hypothalamus and was thus flagged as a possible therapeutic for obesity151. The exact roles of propionate and butyrate in the liver are also under contention. Butyrate has been shown to robustly promote the mevalonate and fatty acid synthesis pathways by yielding intermediates such as HMG-CoA152,153. Propionate, on the other hand, inhibits the incorporation of acetyl-CoA into fatty acids without reducing cytosolic citrate levels, suggesting that it may compete with acetate for CoA and thereby suppress DNL and drive odd-chain fatty acid synthesis154,155. To make matters more complex, Weitkunat et al. found that the addition of both propionate and high amounts acetate in a long-term high-fat diet decreased the liver TG content and DNL gene expression, suggesting that SCFAs may play a protective role against steatosis156. Therefore, the role of SCFAs in NAFLD is likely context-dependent, and more systematic investigation is required.
Effect of fructose on colon integrity
In addition to supplying lipogenic substrates via the microbiome, fructose itself causes intestinal damage to induce NAFLD/NASH. Fructose feeding has been shown to elevate plasma endotoxin levels, presumably due to increased oxidative stress and decreased intestinal junction integrity157. Bacterial endotoxins induce adipocyte lipolysis via Toll-like receptor 4 (TLR4) signaling and increase serum-free fatty acids158. Elevated free fatty acid levels have been linked to impaired insulin signaling159, and serum fatty acids that enter the liver can be incorporated into TGs, contributing to steatosis and insulin resistance69,160,161. A recent study further supported the notion that fructose-fed mice developed NAFLD/NASH phenotypes as a result of intestinal barrier deterioration and TLR signaling162. Antibiotic treatment blocked intestinal tight junction degeneration and lowered inflammatory hepatic cytokines and chemokines. Interestingly, antibiotics also suppressed DNL induction. Gut-mediated inflammation and endotoxemia may also explain NASH development. The progression of NAFLD into NASH has been described in the previous sections by the ‘multiple hit’ hypothesis, in which the first hits of lipid accumulation and insulin resistance are exacerbated by inflammation, leading to further hepatic injury. It is thus possible that fructose-induced gut permeability increases the liver’s susceptibility to these ‘second hits’ of systemic inflammation from serum endotoxins.
However, the human relevance of fructose-induced gut leakiness and microbial dysbiosis is still controversial. A recent clinical study showed that obese participants given a daily dose of 75 g of fructose over a course of a few weeks displayed no gut permeability or endotoxemia126. It is possible that the concentration of fructose administered in animal studies is beyond the concentrations received by typical human consumption, causing chronic and systemic changes. Conversely, these phenotypes may also be seen in humans if high fructose consumption proceeds long enough. Therefore, while understanding of the relationship between the microbiome, fructose, and hepatotoxicity in animal models has increased, more human data are necessary to draw the same conclusions regarding human NAFLD pathologies.
Is dietary fiber, the major source of acetate produced by the microbiota, a friend or foe?
As described above, acetate is the preferential substrate of hepatic DNL. Even in the absence of fructose, copious acetate is produced by the microbial fermentation of dietary fibers. Dietary fibers, carbohydrate polymers with three or more carbohydrate units (primarily glucose or fructose), are naturally found in fruits, vegetables, and grains163,164. Some dietary fibers are resistant to digestive enzymes and therefore neither hydrolyzed nor absorbed until they reach the colon. Depending on their physicochemical characteristics, dietary fibers are subdivided into insoluble and soluble dietary fibers165. Insoluble fibers, such as cellulose and lignin, are barely fermented by the gut microbiota and thus have a fecal bulking effect166. In contrast, soluble fibers, such as inulin, β-glucan, pectins, and maltodextrins, are readily fermented to produce SCFAs167.
The concentration of SCFAs produced by the gut microbiota is largely influenced by the composition and type of dietary fibers in food168. In general, a diet containing more soluble fiber induces a higher concentration of acetate in fecal samples169, and this acetate readily feeds hepatic lipogenesis170,171. The roles of fiber-derived propionate and butyrate are less understood, but their presence in excessive amounts can induce insulin resistance172 or global gene alterations in colonocytes or hepatocytes, as butyrate can inhibit histone deacetylases (HDACs)173,174,175,176,177. Butyrate may thus provide a mode of epigenetic control. In colonocytes, the accumulation of butyrate has been shown to have a therapeutic effect as an HDAC inhibitor and suppresses colon tumorigenesis178,179. However, whether fiber-derived butyrate can reach millimolar concentrations to have any effects in hepatocytes is unclear.
Despite the potentially detrimental effect of dietary fibers through increased hepatic lipogenesis and perturbation of signaling pathways, certain types of dietary fibers reduce the risk of metabolic diseases180,181. In animal studies with a fructose-rich diet, supplementation with inulin significantly reduced blood TG and cholesterol levels182,183. Similar effects were also observed in clinical studies. In a dietary intervention study, increasing inulin intake reduced blood TG levels and fat mass in obese children184. Twelve healthy participants who consumed cereal containing high amounts of inulin for four weeks showed significantly lower plasma TG and cholesterol levels than the control group185.
To determine the causal relationship between fiber fermentation and improved metabolism, several studies have focused on the impact of an SCFA-enriched diet. Supplementation with propionate and butyrate improved glucose homeostasis in both clinical and animal studies149,186. On the other hand, in type 2 diabetic rats, acetate supplementation reduced hepatic TG accumulation and improved glucose tolerance by decreasing SREBP-1187. SCFA supplementation has also been implicated in the prevention of CVD. Acetate significantly reduced blood pressures, cardiac fibrosis, and left ventricular hypertrophy in a mouse model of CVD induced with deoxycorticosterone, and a high-fiber diet generated consistent improvement188. This protective action of dietary fibers and acetate likely involves downregulation of CVD-associated genes, such as early growth response protein 1 (Egr1).
However, other recent studies have indicated that dietary fibers can be detrimental. In a randomized clinical study, Chambers et al. compared the effect of inulin or inulin-propionate ester consumption in NAFLD patients. In contrast to the authors’ prediction, after 42 days of 20 g/day inulin supplementation, the subjects showed significantly increased intrahepatocellular lipid accumulation. The provision of inulin-propionate ester did not have this effect. Given that NAFLD patients exhibit high levels of hepatic DNL, the authors speculated that acetate derived from the high-inulin diet provided excessive carbon for increased DNL, while propionate supplementation suppressed it by competing with acetate for CoA189. A high-inulin diet was also shown to worsen liver cancer. Feeding TLR5-deficient mice a 7.5% inulin diet caused cholestasis, hepatic inflammation, and hepatocellular carcinoma190. Germ-free or antibiotic-treated mice did not show these phenotypes, indicating that microbiota dysbiosis was involved. This intriguing idea of context-dependent fiber effects on lipogenesis and liver pathology merits further investigation.
To conclude, despite the widespread belief in the beneficial effects of dietary fibers, the composition of dietary fibers and health status of the individual (e.g., insulin resistance, NAFLD, microbiota dysbiosis) are likely critical factors that determine the impact of dietary fibers on metabolic health. This notion supports the increasingly popular notion of personalized medicine and diet. Future studies using isotope tracing and metabolomics to define the metabolic fates of various dietary fibers and their byproducts in diverse pathophysiological contexts will expand our understanding of the link between fibers, their fermentation products, and NAFLD onset.
Concluding remarks
A recent alarming increase in NAFLD patients and patients with associated detrimental metabolic diseases has sparked extensive research in the field. Numerous dietary and genetic animal models have been prepared to recapitulate the human pathology, and the use of these models has identified several new metabolic pathways involved in the disease process. Among them, increased hepatic lipogenesis driven by a high-carbohydrate diet and microbial metabolism/dysbiosis have been repeatedly reported in NAFLD. Accordingly, targeting hepatic lipogenesis via inhibition of a specific lipogenic enzyme or limiting the supply of lipogenic carbon substrates has been performed in clinical trials and achieved some promising results. In addition to the further development of therapeutic strategies, a more detailed understanding of the complex mechanisms of NAFLD pathogenesis in various genetic and dietary backgrounds (e.g., >100 outbred mouse strains or large-scale human dietary intervention studies) will explain why some people are more susceptible to NAFLD than others. This will also expedite the development of personalized dietary guidelines and pharmaceutical interventions. Finally, identifying accurate and early diagnostic markers of NAFLD in the blood and/or fecal samples is another crucial future research avenue.
References
Kawano, Y. & Cohen, D. E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 48, 434–441 (2013).
Young, S. et al. Prevalence and profile of nonalcoholic fatty liver disease in lean adults: systematic review and meta-analysis. Hepatol. Commun. 4, 953–972 (2020).
Zhou, J. et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology 71, 1851–1864 (2020).
Ogden, C. L., Flegal, K. M., Carroll, M. D. & Johnson, C. L. Prevalence and trends in overweight among US children and adolescents, 1999–2000. JAMA 288, 1728–1732 (2002).
Tso, P., Pitts, V. & Granger, D. N. Role of lymph flow in intestinal chylomicron transport. Am. J. Physiol. 249, G21–G28. (1985).
Postic, C. & Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J. Clin. Invest. 118, 829–838 (2008).
Softic, S., Cohen, D. E. & Kahn, C. R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 61, 1282–1293 (2016).
Kaplan, R. S., Mayor, J. A., Johnston, N. & Oliveira, D. L. Purification and characterization of the reconstitutively active tricarboxylate transporter from rat liver mitochondria. J. Biol. Chem. 265, 13379–13385 (1990).
Steensels, S. et al. Acyl-coenzyme A thioesterase 9 traffics mitochondrial short-chain fatty acids toward de novo lipogenesis and glucose production in the liver. Hepatology 72, 857–872 (2020).
Guynn, R. W., Veloso, D. & Veech, R. L. The concentration of malonyl-coenzyme A and the control of fatty acid synthesis in vivo. J. Biol. Chem. 247, 7325–7331 (1972).
Shimomura, I. et al. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl Acad. Sci. USA 96, 13656–13661 (1972).
Horton, J. D., Bashmakov, Y., Shimomura, I. & Shimano, H. Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc. Natl Acad. Sci. USA 95, 5987–5992 (1998).
Towle, H. C., Kaytor, E. N. & Shih, H. M. Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu. Rev. Nutr. 17, 405–433 (1997).
Foretz, M. et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol. Cell. Biol. 19, 3760–3768 (1999).
Liang, G. et al. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 277, 9520–9528 (2002).
Shimomura, I., Shimano, H., Horton, J. D., Goldstein, J. L. & Brown, M. S. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J. Clin. Invest. 99, 838–845 (1997).
Sheng, Z., Otani, H., Brown, M. S. & Goldstein, J. L. Independent regulation of sterol regulatory element-binding proteins 1 and 2 in hamster liver. Proc. Natl Acad. Sci. USA 92, 935–938 (1995).
Smith, G. I. et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Invest. 130, 1453–1460 (2020).
Foufelle, F. et al. Glucose stimulation of lipogenic enzyme gene expression in cultured white adipose tissue. A role for glucose 6-phosphate. J. Biol. Chem. 267, 20543–20546 (1992).
Prip-Buus, C. et al. Induction of fatty-acid-synthase gene expression by glucose in primary culture of rat hepatocytes. Depend. glucokinase Act. Eur. J. Biochem. 230, 309–315 (1995).
Kim, J. B. et al. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Invest. 101, 1–9 (1998).
Dentin, R. et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J. Biol. Chem. 279, 20314–20326 (2004).
Kim, M. et al. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight 2, e96703 (2017).
Kabashima, T., Kawaguchi, T., Wadzinski, B. E. & Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl Acad. Sci. USA 100, 5107–5112 (2003).
Herman, M. A. & Kahn, B. B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J. Clin. Invest. 116, 1767–1775 (2006).
Iizuka, K., Bruick, R. K., Liang, G., Horton, J. D. & Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl Acad. Sci. USA 101, 7281–7286 (2004).
Postic, C., Dentin, R., Denechaud, P. D. & Girard, J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu. Rev. Nutr. 27, 179–192 (2007).
Ferré, P. & Foufelle, F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 12, 83–92 (2010).
Linden, A. G. et al. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 59, 475–487 (2018).
Jang, C. et al. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 27, 351–361.e3 (2018).
Asipu, A., Hayward, B. E., O’Reilly, J. & Bonthron, D. T. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes 52, 2426–2432 (2003).
Sun, S. Z. & Empie, M. W. Fructose metabolism in humans - what isotopic tracer studies tell us. Nutr. Metab. 9, 89 (2012).
Iynedjian, P. B. Mammalian glucokinase and its gene. Biochem. J. 293, 1–13 (1993).
Jensen, T. et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 68, 1063–1075 (2018).
Jang, C. et al. The small intestine shields the liver from fructose-induced steatosis. Nat. Metab. 2, 586–593 (2020).
Raman, M. et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 11, 868–75.e753 (2013).
Zhao, S. et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579, 586–591 (2020).
Koo, H. Y. et al. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim. Biophys. Acta 1782, 341–348 (2008).
Koo, H. Y., Miyashita, M., Cho, B. H. & Nakamura, M. T. Replacing dietary glucose with fructose increases ChREBP activity and SREBP-1 protein in rat liver nucleus. Biochem. Biophys. Res. Commun. 390, 285–289 (2009).
Ishimoto, T. et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl Acad. Sci. USA 109, 4320–4325 (2012).
Lanaspa, M. A. et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Invest. 128, 2226–2238 (2018).
Andres-Hernando, A. et al. Deletion of fructokinase in the liver or in the intestine reveals differential effects on sugar-induced metabolic dysfunction. Cell Metab. 32, 117–127.e3 (2020).
Bantle, J. P., Raatz, S. K., Thomas, W. & Georgopoulos, A. Effects of dietary fructose on plasma lipids in healthy subjects. Am. J. Clin. Nutr. 72, 1128–1134 (2000).
Parks, E. J., Skokan, L. E., Timlin, M. T. & Dingfelder, C. S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 138, 1039–1046 (2008).
Softic, S. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Invest. 127, 4059–4074 (2017).
Futatsugi, K. et al. Discovery of PF-06835919: a potent inhibitor of ketohexokinase (KHK) for the treatment of metabolic disorders driven by the overconsumption of fructose. J. Med. Chem. 63, 13546–13560 (2020).
Lambert, J. E., Ramos-Roman, M. A., Browning, J. D. & Parks, E. J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146, 726–735 (2014).
Parks, E. J., Krauss, R. M., Christiansen, M. P., Neese, R. A. & Hellerstein, M. K. Effects of a low-fat, high-carbohydrate diet on VLDL-triglyceride assembly, production, and clearance. J. Clin. Invest. 104, 1087–1096 (1999).
Fabbrini, E. et al. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 134, 424–431 (2008).
Nakamuta, M. et al. Evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 16, 631–635 (2005).
Kohjima, M. et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 20, 351–358 (2007).
Donnelly, K. L. et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 (2005).
Timlin, M. T. & Parks, E. J. Temporal pattern of de novo lipogenesis in the postprandial state in healthy men. Am. J. Clin. Nutr. 81, 35–42 (2005).
Schwarz, J. M., Neese, R. A., Turner, S., Dare, D. & Hellerstein, M. K. Short-term alterations in carbohydrate energy intake in humans. Striking effects on hepatic glucose production, de novo lipogenesis, lipolysis, and whole-body fuel selection. J. Clin. Invest. 96, 2735–2743 (1995).
Marques-Lopes, I., Ansorena, D., Astiasaran, I., Forga, L. & Martínez, J. A. Postprandial de novo lipogenesis and metabolic changes induced by a high-carbohydrate, low-fat meal in lean and overweight men. J. Clin. Invest. 73, 253–261 (2001).
Adams, L. A. et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129, 113–121 (2005).
Ekstedt, M. et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 44, 865–873 (2006).
Dam-Larsen, S. et al. Final results of a long-term, clinical follow-up in fatty liver patients. Scand. J. Gasteroenterol. 44, 1236–1243 (2009).
Younossi, Z. M., Gramlich, T., Matteoni, C. A., Boparai, N. & McCullough, A. J. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin. Gastroenterol. Hepatol. 2, 262–265 (2004).
Gaggini, M. et al. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 5, 1544–1560 (2013).
Lomonaco, R. et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 55, 1389–1397 (2012).
Shibata, M., Kihara, Y., Taguchi, M., Tashiro, M. & Otsuki, M. Nonalcoholic fatty liver disease is a risk factor for type 2 diabetes in middle-aged Japanese men. Diabetes Care 30, 2940–2944 (2007).
Chon, C. W. et al. Effect of nonalcoholic Fatty liver disease on the development of type 2 diabetes in nonobese, nondiabetic Korean men. Gut Liver. 6, 368–373 (2012).
Marchesini, G. et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 107, 450–455 (1999).
Eguchi, Y. et al. Visceral fat accumulation and insulin resistance are important factors in nonalcoholic fatty liver disease. J. Gastroenterol. 41, 462–469 (2006).
Perry, R. J. et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 18, 740–748 (2013).
Memon, R. A., Grunfeld, C., Moser, A. H. & Feingold, K. R. Fatty acid synthesis in obese insulin resistant diabetic mice. Horm. Metab. Res. 26, 85–87 (1994).
Suzuki, T., Muramatsu, T., Morioka, K., Goda, T. & Mochizuki, K. ChREBP binding and histone modifications modulate hepatic expression of the Fasn gene in a metabolic syndrome rat model. Nutrition 31, 877–883 (2015).
Monetti, M. et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 6, 69–78 (2007).
Tilg, H. & Moschen, A. R. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52, 1836–1846 (2010).
Listenberger, L. L. et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl Acad. Sci. USA 100, 3077–3082 (2003).
Yamaguchi, K. et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 45, 1366–1374 (2007).
Hui, E., Xu, A., Bo Yang, H. & Lam, K. S. Obesity as the common soil of non-alcoholic fatty liver disease and diabetes: role of adipokines. J. Diabetes Investig. 4, 413–425 (2013).
Alkhouri, N., Dixon, L. J. & Feldstein, A. E. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 3, 445–451 (2009).
Arner, P. Insulin resistance in type 2 diabetes: role of fatty acids. Diabetes Metab. Res. Rev. 18, S5–S9 (2002).
Albu, J. B. et al. Systemic resistance to the antilipolytic effect of insulin in black and white women with visceral obesity. Am. J. Physiol. 277, E551–E560 (1999).
Shoelson, S. E., Herrero, L. & Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 132, 2169–2180 (2007).
Feng, X., Zhang, L., Xu, S. & Shen, A. Z. ATP-citrate lyase (ACLY) in lipid metabolism and atherosclerosis: an updated review. Prog. Lipid Res. 77, 101006 (2020).
Zaidi, N., Swinnen, J. V. & Smans, K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. 72, 3709–3714 (2012).
Zhao, S. et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep. 17, 1037–1052 (2016).
Zaidi, N., Royaux, I., Swinnen, J. V. & Smans, K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol. Cancer Ther. 11, 1925–1935 (2012).
Bauer, D. E., Hatzivassiliou, G., Zhao, F., Andreadis, C. & Thompson, C. B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322 (2005).
Hatzivassiliou, G. et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 8, 311–321 (2005).
Migita, T. et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 68, 8547–8554 (2008).
Carrer, A. et al. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 9, 416–435 (2019).
Baardman, J. et al. Macrophage ATP citrate lyase deficiency stabilizes atherosclerotic plaques. Nat. Commun. 11, 6296 (2020).
Fukuda, H., Katsurada, A. & Iritani, N. Effects of nutrients and hormones on gene expression of ATP citrate-lyase in rat liver. Eur. J. Biochem. 209, 217–222 (1992).
Kim, Y. M. et al. Sterol regulatory element-binding protein (SREBP)-1-mediated lipogenesis is involved in cell senescence. J. Biol. Chem. 285, 29069–29077 (2010).
Kim, M. S. et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J. Clin. Invest. 126, 4372–4386 (2016).
Wellen, K. E. et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009).
Sivanand, S. et al. Nuclear Acetyl-CoA production by ACLY promotes homologous recombination. Mol. Cell. 67, 252–265.e6 (2017).
Berwick, D. C., Hers, I., Heesom, K. J., Moule, S. K. & Tavare, J. M. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 277, 33895–33900 (2002).
Potapova, I. A., El-Maghrabi, M. R., Doronin, S. V. & Benjamin, W. B. Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the enzyme activity. Allosteric activation of ATP:citrate lyase by phosphorylated sugars. Biochemistry 39, 1169–1179 (2000).
White, P. J. et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 27, 1281–1293.e7 (2018).
Lin, R. et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell. 51, 506–518 (2013).
Guo, L. et al. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J. Biol. Chem. 294, 11805–11816 (2019).
Li, K. et al. Hrd1-mediated ACLY ubiquitination alleviate NAFLD in db/db mice. Metabolism 114, 154349 (2021).
Zhang, C. et al. Cullin3-KLHL25 ubiquitin ligase targets ACLY for degradation to inhibit lipid synthesis and tumor progression. Genes Dev. 30, 1956–1970 (2016).
Joshi-Barve, S., Barve, S. S., Butt, W., Klein, J. & McClain, C. J. Inhibition of proteasome function leads to NF-kappaB-independent IL-8 expression in human hepatocytes. Hepatology 38, 1178–1187 (2003).
Pinkosky, S. L. et al. AMP-activated protein kinase and ATP-citrate lyase are two distinct molecular targets for ETC-1002, a novel small molecule regulator of lipid and carbohydrate metabolism. J. Lipid Res. 54, 134–151 (2013).
Pinkosky, S. L. et al. Liver-specific ATP-citrate lyase inhibition by bempedoic acid decreases LDL-C and attenuates atherosclerosis. Nat. Commun. 7, 13457 (2016).
Korzh, O. Defining the role of bempedoic acid in lowering low-density lipoprotein cholesterol. J. Appl. Pharm. Sci. 10, 130–139 (2020).
Banach, M. et al. Efficacy of bempedoic acid: a pooled analysis of 4 pivotal phase 3 clinical trials. Circulation 140, 12268 (2019).
Beigneux, A. P. et al. ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 279, 9557–9564 (2004).
Tumanov, S., Bulusu, V., Gottlieb, E. & Kamphorst, J. J. A rapid method for quantifying free and bound acetate based on alkylation and GC-MS analysis. Cancer Metab. 4, 17 (2016).
Fernandez, S. et al. Adipocyte ACLY facilitates dietary carbohydrate handling to maintain metabolic homeostasis in females. Cell Rep. 27, 2772–2784.e6 (2019).
Xu, H. et al. Acyl-CoA synthetase short-chain family member 2 (ACSS2) is regulated by SREBP-1 and plays a role in fatty acid synthesis in caprine mammary epithelial cells. J. Cell. Physiol. 233, 1005–1016 (2018).
Bulusu, V. et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 18, 647–658 (2017).
Yamashita, H., Kaneyuki, T. & Tagawa, K. Production of acetate in the liver and its utilization in peripheral tissues. Biochim. Biophys. Acta 1532, 79–87 (2001).
Fujino, T., Kondo, J., Ishikawa, M., Morikawa, K. & Yamamoto, T. T. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J. Biol. Chem. 276, 11420–11426 (2001).
Yamamoto, J. et al. A Kruppel-like factor KLF15 contributes fasting-induced transcriptional activation of mitochondrial acetyl-CoA synthetase gene AceCS2. J. Biol. Chem. 279, 16954–16962 (2004).
Sakakibara, I. et al. Fasting-induced hypothermia and reduced energy production in mice lacking acetyl-CoA synthetase 2. Cell Metab. 9, 191–202 (2009).
Schug, Z. T. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 27, 57–71 (2015).
Cai, L., Sutter, B. M., Li, B. & Tu, B. P. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell. 42, 426–437 (2011).
Takahashi, H., McCaffery, J. M., Irizarry, R. A. & Boeke, J. D. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell. 23, 207–217 (2006).
Mizzen, C. A. & Allis, C. D. Linking histone acetylation to transcriptional regulation. Cell Mol. Life Sci. 54, 6–20 (1998).
Mews, P. et al. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386 (2017).
Li, X. et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol. Cell. 66, 684–697.e9 (2017).
Huang, Z. et al. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl Acad. Sci. USA115, E9499–E9506 (2018).
Ikeda, Y. et al. Transcriptional regulation of the murine acetyl-CoA synthetase 1 gene through multiple clustered binding sites for sterol regulatory element-binding proteins and a single neighboring site for Sp1. J. Biol. Chem. 276, 34259–34269 (2001).
Moffett, J. R., Puthillathu, N., Vengilote, R., Jaworski, D. M. & Namboodiri, A. M. Acetate revisited: a key biomolecule at the nexus of metabolism, epigenetics and oncogenesis-part 1: Acetyl-CoA, acetogenesis and Acyl-CoA short-chain synthetases. Front. Physiol. 11, 580167 (2020).
Gao, X. et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 7, 11960 (2016).
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Beisner, J., Gonzalez-Granda, A., Basrai, M., Damms-Machado, A. & Bischoff, S. C. Fructose-induced intestinal microbiota shift following two types of short-term high-fructose dietary phases. Nutrients 12, 3444 (2020).
Rumessen, J. J. & Gudmand-Høyer, E. Absorption capacity of fructose in healthy adults. Comparison with sucrose and its constituent monosaccharides. Gut 27, 1161–1168 (1986).
Aleman, J. O. Excess Dietary Fructose Does Not Alter Gut Microbiota or Permeability in Humans: A Randomized Controlled Pilot Study. Preprint at https://www.medrxiv.org/content/10.1101/2020.11.23.20235515v1 (2020).
Wang, R. et al. Genetically obese human gut microbiota induces liver steatosis in germ-free mice fed on normal diet. Front. Microbiol. 9, 1602 (2018).
Le Roy, T. et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794 (2013).
Bäckhed, F. et al. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl Acad. Sci. USA 101, 15718–15723 (2004).
Kaden-Volynets, V. et al. Lack of liver steatosis in germ-free mice following hypercaloric diets. Eur. J. Nutr. 58, 1933–1945 (2019).
Kindt, A. et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat. Commun. 9, 3760 (2018).
Bergheim, I. et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J. Hepatol. 48, 983–992 (2008).
De Minicis, S. et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 59, 1738–1749 (2014).
Chen, Y. H. et al. Gnotobiotic mice inoculated with Firmicutes, but not Bacteroidetes, deteriorate nonalcoholic fatty liver disease severity by modulating hepatic lipid metabolism. Nutr. Res. 69, 20–29 (2019).
Brütting, C., Lara Bisch, M., Brandsch, C., Hirche, F. & Stangl, G. I. Impact of dietary propionate on fructose-induced changes in lipid metabolism, gut microbiota and short-chain fatty acids in mice. Int. J. Food Sci. Nutr. 1–13; advance online publication 4 Jun 2020 https://doi.org/10.1080/09637486.2020.1773415.
Dumas, M. E. et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl Acad. Sci. USA 103, 12511–12516 (2006).
Ye, J. Z. et al. Dynamic alterations in the gut microbiota and metabolome during the development of methionine-choline-deficient diet-induced nonalcoholic steatohepatitis. World J. Gastroenterol. 24, 2468–2481 (2018).
Schwiertz, A. et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18, 190–195 (2010).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Murphy, E. F. et al. Composition and energy harvesting capacity of the gut microbiota: relationship to diet, obesity and time in mouse models. Gut 59, 1635–1642 (2010).
Canfora, E. E., Jocken, J. W. & Blaak, E. E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 11, 577–591 (2015).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Laurent, C. et al. Effect of acetate and propionate on fasting hepatic glucose production in humans. Eur. J. Clin. Nutr. 49, 484–491 (1995).
Wolever, T. M., Brighenti, F., Royall, D., Jenkins, A. L. & Jenkins, D. J. Effect of rectal infusion of short chain fatty acids in human subjects. Am. J. Gastroenterol. 84, 1027–1033 (1989).
den Besten, G. et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G900–G910 (2013).
Samuel, B. S. et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl Acad. Sci. USA 105, 16767–16772 (2008).
Hong, Y. H. et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 146, 5092–5099 (2005).
Wang, J. et al. Hepatic regulator of G protein signaling 5 ameliorates nonalcoholic fatty liver disease by suppressing transforming growth factor beta-activated kinase 1-c-Jun-N-terminal kinase/p38 signaling. Hepatology. Advance online publication, 19 Mar 2020 https://doi.org/10.1002/hep.31242).
Lin, H. V. et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 7, e35240 (2012).
Gao, Z. et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517 (2009).
Frost, G. et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 5, 3611 (2014).
Velázquez, O. C., Jabbar, A., DeMatteo, R. P. & Rombeau, J. L. Butyrate inhibits seeding and growth of colorectal metastases to the liver in mice. Surgery 120, 440–448 (1996).
Wächtershäuser, A., Akoglu, B. & Stein, J. HMG-CoA reductase inhibitor mevastatin enhances the growth inhibitory effect of butyrate in the colorectal carcinoma cell line Caco-2. Carcinogenesis 22, 1061–1067 (2001).
Demigné, C. et al. Effect of propionate on fatty acid and cholesterol synthesis and on acetate metabolism in isolated rat hepatocytes. Effect of propionate on fatty acid and cholesterol synthesis and on acetate metabolism in isolated rat hepatocytes. Br. J. Nutr. 74, 209–219 (1995).
Weitkunat, K. et al. Odd-chain fatty acids as a biomarker for dietary fiber intake: a novel pathway for endogenous production from propionate. Am. J. Clin. Nutr. 105, 1544–1551 (2017).
Weitkunat, K. et al. Importance of propionate for the repression of hepatic lipogenesis and improvement of insulin sensitivity in high-fat diet-induced obesity. Mol. Nutr. Food Res. 60, 2611–2621 (2016).
Cho, Y. E., et al. Fructose Promotes Leaky Gut, Endotoxemia, and Liver Fibrosis Through Ethanol-Inducible Cytochrome P450-2E1-Mediated Oxidative and Nitrative Stress. Hepatology. Advance online publication, 8 Apr. 2019 https://doi.org/10.1002/hep.30652).
Zu, L. et al. Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. J. Biol. Chem. 284, 5915–5926 (2009).
Gan, L., Xiang, W., Xie, B. & Yu, L. Molecular mechanisms of fatty liver in obesity. Front. Med. 9, 275–287 (2015).
Belfort, R. et al. Dose-response effect of elevated plasma free fatty acid on insulin signaling. Diabetes 54, 1640–1648 (2005).
Sellmann, C. et al. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem. 26, 1183–1192 (2015).
Todoric, J. et al. Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat. Metab. 2, 1034–1045 (2020).
Makki, K., Deehan, E. C., Walter, J. & Bäckhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 23, 705–715 (2018).
Holscher, H. D. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 8, 172–184 (2017).
Deehan, E. C., et al. Modulation of the gastrointestinal microbiome with nondigestible fermentable carbohydrates to improve human health. in Bugs as Drugs: Therapeutic Microbes for the Prevention and Treatment of Disease (eds. Britton, R. A. & Cani, P. D.) 453–483 (ASM Press, 2018).
McRorie, J. W. Jr. Psyllium is not fermented in the human gut. Neurogastroenterol. Motil. 27, 1681–1682 (2015).
Sims, I. M. & Monro, J. A. Fiber: composition, structures, and functional properties. Adv. Food Nutr. Res. 68, 81–99 (2013).
Bliss, D. Z., Weimer, P. J., Jung, H. J. & Savik, K. In vitro degradation and fermentation of three dietary fiber sources by human colonic bacteria. J. Agric. Food Chem. 61, 4614–4621 (2013).
Baxter, N. T. et al. Dynamics of human gut microbiota and short-chain fatty acids in response to dietary interventions with three fermentable fibers. mBio 10, e02566–18 (2019).
De Vadder, F. et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156, 84–96 (2014).
Koh, A., De Vadder, F., Kovatcheva-Datchary, P. & Bäckhed, F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345 (2016).
Tirosh, A. et al. The short-chain fatty acid propionate increases glucagon and FABP4 production, impairing insulin action in mice and humans. Sci. Transl. Med. 11, eaav0120 (2019).
Carrer, A. et al. Impact of a high-fat diet on tissue Acyl-CoA and histone acetylation levels. J. Biol. Chem. 292, 3312–3322 (2017).
Vinolo, M. A. et al. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J. Nutr. Biochem. 22, 849–855 (2011).
Schilderink, R. et al. The SCFA butyrate stimulates the epithelial production of retinoic acid via inhibition of epithelial HDAC. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G1138–G1146 (2016).
Silva, L. G., Ferguson, B. S., Avila, A. S. & Faciola, A. P. Sodium propionate and sodium butyrate effects on histone deacetylase (HDAC) activity, histone acetylation, and inflammatory gene expression in bovine mammary epithelial cells. J. Anim. Sci. 96, 5244–5252 (2018).
Krautkramer, K. A. et al. Diet-microbiota interactions mediate global epigenetic programming in multiple host tissues. Mol. Cell. 64, 982–992 (2016).
Donohoe, D. R. et al. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell. 48, 612–626 (2012).
Donohoe, D. R. et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 4, 1387–1397 (2014).
Beylot, M. Effects of inulin-type fructans on lipid metabolism in man and in animal models. Br. J. Nutr. 93, S163–S168 (2005).
Menni, C. et al. Gut microbiome diversity and high-fibre intake are related to lower long-term weight gain. Int. J. Obes. 41, 1099–1105 (2017).
Delzenne, N. M. & Kok, N. Effects of fructans-type prebiotics on lipid metabolism. Am. J. Clin. Nutr. 73, 456s–458s (2001).
Delzenne, N. M., Daubioul, C., Neyrinck, A., Lasa, M. & Taper, H. S. Inulin and oligofructose modulate lipid metabolism in animals: review of biochemical events and future prospects. Br. J. Nutr. 87, S255–S259 (2002).
Nicolucci, A. C. et al. Prebiotics reduce body fat and alter intestinal microbiota in children who are overweight or with obesity. Gastroenterology 153, 711–722 (2017).
Brighenti, F., Casiraghi, M. C., Canzi, E. & Ferrari, A. Effect of consumption of a ready-to-eat breakfast cereal containing inulin on the intestinal milieu and blood lipids in healthy male volunteers. Eur. J. Clin. Nutr. 53, 726–733 (1999).
Venter, C. S., Vorster, H. H. & Cummings, J. H. Effects of dietary propionate on carbohydrate and lipid metabolism in healthy volunteers. Am. J. Gastroenterol. 85, 549–553 (1990).
Yamashita, H. et al. Improvement of obesity and glucose tolerance by acetate in Type 2 diabetic Otsuka Long-Evans Tokushima Fatty (OLETF) rats. Biosci. Biotechnol. Biochem. 71, 1236–1243 (2007).
Marques, F. Z. et al. High-fiber diet and acetate supplementation change the gut microbiota and prevent the development of hypertension and heart failure in hypertensive mice. Circulation 135, 964–977 (2017).
Chambers, E. S. et al. The effects of dietary supplementation with inulin and inulin-propionate ester on hepatic steatosis in adults with non-alcoholic fatty liver disease. Diabetes Obes. Metab. 21, 372–376 (2019).
Singh, V. et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell 175, 679–694 (2018).
Acknowledgements
We thank all the members of the Jang laboratory for their discussion. This work was funded by the American Association for the Study of Liver Diseases (AASLDF 50028) and the Edward Mallinckrodt, Jr. Foundation Award to C.J. We apologize to our colleagues whose work we could not cite due to space restrictions.
Author information
Authors and Affiliations
Contributions
C.J. and K.E.W. conceptualized the scope of the manuscript. G.P., S.J. and C.J. wrote the manuscript. G.P. and S.J. prepared the figure and table. All authors revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, G., Jung, S., Wellen, K.E. et al. The interaction between the gut microbiota and dietary carbohydrates in nonalcoholic fatty liver disease. Exp Mol Med 53, 809–822 (2021). https://doi.org/10.1038/s12276-021-00614-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00614-x
This article is cited by
-
Dietary sodium acetate and sodium butyrate improve high-carbohydrate diet utilization by regulating gut microbiota, liver lipid metabolism, oxidative stress, and inflammation in largemouth bass (Micropterus salmoides)
Journal of Animal Science and Biotechnology (2024)
-
Acetyl-CoA metabolism in cancer
Nature Reviews Cancer (2023)
