
Abstract
Aging is associated with cellular senescence followed by bone loss leading to bone fragility in humans. However, the regulators associated with cellular senescence in aged bones need to be identified. Hypoxia-inducible factor (HIF)−2α regulates bone remodeling via the differentiation of osteoblasts and osteoclasts. Here, we report that HIF-2α expression was highly upregulated in aged bones. HIF-2α depletion in male mice reversed age-induced bone loss, as evidenced by an increase in the number of osteoblasts and a decrease in the number of osteoclasts. In an in vitro model of doxorubicin-mediated senescence, the expression of Hif-2α and p21, a senescence marker gene, was enhanced, and osteoblastic differentiation of primary mouse calvarial preosteoblast cells was inhibited. Inhibition of senescence-induced upregulation of HIF-2α expression during matrix maturation, but not during the proliferation stage of osteoblast differentiation, reversed the age-related decrease in Runx2 and Ocn expression. However, HIF-2α knockdown did not affect p21 expression or senescence progression, indicating that HIF-2α expression upregulation in senescent osteoblasts may be a result of aging rather than a cause of cellular senescence. Osteoclasts are known to induce a senescent phenotype during in vitro osteoclastogenesis. Consistent with increased HIF-2α expression, the expression of p16 and p21 was upregulated during osteoclastogenesis of bone marrow macrophages. ChIP following overexpression or knockdown of HIF-2α using adenovirus revealed that p16 and p21 are direct targets of HIF-2α in osteoclasts. Osteoblast-specific (Hif-2αfl/fl;Col1a1-Cre) or osteoclast-specific (Hif-2αfl/fl;Ctsk-Cre) conditional knockout of HIF-2α in male mice reversed age-related bone loss. Collectively, our results suggest that HIF-2α acts as a senescence-related intrinsic factor in age-related dysfunction of bone homeostasis.
Similar content being viewed by others

Introduction
Bone homeostasis is maintained by tight regulation of the interplay between two specific cell types: bone-forming osteoblasts and bone-resorbing osteoclasts1. Imbalances in bone homeostasis cause severe bone diseases, including osteoporosis, the most prevalent chronic disease in aging adults. Bone homeostasis is affected by various factors, such as heart/vascular disease, estrogen deficiency, diabetes, and aging2. Aging is the main risk factor for age-related osteoporosis, which is characterized predominantly by decreased bone mineral density. The consequent bone structure fragility is a primary cause of osteoporotic fracture in elderly individuals3,4.
Bone-forming osteoblasts have a short lifespan and are constantly being replaced5. Osteoblast differentiation is governed by osteoblast-specific transcription factors, including runt-related transcription factor-2 (RUNX2)6 and osterix7. Differentiation consists of a sequence of events, including lineage commitment (osteoprogenitors), proliferation and matrix synthesis (preosteoblasts), and matrix maturation and mineralization (mature osteoblasts). Mature osteoblasts are responsible for the assembly of matrix proteins, including collagen type I (COL1A1), bone integrin-binding sialoprotein (IBSP), osteopontin (OPN) and osteocalcin (OCN)8, and are characterized by an increase in alkaline phosphatase (ALP) activity9. Age-related bone loss was shown to be caused by a deficit in bone formation10. Osteoblast dysfunction is the main cause of age-related bone loss in humans and results from two distinct types of mechanisms. Extrinsic mechanisms are mediated by changes in the bone microenvironment associated with hormones and growth factors, whereas intrinsic mechanisms are mediated by osteoblast senescence following reductions in the number, lifespan, and differentiation of osteoblasts11.
Age-related bone loss has also been associated with increases in the number of bone-resorbing osteoclasts12. Osteoclasts differentiate from hematopoietic progenitors of the monocyte-macrophage lineage in response to stimulation by macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL)13. Osteoclasts develop into multinucleated giant cells through a sequence of steps that include proliferation, differentiation, and maturation14,15. Importantly, osteoclasts exhibit characteristics of senescence, such as the expression of several different cyclin-dependent kinase (CDK) inhibitors, INK4A (p16) and CDKN1A (p21), during osteoclast differentiation16,17. Osteocytes in the bones of aged mice are postmitotic and show elevated levels of p1612,18, whereas senescent osteoblast progenitors exhibit high levels of p21 but not p1619. Selective elimination of p16-expressing cells in mouse models increases bone mass18, and stromal cell cultures obtained from the bone marrow of old mice are better at promoting osteoclastogenesis than cells from young mice20, supporting the finding that senescent cells contribute to age-related bone loss.
Hypoxia-inducible factor (HIF), a heterodimeric transcription factor consisting of an oxygen-regulated α-subunit and a constitutively expressed β-subunit, plays important roles in bone formation by regulating the differentiation and activation of osteoblasts and osteoclasts21. Under normal oxygen tension, the α-subunit of HIF is hydroxylated by oxygen-sensitive prolyl hydroxylase, which is recognized by the von Hippel-Lindau protein, a component of the E3 ubiquitin ligase complex, and targeted for degradation22. Unlike HIF-1α, which is structurally very similar to HIF-2α but has many different functions23, HIF-2α can accumulate under normoxic conditions in the context of pathologies such as osteoporosis24 and arthritis25,26,27 and can also regulate bone remodeling24. HIF-2α depletion downregulates RANKL expression in fibroblast-like synoviocytes from the rheumatoid arthritis synovium25. In addition, HIF-2α regulates osteoblast and osteoclast differentiation via the expression of essential transcription factors involved in the differentiation of these cells and influences the interplay between these cell types24, suggesting that HIF-2α plays a catabolic role in bone remodeling. In this study, we tried to examine whether age-related upregulation of HIF-2α expression in osteoblasts and osteoclasts contributes to the differentiation of these cells, senescence progression and age-related bone loss.
Materials and methods
Mice
Male C57BL/6 J mice aged 1.5, 4, 12, and 20 months were purchased from Damool Science Animal Laboratory (Daejeon, Korea). Heterozygous Hif-2α KO (Hif-2α+/−) mice (#003266) and Hif-2αfl/fl mice (#008407) were obtained from Jackson Laboratory (Sacramento, CA, USA). Ctsk-Cre mice were obtained from the Rodent Model Resource Center (RMRC13132, Taipei, Taiwan), and Col1a1-Cre mice were kindly provided by Dr. Je-Yong Choi (Kyungpook National University, Daegu, Korea). To generate osteoblast- and osteoclast-specific Hif-2α-KO mice, Hif-2αfl/fl mice were backcrossed with Col1a1-Cre and Ctsk-Cre mice, respectively. Specific deletion of Hif-2α in targeted tissues in those mice was demonstrated in a previous study24. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Chonnam National University (Gwangju, Korea).
Micro (μ)CT analysis and bone histomorphometry
Mouse femurs were fixed in 10% neutral buffered formalin and examined using μCT as previously described24. Trabecular morphometry was analyzed by measuring bone mineral density (BMD), bone volume/tissue volume (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), trabecular number (Tb.N), and cortical thickness (Ct.th). Three-dimensional surface rendering images were generated using Mimics 14.0 imaging software (Materialise, Plymouth, MI, USA). Histomorphometric parameters were analyzed by OsteoMeasure software (Osteometrics, Inc., Decatur, GA, USA). Paraffin sections were stained with hematoxylin and eosin (H&E) and BV/TV, the number of osteoblasts/bone perimeter (N.Ob/B.Pm), and the osteoblast surface/bone surface (Ob.S/BS) were analyzed. To investigate osteoclast parameters, the sections were stained with tartrate-resistant acid phosphate (TRAP) and the number of osteoclasts/bone perimeter (N.Oc/B.Pm) and the osteoclast surface/bone surface (Oc.S/BS) were measured.
Osteoblast differentiation of primary cultured preosteoblasts
For primary culture of calvarial preosteoblasts, calvarial bones from 3-day-old pups were enzymatically digested twice with DMEM comprising 0.1% type II collagenase (Sigma–Aldrich, St. Louis, MO, USA) as described previously24. To induce cellular senescence in the proliferation stage, osteoblasts stimulated with doxorubicin on differentiation day 2 were treated with a potent HIF-2α inhibitor, TC-S 7009 (R&D Systems, Minneapolis, MN, USA) on differentiation day 4, and cells were harvested after 3 days. Cellular senescence in the matrix stage was induced by doxorubicin on differentiation day 9, and TC-S 7009 was administered on differentiation day 11 for 3 days. Then, the cells were harvested on day 14 of differentiation. A schematic diagram showing the time point at which each chemical compound was administered is provided in Supplementary Fig. 5. To overexpress Hif-2α in vitro, primary calvarial preosteoblasts were infected with adenovirus expressing Hif-2α at the indicated multiplicity of infection (MOI) on differentiation day 2. Doxorubicin is a commonly used chemotherapy drug that can induce DNA cross-linking and lead to cell apoptosis, autophagy, and/or senescence28. The concentration of doxorubicin used in this experiment did not cause cell apoptosis.
Osteoclast differentiation of bone marrow macrophages (BMMs)
Mouse bone marrow cells were isolated and harvested as previously described24. Adherent BMMs (4 × 104 cells per well in 48-well plates) were maintained in complete α-MEM containing 30 ng/ml M-CSF (PeproTech) for 24 h. Then, the medium was replaced with medium comprising 100 ng/ml RANKL (PeproTech), and the cells culture for 5 days. To ectopically overexpress HIF-2α, adenovirus encoding Hif-2α was applied at the indicated MOI on day 1. TRAP (Sigma–Aldrich) staining was performed on day 5 according to the manufacturer’s instructions.
ALP and ARS staining
For ALP staining, cells were fixed with 4% paraformaldehyde for 2 min, washed with deionized water and stained with 5-bromo-4-chloro-3-indolyl phosphate (BCIP®)/nitro blue tetrazolium (NBT) liquid substrate solution (Sigma–Aldrich) for 15 min in a dark room. For mineralization assays, the cells were fixed with 10% formalin for 15 min and stained with 2% Alizarin red S (ARS, Sigma–Aldrich) solution for 45 min at room temperature.
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative real-time (qRT)-PCR
Mouse bone tissues were immediately frozen in liquid nitrogen and stored at −70 °C until use. Total RNA was isolated from osteoblasts, osteoclasts, and femoral bone tissues using TRI reagent (Molecular Research, Cincinnati, OH, USA) and reverse-transcribed with TOPscript RT DryMIX (Enzynomics, Daejeon, Korea). cDNA was subjected to PCR using AmpOneTM Tap DNA Polymerase Mix (GeneAll, Seoul, Korea) and appropriate primers. qPCR was performed using a StepOnePlus Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA, USA) and SYBR Premix Ex Taq (TaKaRa Bio, Kyoto, Japan). The individual transcript levels of each individual target were normalized to those of Gapdh and are expressed as the fold change relative to the levels in the indicated controls. The primer sequences and sizes of the amplicons are listed in Supplementary Table 1.
Western blotting
Cells were lysed in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM NaF, 1% NP-40, 0.2% SDS, and 0.5% deoxycholate) supplemented with protease and phosphatase inhibitor cocktails (Roche, Basel, Switzerland). Proteins were separated by SDS-PAGE, and the blots were incubated with rabbit anti-HIF-2α (NB100-122; Novus Biologicals, CO, USA) at 4 °C overnight. After washing, the membranes were incubated with HRP-conjugated anti-rabbit IgG, and images were obtained using ImageSaver6 software via the EZ-capture MG system (ATTO, Tokyo, Japan). The protein bands were quantified using a CS analyzer 4 (ATTO), and protein levels were normalized relative to the level of β-actin (Sigma–Aldrich) in the same sample.
Immunohistochemistry and immunofluorescence microscopy
Sagittal Section (5-μm thick) were placed on slides for immunohistochemical staining. The sections were incubated in 3% H2O2 for 10 min to block endogenous peroxidase activity and incubated in 0.1% trypsin for 30 min at 37 °C for antigen retrieval. After blocking with 1% BSA for 30 min, the slides were reacted with rabbit anti-HIF-2α (Novus Biologicals), mouse anti-p16 (MA5-17142, Invitrogen, Carlsbad, CA, USA), and mouse anti-p21 (AHZ0422, Invitrogen) antibodies, stained using EnVision+System-HRP (Dako, Denmark) and AEC+ substrate and counterstaining with hematoxylin (Dako). The primary antibodies used for double immunofluorescence staining of mouse tissues included mouse anti-HIF-2α (sc13596; Santa Cruz Biotech., Dallas, TX, USA), rabbit anti-OCN (AB10911; Millipore, Burlington, MA, USA), and rabbit anti-cathepsin K (ab19027; Abcam, Cambridge, MA, USA) antibodies. Proteins were visualized using Alexa 488- or Alexa 594-conjugated secondary antibodies (Thermo Fisher Scientific). Nuclei were visualized with DAPI.
Enzyme-linked immunosorbent assay (ELISA)
Serum OCN and CTX-1 levels were measured using a mouse Gla-OCN High Sensitive EIA Kit (MK127; TaKaRa Bio) and CTX-1 EIA Kit (NBP2-69074; Novus Biologicals). All procedures were performed according to the manufacturers’ instructions.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using a Magna ChIP Kit (Millipore) as previously described24. Briefly, cells were fixed with 1% formaldehyde to crosslink the DNA and protein complexes. After the lysates were sonicated, soluble chromatin complexes were incubated with rabbit anti-HIF-2α antibody or anti-IgG at 4 °C overnight. The precipitated DNA segment of the p16 or p21 promoter was amplified by RT-PCR. The sequences of the primers used for the ChIP assay are listed in Supplementary Table 1.
Statistical analysis
All experiments were repeated at least three times. The results are reported as the mean ± SD. μCT and histomorphometric parameters are presented in bar graphs with scatter plots. All statistical analyses were performed using GraphPad Prism version 7 (GraphPad, Inc., San Diego, CA). All quantitative data were first tested for normal distribution using the Shapiro-Wilk test. Then, the means of quantitative variables were compared between two groups by two-tailed Student’s t-test or between three or more independent groups by analysis of variance (ANOVA) followed by Bonferroni’s post-hoc test (multicomparison), as appropriate. n is the number of independent experiments or mice. The results were considered statistically significant when the p-value was less than 0.05.
Results
HIF-2α expression is upregulated in skeletal bones with aging
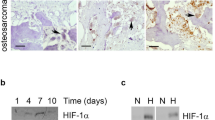
Hif-2α expression was markedly higher in the aged bones of 20-month-old male mice than in the bones of younger mice and exhibited the same pattern of expression as the aging-related markers p16 and p21 (Fig. 1a). Because bone tissue is a complex system that consists of diverse cell types, including osteoblasts, osteocytes and osteoclasts along with various supporting cells, including hematopoietic cells, the upregulated Hif-2α expression shown in Fig. 1a, b might be associated with various cell types. Here, we focused on a representative bone remodeling process maintained by the resorption of osteoclasts and performed by osteoblasts. The expression of the osteoblast markers Ocn and Runx2 was lower in the bones of aged mice than in those of younger mice, while the expression of Trap, a marker of osteoclasts, was dramatically increased in aged bones (Fig. 1a). Consistent with the level of Hif-2α mRNA, HIF-2α protein levels were significantly higher in the bones aged mice than in those of younger mice (Fig. 1b). To evaluate bone turnover in aged mice, the levels of biochemical markers in the serum were compared between mice at different ages. Serum OCN levels were lower but the level of the bone resorption-specific biomarker serum CTX-1 was higher in 12- and 20-month-old male mice than in 1.5- and 4-month-old male mice (Fig. 1c). To confirm the in vivo involvement of HIF-2α expression and bone turnover in aged bone cells, the numbers of HIF-2α-expressing osteoblasts (Fig. 1d) and osteoclasts (Fig. 1e) were quantified histochemically in young (4-month-old) and aged (12-month-old) mice. The number of HIF-2α-positive osteoblasts were higher in the bone tissue of aged mice than in that of younger mice (Fig. 1d). The number of HIF-2α-expressing osteoclasts was increased in aged mice (Fig. 1e). These findings indicated that HIF-2α expression was upregulated in both osteoblasts and osteoclasts upon aging.

a Detection of the indicated mRNAs in the long bones of 1.5-, 4-, and 20-month-old mice. The levels of mRNAs encoded by Hif-2α, osteoblast marker genes (Ocn and Runx2), an osteoclast marker gene (Trap) and senescence marker genes (p16 and p21) were determined by RT-PCR and quantified by qRT-PCR (n ≥ 4). b The protein levels of HIF-2α extracted from mice of the indicated age determined by western blot analysis (left panel) and quantified by a CS analyzer 4 (right panel) (n = 5). c ELISA-based measurements of the serum concentrations of OCN (n = 5) and CTX-1 (n = 5) in 1.5-, 4-, 12- and 20-month-old mice. d, e Immunostaining of osteoblasts (d) and osteoclasts (e) from the bone tissues of young (4-month-old) and old (12-month-old) mice for HIF-2α. The dotted lines indicate osteoblasts, and the arrowheads indicate osteoclasts. Scale bar: 25 μm. Quantification of HIF-2α-positive cells is shown in the right panel (n = 8). M = month. The values are presented as the means ± SDs (*P < 0.05, **P < 0.01, and ***P < 0.005).
Age-induced osteoporotic bone loss is reversed in heterozygous Hif-2α knockout (KO) mice
The association between upregulation of HIF-2α expression and aged-related bone loss was investigated by examining the bone microarchitecture of 12-month-old male heterozygous Hif-2α KO (Hif-2α+/−) and wild-type (Hif-2α+/+) mice using X-ray microcomputed tomography (μCT). Immunohistochemical staining with anti-HIF-2α and anti-OCN antibodies confirmed that Hif-2α was depleted of osteoblasts in Hif-2α KO mice (Fig. 2a and Supplementary Fig. 1a). Since OCN is expressed only in osteoblasts, osteoblasts can be distinguished from bone lining cells, which do not express OCN29. Knockdown of HIF-2α expression in the osteoclasts of Hif-2α KO mice was also verified by immunohistochemistry (Fig. 2b). Consistent with our previous findings24, quantitative results of bone parameters indicated that the bone mass and trabecular bone percentages were higher in 4-month-old Hif-2α KO mice than in age-matched WT mice (Supplementary Fig. 1b). Quantitative μCT analyses showed that trabecular bone volume was significantly decreased in aged mice (12 months old) compared to young mice (4 months old) (Supplementary Fig. 1b) and that cancellous trabeculae in the femoral bone were better preserved in Hif-2α+/− mice than in WT littermates (Fig. 2c). Trabecular bone volume was higher in Hif-2α+/− mice than in WT mice, as measured by BMD and BV/TV (Fig. 2c). Other bone parameters, including Tb.Th and Tb.N, were higher, whereas Tb.Sp was lower, in Hif-2α+/− mice than in WT mice (Fig. 2c). No differences in cortical thickness were detected between Hif-2α+/− mice and age-matched WT mice at any age (Supplementary Fig 1b). The cause of this discrepancy may be the fact that cortical bone is generally less metabolically active than trabecular bone30,31. Therefore, we focused on analyzing trabecular bone physiology. Bone histomorphometric analyses showed that the bone trabecular percentage was higher and that the number of osteoclasts was lower in heterozygous Hif-2α KO mice than in WT mice (Fig. 2d). BV/TV, N.Ob/B.Pm, and Ob.S/BS were increased in Hif-2α+/− mice, whereas N.Oc/B.Pm and Oc.S/BS, which are parameters associated with bone resorption, were decreased in Hif-2α+/− mice (Fig. 2d). These data indicated that HIF-2α deficiency retarded bone destruction in aged mice.

a, b Representative images of osteoblasts (a) and osteoclasts (b) from the bone tissues of WT (Hif-2α+/+) and Hif-2α+/− mice stained with an anti-HIF-2α antibody. The dotted lines indicate osteoblasts, and the arrowheads indicate osteoclasts. Scale bar: 25 μm. Quantification of HIF-2α-positive osteoblasts and osteoclasts is shown in the right panels (n = 8). c, d Analysis of femoral trabecular bones from 12-month-old Hif-2α+/− and WT mice. Representative images of µCT reconstructions of femoral trabecular and cortical bones (c) and H&E and TRAP staining of trabecular bones (Scale bar: 100 μm; d). Bone mineral density (BMD), bone volume per tissue volume (BV/TV), trabecular bone thickness (Tb.Th), trabecular separation (Tb.Sp), and trabecular number (Tb.N) determined from µCT measurements (n = 8; c). BV/TV, the number of osteoblastic cells per bone perimeter (N.Ob/B.Pm), the osteoblast surface normalized to the bone surface (Ob.S/BS), the number of osteoclastic cells per bone perimeter (N.Oc/B.Pm), and the osteoclast surface normalized to the bone surface (Oc.S/BS) were assessed by bone histomorphometric analyses of the metaphyseal regions of femurs (n = 8; d). The values are presented as the means ± SDs (*P < 0.05, **P < 0.01, and ***P < 0.005).
Upregulation of HIF-2α expression is associated with osteoblast dysfunction upon aging
The effect of increased HIF-2α expression in osteoblasts during age-related bone loss was investigated by assessing the effects of HIF-2α on osteoblast differentiation and senescence. OCN was found to be rarely expressed in HIF-2α-expressing osteoblasts from the bones of 12-month-old mice (Supplementary Fig. 2), suggesting that increased HIF-2α expression in aged bones might be associated with osteoblast senescence, resulting in an imbalance in bone homeostasis and age-mediated bone loss. To assess this hypothesis, doxorubicin-induced senescence during mouse osteoblast differentiation was analyzed. Two different experimental conditions were tested to determine the stage of osteoblast differentiation affected by doxorubicin-induced senescence. Generally, differentiation from preosteoblasts to mature osteoblasts is divided into two stages, proliferation, which occurs in the first 7 days, and matrix maturation, which occurs over the subsequent 7 days, depending on the duration of culture32,33. The protein expression of HIF-2α increased during osteoblast proliferation, peaked during matrix maturation and was observed until day 14 (data not shown). The addition of doxorubicin during both phases effectively blocked L-ascorbic acid (L-AA) and β-glycerophosphate (β-GP)-induced differentiation of preosteoblasts, as determined by measurement of ALP activity and ARS staining (Fig. 3a and Supplementary Fig. 3a). Induction of doxorubicin-mediated osteoblast senescence, as determined by increased p21 expression but not p16 expression, resulted in the upregulation of Hif-2α expression and the downregulation of expression of the osteoblastic marker genes Ocn and Runx2 (Fig. 3b and Supplementary Fig. 3b, c). To evaluate whether overexpression of HIF-2α during osteoblast differentiation affects the progression of senescence, calvarial preosteoblasts were infected with adenovirus expressing Hif-2α (Ad-Hif-2α), which significantly inhibited the expression of Ocn and Runx2 (Fig. 3c and Supplementary Fig. 3d). However, overexpression of Hif-2α did not affect the expression of the senescence marker genes p16 and p21, indicating that Hif-2α expression did not induce senescence (Fig. 3c and Supplementary Fig. 3d). This result was additionally confirmed by osteoblast-specific Hif-2α knockout by Ad-Cre infection in osteoblasts obtained from Hif-2αfl/fl mice. Consistent with the alteration of the expression of genes, including Hif-2α, Ocn, and Runx2, the expression of the senescence marker genes p16 and p21 was not affected (Fig. 3d). TC-S 7009 is a potent inhibitor of HIF-2α activity, and its inhibitory effect was confirmed by its ability to suppress IL-1β- and Ad-Hif-2α-induced HIF-2α activation in a reporter gene assay using a 4×HRE-containing luciferase vector construct (Supplementary Fig. 4). TC-S 7009-mediated inhibition of HIF-2α activity during the osteoblast proliferation phase failed to recover osteoblast differentiation inhibited by senescence (Supplementary Fig. 5a, b). However, HIF-2α inhibition during matrix maturation resulted in significant reversal of the doxorubicin-mediated reduction in Ocn and Runx2 expression without altering p16 expression (Fig. 3e and Supplementary Fig. 5c). Moreover, doxorubicin-mediated inhibition of osteoblast differentiation was reversed by TC-S 7009-induced inhibition of HIF-2α, as determined by ALP and ARS staining (Fig. 3f and Supplementary Fig. 5d). These findings indicated that increased HIF-2α expression was an effect of aging rather than a cause of osteoblast senescence in aged bones. However, inhibition of HIF-2α was able to restore age-related impaired osteoblast matrix maturation by increasing the expression of genes such as Ocn and Runx2.

a, b Doxorubicin-induced inhibition of osteoblast differentiation. Primary calvarial preosteoblasts from newborn mice were cultured in osteogenic differentiation medium (DM) containing 50 μg/ml L-AA and 5 mM β-GP for 7 days. Before harvesting cells on day 14, 0.01 μg/ml doxorubicin was added to the differentiation medium for 1, 3, or 5 days. Representative images of alkaline phosphatase (ALP) and alizarin red S (ARS) staining in primary calvarial preosteoblasts cultured in control medium (CM) or differentiation medium in the absence or presence of 0.01 μg/ml doxorubicin are shown (a). The expression levels of Hif-2α, Ocn, Runx2, p16, and p21 were analyzed by qRT-PCR (n = 5; b). c Quantification of mRNA levels by qRT-PCR. The transcript levels of the indicated genes were measured in primary calvarial preosteoblasts infected with Ad-C at an MOI of 800 or Ad-Hif-2α at the indicated MOI (n ≥ 4). d The mRNA levels of Hif-2α, Ocn, Runx2, p16, and p21. Osteoblasts isolated from Hif-2αfl/fl mice were infected with Ad-C or Ad-Cre in the presence of differentiation medium (n ≥ 4). e, f Effect of HIF-2α inhibition on osteoblast matrix maturation. Doxorubicin-treated osteoblasts were treated with the potent HIF-2α inhibitor TC-S 7009 and incubated for 3 days. The levels of Ocn, Runx2, p16, and p21 mRNAs were measured by qRT-PCR (n = 5; e). Assessment of osteoblast differentiation and mineral deposition by ALP and ARS staining (f). The values are presented as the means ± SDs (*P < 0.05, **P < 0.01, and ***P < 0.005; NS not significant).
Osteoblast-specific depletion of HIF-2α increases bone mass in aged mice
To further investigate the association between HIF-2α expression upregulation in osteoblasts and age-mediated osteoporotic bone loss, male osteoblast-specific Hif-2α conditional KO (Hif-2α;fl/flCol1a1-Cre) mice were analyzed. HIF-2α was specifically depleted in the osteoblasts of these mice (Fig. 4a). μCT images showed that aging-induced trabecular bone loss in 12-month-old Hif-2α;fl/flCol1a1-Cre mice (Supplementary Fig. 6) was much lower than that in age-matched Hif-2αfl/fl mice (Fig. 4b). Quantitative analyses showed that BMD, BV/TV, Tb.Th and Tb.N were higher and that Tb.Sp was lower in osteoblast-specific Hif-2α-deficient mice than in Hif-2αfl/fl mice (Fig. 4b). Consistent with these μCT images, H&E staining and bone histomorphometric parameters, such as BV/TV, N.Ob/B.Pm and Ob.S/BS also showed that the trabecular percentage was increased in Hif-2α;fl/flCol1a1-Cre mice (Fig. 4c). The results of TRAP staining and measurement of quantitative parameters associated with bone resorption, such as N.Oc/B.Pm and Oc.S/BS, revealed that bone resorption was decreased in aged Hif-2α;fl/flCol1a1-Cre mice (Fig. 4c). These findings indicated that osteoblast-specific depletion of HIF-2α delayed age-induced osteoporotic bone loss.

a Representative images of HIF-2α in osteoclasts (left panel) and osteoblasts (right panel) from Hif-2αfl/fl and Hif-2α;fl/flCol1a1-Cre mice. The dotted lines indicate osteoblasts. Scale bar: 25 μm. b, c Analyses of femoral trabecular bones from 12-month-old Hif-2αfl/fl and Hif-2α;fl/flCol1a1-Cre mice. Representative images of µCT reconstructions of trabecular bones (b) and H&E and TRAP staining (c) are presented. BMD, BV/TV, Tb.Th, Tb.Sp, and Tb.N were determined from µCT measurements (n = 8; b). Determination of BV/TV, N.Ob/B.Pm, Ob.S/BS, N.Oc/B.Pm and Oc.S/BS from bone histomorphometric analysis of the metaphyseal regions of femurs (n = 8; c). Scale bar: 100 μm. The values are presented as the means ± SDs (*P < 0.05; **P < 0.01, and ***P < 0.005).
Aging-associated upregulation of HIF-2α expression accelerates osteoclast function and senescence
Because age-mediated disruption of bone homeostasis is also caused by increased osteoclast activity34, the effect of aging on HIF-2α expression in osteoclasts was assessed. HIF-2α was strongly expressed in CTSK-positive osteoclasts in the bones of 12-month-old mice (Supplementary Fig. 7a). Hif-2α expression was increased on days 3–5 of in vitro RANKL-induced osteoclast differentiation of BMMs, consistent with the increases in p16 and p21 expression (Fig. 5a and Supplementary Fig. 7b). Immunohistochemistry also showed that the expression of p16 and p21 was higher in osteoclasts from the bones of 12-month-old mice than those from the bones of younger mice (Fig. 5b). Assessment of the direction contribution of HIF-2α to the aging of osteoclasts showed that osteoclastogenesis was significantly enhanced by transduction with Ad-Hif-2α, as evidenced by an increase in the number of TRAP-positive multinucleated cells (Fig. 5c). Interestingly, p16 and p21 expression was concomitantly enhanced by the overexpression of Hif-2α during RANKL-induced osteoclast differentiation of BMMs (Fig. 5d and Supplementary Fig. 7c). Conversely, HIF-2α knockdown in BMMs obtained from Hif-2αfl/fl mice by Ad-Cre infection led to the downregulation of p16 and p21 expression (Fig. 5e). To determine whether p16 and p21 are specific targets of HIF-2α, putative HIF-2α binding sites [5’-(A/G)CGTG-3’] were identified within the promoter region of these two genes (Fig. 5f, g). ChIP assays using four primer pairs designed to span these putative binding sites revealed that HIF-2α directly regulated the expression of both p16 and p21 (Fig. 5f, g). These findings indicated that increased HIF-2α expression in aged osteoclasts promoted the expression of p16 and p21 and mediated the senescence-related enhancement of the bone resorption activity of osteoclasts.

a The mRNA levels of osteoclast marker genes (Trap), Hif-2α, and senescence markers (p16, p21) during M-CSF/RANKL-induced osteoclastogenesis of bone marrow macrophages (BMMs) (n ≥ 3). b Representative images of p16 and p21 immunostaining in bone osteoclasts from young (4-month-old) and old (12-month-old) mice. Scale bar: 25 μm. M month. c, d TRAP staining (c) and qRT-PCR analysis of Hif-2α, Trap, p16, and p21 expression (n ≥ 4; d) in Hif-2α-overexpressing osteoclasts. BMMs were infected with Ad-C or Ad-Hif-2α and cultured with M-CSF and RANKL for 5 days. Scale bar: 100 μm. e The mRNA levels of Hif-2α, Trap, p16, and p21. Osteoclasts isolated from Hif-2αfl/fl mice were infected with Ad-C or Ad-Cre during osteoclastogenesis (n ≥ 4). f, g Binding of HIF-2α to the promoter regions of p16 (f) and p21 (g). Ad-Hif-2α-infected osteoclasts were subjected to ChIP with an anti-HIF-2α antibody and a primer pair designed to span the putative HIF-2α binding regions [5’-(A/G)CGTG-3’] within the promoters of p16 (n = 3; f) and p21 (n = 3; g). The values are presented as the means ± SDs (*P < 0.05, **P < 0.01, and ***P < 0.005).
Osteoclast-specific depletion of HIF-2α inhibits bone loss in aged mice
The association between HIF-2α expression upregulation in osteoclasts and age-mediated osteoporotic bone loss was further analyzed in osteoclast-specific male Hif-2α conditional KO (Hif-2α;fl/flCtsk-Cre) mice. Depletion of HIF-2α in osteoclasts but not osteoblasts was verified by immunohistochemical staining (Fig. 6a). Immunohistochemistry also showed that p16 (Fig. 6b) and p21 (Fig. 6c) expression was downregulated in Hif-2α-deficient osteoclasts. Quantitative analyses revealed that 12-month-old mice showed much lower trabecular bone mass than 4-month-old mice (Supplementary Fig. 8). Osteoclast-specific depletion of Hif-2α prevented this reduction in trabecular bone loss, as determined by μCT imaging and analyses of quantitative parameters, such as BMD, BV/TV, Tb.Th, Tb.Sp, and Tb.N (Fig. 6d). Bone histomorphometric analyses by H&E and TRAP staining showed that N.Oc/B.Pm and Oc.S/BS was significantly decreased in Hif-2α;fl/flCtsk-Cre mice, whereas N.Ob/B.Pm and Ob.S/BS were increased (Fig. 6e). These in vivo data showed that specific depletion of HIF-2α in osteoclasts delayed age-induced osteoporotic bone loss.

a–c Immunostaining of HIF-2α, p16, and p21 in Hif-2αfl/fl and Hif-2α;fl/flCtsk-Cre mice. Osteoclast-specific depletion of HIF-2α in 12-month-old Hif-2αfl/fl and Hif-2α;fl/flCtsk-Cre mice was assessed by immunohistochemistry with anti-HIF-2α antibody. The dotted lines indicate osteoblasts (Scale bar: 25 μm; a). Immunostaining of p16 (b) and p21 (c) in osteoclasts from Hif-2αfl/fl and Hif-2α;fl/flCtsk-Cre mice was examined by immunofluorescence microscopy. Scale bar: 25 μm. d Quantitative µCT analysis of trabecular bones. BMD, BV/TV, Tb.Th, Tb.Sp, and Tb.N in trabecular bones from 12-month-old Hif-2αfl/fl and Hif-2α;fl/flCtsk-Cre mice (n = 8). e Representative images of H&E and TRAP staining in 12-month-old Hif-2αfl/fl and Hif-2α;fl/flCtsk-Cre mice (n = 8; Scale bar, 100 μm). BV/TV, N.Ob/B.Pm, Ob.S/BS, N.Oc/B.Pm, and Oc.S/BS were determined by bone histomorphometric analyses of the metaphyseal regions of femurs. The values are presented as the means ± SDs (*P < 0.05, **P < 0.01, ***P < 0.005).
Discussion
As aging progresses, bone remodeling, which is normally tightly regulated, may be dysregulated due to imbalances between osteoblast-mediated bone formation and osteoclast-mediated bone resorption. This imbalance can result in an increased risk of fracture, impaired bone healing, and the chronic age-related bone metabolism disease osteoporosis. This study showed that senescence-induced HIF-2α expression upregulation in both osteoblasts and osteoclasts acts as a novel intrinsic mediator of age-related bone loss.
The senescence-related genes p16 and p21 play important roles in the initiation of senescence by leading to cell cycle arrest11. Doxorubicin-induced senescence during osteoblastic differentiation of osteoblast progenitor cells resulted in increased expression of p21 but not p16. In the absence of doxorubicin-mediated induction of senescence, however, both genes were highly expressed in mature osteoclasts. Upregulated HIF-2α expression in p21-expressing senescent osteoblasts inhibited osteoblast differentiation and function, as determined by decreases in Ocn and Runx2 expression. Twist2, a gene directly targeted by HIF-2α, has been shown to be expressed during early stages of osteogenic differentiation and to inhibit the expression of RUNX2, a key transcription factor for osteoblast differentiation, resulting in decreased expression of OCN, which has been implicated in bone mineralization24. TWIST is a negative regulator of RUNX2 expression and function that downregulates OCN expression35, whereas doxorubicin enhances TWIST expression in mouse osteoblastic cells36. The current study hypothesized that upregulated HIF-2α expression in osteoblasts in aged bones suppresses RUNX2 expression by inducing TWIST2 expression upregulation, resulting in the sequential downregulation of OCN expression. Senescent cells can cause tissue dysfunction through the development of senescence-associated secretory phenotypes, which are characterized by the production of extracellular matrix-degrading proteins, proinflammatory cytokines and chemokines37,38. These aging-associated changes in the bone microenvironment may induce increased HIF-2α expression in osteoblasts. Indeed, unlike that of HIF-1α, the expression of HIF-2α can be enhanced by proinflammatory cytokines, including IL-1β and TNFα, in the absence of hypoxia26. In general, the osteoblast differentiation process can be divided into three phases: proliferation, matrix maturation, and mineralization39. We previously reported that HIF-2α is highly expressed until the matrix maturation stage and that HIF-2α expression declines during later stages of osteogenic differentiation24 .The present study showed that doxorubicin inhibited osteoblast differentiation during both the proliferation and matrix maturation stages. In contrast, HIF-2α inhibited the maturation process but not the proliferation, thereby controlling osteoblast differentiation. These unexpected discrepancies may be related to the altered expression of differentiation-associated genes, differences in accessibility of the promoters of HIF-2α target genes during the complicated differentiation process, or the sensitivity of the HIF-2α inhibitor employed. Further studies are needed to assess the catabolic effect of HIF-2α on age-related osteoblast dysfunction in mediating age-related reductions in bone mass.
This study also revealed that the expression levels of HIF-2α, p16, and p21 were increased in osteoclasts and that their expression levels were higher in the bones of aged mice than in those of younger mice. Upregulated HIF-2α expression in osteoclasts promoted the transcription of p16 and p21 by binding directly to hypoxia-responsive elements (HREs; -(A/G)CGTG-) in the promoters of these genes. Thus, low p16 and p21 expression was detected in Hif-2α;fl/flCtsk-Cre mice. Upregulation of HIF-2α expression in senescent osteoclasts predominantly enhances bone-resorbing activity, as evidenced by the fact that inducible elimination of p16-expressing senescent cells in INK-ATTAC transgenic mice is associated with decreases in the number of osteoclasts and bone resorption40. In addition, senescent cell-condition medium enhances osteoclastogenesis and bone-resorbing activity and impairs osteoblast mineralization40. In our current study, the degree of cell senescence was assessed by measuring the expression levels of p16 and p21 because it is well established that these genes are the most well-known effectors of senescence and that their expression levels increase with age in several rodent and human cell lines4,18,41. In addition, Saeed et al. demonstrated that p16 and p21 levels are significantly increased in bone cells from telomerase knockout mice, which are premature aging model mice42. Although these genes act as key effectors of cellular senescence, since the phenomena caused by aging are diverse, additional investigations of other factors are necessary.
Bone is affected by weight load. The spine and legs, which support the human body, show marked changes in shape, such as bending of the bones, with aging. However, in mice, since the body is supported by four limbs, the morphological and physiological changes associated with age-related bone loss in the femur might be more serious than those in vertebrae. In addition, in murine models, weight-bearing bone is highly sensitive to running, which is a weight-bearing exercise43,44. Furthermore, some studies that observed aged bone showed no differences in age-related bone loss between femurs and vertebrae45,46. Therefore, in this study, we measured bone parameters in the femurs, not the spines, of mice.
HIF-2α deficiency in aged male mice markedly reversed age-related bone loss, which is consistent with our previous results showing that HIF-2α depletion in mice increases bone mass by promoting osteoblast differentiation and function and by inhibiting osteoclast differentiation and maturation24. Interestingly, both Hif-2α;fl/flCol1a1-Cre and Hif-2α;fl/flCtsk-Cre male mice showed relatively better preservation of trabeculae than age-matched Hif-2αfl/fl mice due to changes in both osteoblasts and osteoclasts. Osteoblast-mediated bone formation was higher and osteoclast-mediated bone resorption was lower in the bones of 12-month-old Hif-2α;fl/flCol1a1-Cre and Hif-2α;fl/flCtsk-Cre conditional KO mice than in the bones of control mice, which may have been due to interactions between osteoblasts and osteoclasts. Osteoblasts influence osteoclast formation and differentiation through OPG/RANKL/RANK and LGR4/RANKL/RANK signaling47,48. RANKL expressed by osteoblasts can promote osteoclastogenesis by stimulating the RANK receptor on osteoclasts, a process that is regulated by the ratio of RANKL to OPG, a natural antagonist of RANK49. We previously reported that Rankl is a direct target gene of HIF-2α and that RANKL expression induced by HIF-2α overexpression promotes osteoblast-mediated osteoclast differentiation24. We hypothesized that osteoblast-specific HIF-2α depletion might reduce RANKL secretion by osteoblasts, reducing the RANKL/OPG ratio and the number of osteoclasts. Moreover, osteoclasts can affect osteoblast-mediated bone formation via Atp6v0d250, complement component 3a47, semaphorin 4D51, or exosomal miR-214-3p52. Because TRAF6, which acts as a crucial adaptor protein of RANK signaling in osteoclasts, is a gene that is directly regulated by HIF-2α24, we hypothesized that the decreased osteoclastic activity in Hif-2α;fl/flCtsk-Cre mice was caused by a decrease in TRAF6 levels. However, the molecular mechanism responsible for the increased number of osteoblasts in Hif-2α;fl/flCtsk-Cre mice remains to be determined, although it may be due to osteoblast-osteoclast interactions.
Estrogen deficiency in postmenopausal females is another main cause of age-associated net bone loss and subsequently leads to osteoporosis34. We previously used an animal model of OVX-induced osteoporosis to evaluate the catabolic role of HIF-2α in bone remodeling during estrogen deficiency-associated osteoporosis24. Although the molecular mechanisms underlying pathological bone diseases caused by estrogen deficiency and aging differ somewhat, our findings suggest that HIF-2α is a crucial regulator of both estrogen deficiency- and aging-associated osteoporosis. While osteoporosis generally occurs in women, approximately 12% of men aged >50 years old are expected to experience an osteoporotic fracture during their lifetime53. Almost 30% of patients with hip fractures are men, and mortality rates within 1 year after fracture are higher in men than in women54. These findings suggest that some pathophysiological mechanisms of aging-induced osteoporosis are independent of estrogen deficiency. There are two possible mechanisms of estrogen-independent age-induced osteoporosis: vitamin D deficiency and age-related osteoblast dysfunction55. Aging reduces the production of vitamin D in the skin to almost 50% of the normal level and reduces the intestinal concentration of vitamin D receptor, reducing calcium absorption56. Animal studies have shown that the ability of MSCs to differentiate into osteoblasts is impaired by aging57. Furthermore, the senescence-associated secretory phenotype can cause age-related dysfunction in other tissues. For example, aging and cell senescence in bone can induce dysfunction of osteoblast progenitor cells, induce defective bone formation and increase osteoclastogenesis11,58. However, the molecular mechanisms by which senescence affects osteoblasts and disturbs bone homeostasis remain unclear.
HIF-2α may play an important role in senescence-mediated osteoporosis. Although several drugs, such as bisphosphonates and parathyroid hormone, are currently used to treat osteoporosis59,60, they are ineffective against osteoporosis that has already developed. In addition, few therapeutic targets have been identified for treating osteoporotic bone loss in elderly individuals. Targeting senescence-related intrinsic factors involved in bone homeostatic dysfunction may contribute to the development of a novel class of antiaging therapies. Our current results suggest that HIF-2α is a key regulator of age-related bone loss, as well as the pathogenesis of osteoporosis. Treatment with a potent HIF-2α inhibitor resulted in the recovery of osteoblast function reduced by doxorubicin-mediated senescence. These findings indicate that inhibition of HIF-2α in aged bones not only results in the reversal of age-related bone loss that has already occurred by restoring osteoblast function but also prevents age-mediated osteoporosis by inhibiting osteoclast differentiation and maturation.
References
Mellis, D. J., Itzstein, C., Helfrich, M. H. & Crockett, J. C. The skeleton: a multi-functional complex organ. The role of key signalling pathways in osteoclast differentiation and in bone resorption. J. Endocrinol. 211, 131–143 (2011).
Khan, M. P. et al. Pathophysiological mechanism of bone loss in type 2 diabetes involves inverse regulation of osteoblast function by PGC-1α and skeletal muscle atrogenes: AdipoR1 as a potential target for reversing diabetes-induced osteopenia. Diabetes 64, 2609–2623 (2015).
Hwang, E. S., Yoon, G. & Kang, H. T. A comparative analysis of the cell biology of senescence and aging. Cell. Mol. Life Sci. 66, 2503–2524 (2009).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Weinstein, R. S., Jilka, R. L., Parfitt, A. M. & Manolagas, S. C. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Invest. 102, 274–282 (1998).
Ducy, P. CBFA1: A molecular switch in osteoblast biology. Dev. Dyn. 219, 461–471 (2000).
Nakashima, K. et al. The novel zinc finger-containing transcription factor osterix is equired for osteoblast differentiation and bone formation. Cell 108, 17–29 (2002).
Rutkovskiy, A., Stensløkken, K. O. & Vaage, I. J. Osteoblast differentiation at a glance. Med. Sci. Monit. Basic Res. 22, 95–106 (2016).
Karsenty, G. & Oury, F. The central regulation of bone mass, the first link between bone remodeling and energy metabolism. J. Clin. Endocrinol. Metab. 95, 4795–4801 (2010).
Pietschmann, P. et al. Bone structure and metabolism in a rodent model of male senile osteoporosis. Exp. Gerontol. 42, 1099–1108 (2007).
Kassem, M. & Marie, P. J. Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell 10, 191–197 (2011).
Piemontese, M. et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight 2, e93771 (2017).
Wada, T., Nakashima, T., Hiroshi, N. & Penninger, J. M. RANKL–RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 12, 17–25 (2006).
Vignery, A. Macrophage fusion: the making of osteoclasts and giant cells. J. Exp. Med. 202, 337–340 (2005).
Karsenty, G., Kronenberg, H. M. & Settembre, C. Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 25, 629–648 (2009).
Chen, W. et al. Novel pycnodysostosis mouse model uncovers cathepsin K function as a potential regulator of osteoclast apoptosis and senescence. Hum. Mol. Genet. 16, 410–423 (2007).
Cong, Q. et al. p38α MAPK regulates proliferation and differentiation of osteoclast progenitors and bone remodeling in an aging-dependent manner. Sci. Rep. 7, 45964 (2017).
Farr, J. N. et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 31, 1920–1929 (2016).
Kim, H. N. et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 16, 693–703 (2017).
Kim, H. N. et al. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 18, e12923 (2019).
Wang, Y. et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Invest. 117, 1616–1626 (2007).
Ivan, M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 (2001).
Dengler, V. L., Galbraith, M. & Espinosa, J. M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 49, 1–15 (2014).
Lee, S. Y. et al. Controlling hypoxia-inducible factor-2α is critical for maintaining bone homeostasis in mice. Bone Res. 7, 14 (2019).
Ryu, J. H. et al. Hypoxia-inducible factor-2α is an essential catabolic regulator of inflammatory rheumatoid arthritis. PLoS Biol. 12, e1001881 (2014).
Yang, S. et al. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 16, 687–693 (2010).
Choi, W. S. et al. The CH25H-CYP7B1-RORα axis of cholesterol metabolism regulates osteoarthritis. Nature 566, 254–258 (2019).
Hu, X. & Zhang, H. Doxorubicin-induced cancer cell senescence shows a time delay effect and is inhibited by epithelial-mesenchymal transition (EMT). Med. Sci. Monit. 25, 3617–3623 (2019).
Everts, V. et al. The bone lining cell: its role in cleaning Howship’s lacunae and initiating bone formation. J. Bone Miner. Res. 17, 77–90 (2002).
Cao, J. J., Gregoire, B. R. & Gao, H. High-fat diet decreases cancellous bone mass but has no effect on cortical bone mass in the tibia in mice. Bone 44, 1097–1104 (2009).
Ott, S. M. Cortical or trabecular bone: what’s the difference? Am. J. Nephrol. 47, 373–375 (2018).
Taylor, S. E. B., Shah, M. & Orriss, I. R. Generation of rodent and human osteoblasts. Bonekey Rep. 3, 585 (2014).
Infante, A. & Rodríguez, C. I. Osteogenesis and aging: lessons from mesenchymal stem cells. Stem Cell Res. Ther. 9, 244 (2018).
Henriksen, K., Bollerslev, J., Everts, V. & Karsdal, M. A. Osteoclast activity and subtypes as a function of physiology and pathology–implications for future treatments of osteoporosis. Endocr. Rev. 32, 31–63 (2011).
Kronenberg, H. M. Twist genes regulate Runx2 and bone formation. Dev. Cell 6, 317–318 (2004).
Glackin, C. A., Murray, E. J. & Murray, S. S. Doxorubicin inhibits differentiation and enhances expression of the helix-loop-helix genes Id and mTwi in mouse osteoblastic cells. Biochem. Int. 28, 67–75 (1992).
Coppé, J. P., Desprez, P. Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118 (2010).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Jonason, J. H. & O’Keefe, R. J. Isolation and culture of neonatal mouse calvarial osteoblasts. Methods Mol. Biol. 1130, 295–305 (2014).
Farr, J. N. et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 23, 1072–1079 (2017).
Ma, Y. et al. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 17, e12709 (2018).
Hamid, S. et al. Telomerase-deficient mice exhibit bone loss owing to defects in osteoblasts and increased osteoclastogenesis by inflammatory microenvironment. J. Bone Miner. Res. 26, 1494–1505 (2011).
Bourrin, S., Palle, S., Pupier, R., Vico, L. & Alexandre, C. Effect of physical training on bone adaptation in three zones of the rat tibia. J. Bone Miner. Res. 10, 1745–1752 (1995).
Iwamoto, J., Takeda, T. & Sato, Y. Effect of treadmill exercise on bone mass in female rats. Exp. Anim. 54, 1–6 (2005).
Shum, L. C. et al. Cyclophilin D knock-out mice show enhanced resistance to osteoporosis and to metabolic changes observed in aging bone. PLoS ONE 11, e0155709 (2016).
Li, C. J. et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Invest. 125, 1509–1522 (2015).
Matsuoka, K., Park, K. A., Ito, M., Ikeda, K. & Takeshita, S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J. Bone Miner. Res. 29, 1522–1530 (2014).
Wang, L. et al. Osteoblast-induced osteoclast apoptosis by fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ. 22, 1654–1664 (2015).
Lacey, D. L. et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93, 165–176 (1998).
Lee, S. H. et al. v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 12, 1403–1409 (2006).
Negishi-Koga, T. et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 17, 1473–1480 (2011).
Li, D. et al. Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat. Commun. 7, 10872 (2016).
Schuit, S. C. E. et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam Study. Bone 34, 195–202 (2004).
Center, J. R., Nguyen, T. V., Schneider, D., Sambrook, P. N. & Eisman, J. A. Mortality after all major types of osteoporotic fracture in men and women: an observational study. Lancet 353, 878–882 (1999).
Carmeliet, G., Dermauw, V. & Bouillon, R. Vitamin D signaling in calcium and bone homeostasis: a delicate balance. Best. Pract. Res. Clin. Endocrinol. Metab. 29, 621–631 (2015).
Hill, T. R., Granic, A. & Aspray, T. J. Vitamin D and ageing. Subcell. Biochem. 90, 191–220 (2018).
Nishikawa, K. et al. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J. Clin. Invest. 120, 3455–3465 (2010).
Marie, P. J. Bone cell senescence: mechanisms and perspectives. J. Bone Miner. Res. 29, 1311–1321 (2014).
Eastell, R., Walsh, J. S., Watts, N. B. & Siris, E. Bisphosphonates for postmenopausal osteoporosis. Bone 49, 82–88 (2011).
Baron, R. & Hesse, E. Update on bone anabolics in osteoporosis treatment: Rationale, current status, and perspectives. J. Clin. Endocrinol. Metab. 97, 311–325 (2012).
Acknowledgements
This work was supported by grants from the National Research Foundation of Korea (NRF) through the Korean government (MSIT) (2019R1A5A2027521 and NRF-2018R1A2B2006033); the Bio & Medical Technology Development Program of the NRF, funded by the Korean government (MSIT) (2019M3A9A8065758); and the Korea Health Technology R&D project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare (HI14C3484).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, S.Y., Park, K.H., Lee, G. et al. Hypoxia-inducible factor-2α mediates senescence-associated intrinsic mechanisms of age-related bone loss. Exp Mol Med 53, 591–604 (2021). https://doi.org/10.1038/s12276-021-00594-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00594-y
This article is cited by
-
Differential but complementary roles of HIF-1α and HIF-2α in the regulation of bone homeostasis
Communications Biology (2024)
-
Intermittent hypoxia induces hepatic senescence through promoting oxidative stress in a mouse model
Sleep and Breathing (2024)
-
Mycotoxins and cellular senescence: the impact of oxidative stress, hypoxia, and immunosuppression
Archives of Toxicology (2023)
-
Hedgehog signaling regulates bone homeostasis through orchestrating osteoclast differentiation and osteoclast–osteoblast coupling
Cellular and Molecular Life Sciences (2023)
