
Abstract
Caspase-2 was discovered almost three decades ago. It was one of the first two mammalian homologs of CED-3, the other being interleukin 1β-converting enzyme (ICE/caspase-1). Despite high similarity with CED-3 and its fly and mammalian counterparts (DRONC and caspase-9, respectively), the function of caspase-2 in apoptosis has remained enigmatic. A number of recent studies suggest that caspase-2 plays an important role in the regulation of p53 in response to cellular stress and DNA damage to prevent the proliferation and accumulation of damaged or aberrant cells. Here, we review these recent observations and their implications in caspase-2-mediated cellular death, senescence, and tumor suppression.
Similar content being viewed by others


Introduction
Cysteine aspartate-specific proteases (caspases) are key drivers of inflammation and cell death. They mediate the processing and activation of proinflammatory cytokines or cleave several proteins during apoptosis to facilitate the programmed disassembly of cells1,2. Caspases also function in other pathways, including cell proliferation and differentiation. In general, caspases can be functionally split into two main groups. For example, mammalian caspase-2, -3, -6, -7, -8, -9, and -10 are apoptotic caspases, whereas caspase-1, -4, -5, -11, and -12 are involved in inflammation (or inflammation-induced cell death)1,2. In contrast to the other caspases, caspase-14 is mostly expressed and activated in the differentiating and cornifying layers of the epidermis and plays a crucial role in epidermal barrier formation2. The caspases implicated in apoptotic cell death can be further divided into initiators and effectors. As the term implies, initiator caspases are activated first in response to apoptotic signals, and once activated, they mediate the activation of effector caspases through proteolytic cleavage events. Effector caspases then cleave several hundred potential targets in the cell undergoing apoptotic dismantling. Initiator caspases are autoactivated2,3. They contain long prodomains near the N-terminus that comprise either death effector domains (DEDs; caspase-8 and -10) or caspase-recruitment domains (CARDs; caspase-2, -9, -1, and -11)2,4. These domains mediate protein dimerization and/or recruitment into larger complexes, such as apoptosomes5 and death-inducing signaling complexes (DISCs), to facilitate their activation6. The main effector caspases, caspases-3 and -7, do not possess the ability to autoactivate and their processing must be induced by initiator caspases2.
Apoptotic caspase activation occurs via two main pathways. The mitochondrial pathway (BCL-2-regulated or the intrinsic pathway) is activated in response to various forms of cellular stress, including DNA damage, whereas the tumor necrosis factor (TNF) family of ligands initiates the alternative (extrinsic) pathway of apoptosis by binding to their cognate receptors (Fig. 1)1,3,6. The mitochondrial pathway involves the proapoptotic BCL-2 family members BAX and BAK7, which induce mitochondrial outer membrane permeabilization (MOMP)8,9 and promote cytochrome c release. The apoptotic protease-activating factor-1 (APAF-1) associates with cytochrome c in a multimeric apoptosome to activate caspase-95. In the extrinsic pathway, apoptosis signaling via TNF family members is initiated following ligand-dependent activation of TNF family death receptors (such as FAS, TNFR, TRAILR1, and TRAILR2)6. The cytoplasmic domains of active multimeric receptors recruit and activate caspase-8 or -10 via DISC, which consists of FAS-associated death domain protein (FADD) and/or TNFR-associated death domain protein (TRADD), as well as other proteins (e.g., TRAFs and RIP1) that modulate death receptor signaling10. Activated caspase-8 can activate caspase-3 and caspase-711 and cleave BID to a truncated form (tBID), which engages the mitochondrial pathway to amplify the apoptotic response through MOMP12 (Fig. 1).

Core apoptotic machinery in C. elegans and in mammals that drives caspase activation is shown. In mammals, there are two main apoptosis pathways. In the intrinsic pathway, various cellular insults result in MOMP and cytochrome c release from mitochondria, which drives apoptosome-dependent caspase-3 activation. In the extrinsic pathway, TNF family members activate caspase-8 via DISC. Activated caspase-8 and caspase-9 mediate the processing and activation of effector caspases, of which caspase-3 is the most prominent. The figure was created with BioRender.com.
The focus of this review is caspase-2, a CARD-containing caspase. It is the most evolutionarily conserved member of the caspase family, with 31% identity (55% similarity) with the C. elegans caspase CED-3 and 25% identity (40% similarity) with the only Drosophila initiator caspase: DRONC13,14,15. Caspase-2 is expressed in almost all metazoan cells and is rapidly processed in response to various intrinsic and extrinsic apoptotic signals3,13,16,17,18,19,20,21,22,23. Similar to other initiator caspases, caspase-2 activation occurs via CARD-mediated homodimerization and autoprocessing24,25,26,27 (Fig. 2). Caspase-2 is also recruited to a large multiprotein complex called the PIDDosome28 (see below). Caspase-2 is the only caspase with a classical nuclear localization signal that mediates its predominant localization to the nucleus29. While caspase-2 is implicated in many cell death and non-cell death functions30, here, we discuss recent data that link caspase-2 to p53 stability and its various functions, including cell cycle regulation and tumor suppression.

In the primary structure, the location of zymogen cleavage sites, CARD, p19, p12, nuclear localization signal (NLS), and the catalytic Cys residue are shown. The putative ubiquitination and phosphorylation sites are also indicated. Various biochemical steps that lead to mature dimeric caspase-2 are shown. A ribbon diagram displays the dimeric structure of caspase-2 with two substrate binding pockets, including catalytic Cys (PDB ID: 3R6G)32. The figure was created with BioRender.com.
Caspase-2 activation
Through its CARD, overexpressed caspase-2 can rapidly oligomerize, which is sufficient for its activation24,26,29. Caspase-2, a zymogen with basal enzyme activity that is promoted by dimerization, is further activated by its proteolytic processing into two subunits, p19 and p1231 (Fig. 2). Structural studies suggest that mature caspase-2 forms a p19/p12 homodimer in solution with two active sites32. Biochemical data suggest that caspase-2 can be initially activated without processing and without other proteins24; however, in cell extracts, caspase-2 is recruited to a large complex, which probably mediates its full activation in response to various signals25. One such activation complex is the PIDDosome, which comprises PIDD1 (p53-induced death domain-containing protein 1), RAIDD (RIP-associated ICH-1/CED-3 homologous proteins with a death domain), and caspase-233,34. The PIDDosome is considered to be the main signaling platform for caspase-2 activation in response to DNA damage35,36 and cytokinesis failure37. Somewhat surprisingly, however, PIDD1- or RAIDD-deficient cells and mice show normal caspase-2 activation38,39,40. Caspase-2 can also be activated in response to bacterial toxins in a PIDDosome-independent manner41.
The serine/threonine kinase ATM (ataxia telangiectasia mutated) has been shown to directly promote PIDDosome formation induced by DNA damage35,38,39. ATM phosphorylates Thr788 in the death domain (DD) of PIDD1, leading to conformational changes and allowing binding with RAIDD through DD interactions35,42. More recently, the phosphoprotein nucleophosmin (NPM1) has been shown to control PIDDosome formation specifically in the nucleolus, which activates caspase-2 in response to DNA damage43. The authors also demonstrated a RAIDD-dependent, but PIDD1-independent, platform for caspase-2 activation in the cytoplasm, while both RAIDD and PIDD1 are involved in a nucleolar platform43.
Although many studies have demonstrated the physiological importance of PIDDosome complex as a caspase-2 activation platform, other potential mechanisms of caspase-2 activation have also been reported. As mentioned above, caspase-2 was also shown to be activated in DISC, which includes CD95 (FAS/APO-1) and TNFR144,45. Recently, NLRP3 has also been proposed to be a possible caspase-2 activation platform46. In this study, NLRP3 was translocated to mitochondria upon ER stress, leading to caspase-2 activation following the release of mitochondrial DNA and cytochrome c, resulting in inflammasome activation46. In addition, TRAF2 was shown to promote caspase-2 activation in a complex following cisplatin treatment of cells47. PIDD1 and RAIDD were not essential in this context47.
Caspase-2 activity can be regulated by several factors, including its localization, transcription and posttranslational modifications, including phosphorylation and ubiquitination47,48. A recent study reported that caspase-2 ubiquitination is required to stabilize caspase-2 dimerization and activation in a TRAF2-dependent manner in cultured cells47. Specifically, caspase-2 dimerization and further activation upon cisplatin treatment involved TRAF2-mediated ubiquitination at Lys15, Lys152, and Lys15347.
To date, several phosphorylation sites have been found to be important for caspase-2 regulation in certain contexts. Under nutrient-replete conditions that satisfy the pentose phosphate pathway in Xenopus oocytes, Ser135 (Ser164 in humans) of caspase-2 is phosphorylated by calcium/calmodulin-dependent kinase II (CAMKII), resulting in inhibition of caspase-2 activation49. Caspase-2 activation is also inhibited by phosphorylation at Ser157 by the protein kinase casein kinase-2 (CK2) in TNF-alpha-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis50. A recent study showed that phosphorylation at Thr180 of caspase-2 is p38 mitogen-activated protein kinase (MAPK)-dependent and important in sterol regulatory element-binding protein 2 (SREBP2) regulation in lipid metabolism51. It is unclear whether p38 MAPK phosphorylates Thr180 directly. In the context of the cell cycle and DNA damage response, phosphorylation-dependent control of caspase-2 activity may be most relevant. For example, CDK1-Cyclin B1-dependent phosphorylation at Ser340 inhibits caspase-2 to protect cells from incidental apoptotic death during mitosis52. Recent data indicate that a key mitotic kinase, Aurora kinase B (AURKB), phosphorylates caspase-2 at the highly conserved residue Ser38453. A phosphomimetic mutation at Ser384 blocks caspase-2 catalytic activity, preventing the cleavage of its substrates, BID and MDM2, in cells with mitotic errors. Structural analysis suggests that phosphorylation at Ser384 may prevent substrate binding53. Therefore, phosphorylation prevents cleavage of the key caspase-2 substrates involved in apoptosis and p53 stabilization (see below).
Caspase-2 – cell cycle connection
Possible links between caspase-2, p53 and the cell cycle have been consistently reported. For example, caspase-2-deficient murine embryonic fibroblasts (MEFs) proliferate faster and become immortalized quicker than their wild-type counterparts54,55. Caspase-2 has been shown to interact with cyclin D3, which regulates the G1/S transition during the cell cycle56. Furthermore, as stated above, caspase-2 can be phosphorylated by CDK1-Cyclin B1, a mitosis-promoting kinase, during mitosis52. This phosphorylation inhibits caspase-2 activation, preventing unwanted cell death during mitosis. A role for caspase-2 in mitotic catastrophe was proposed approximately 15 years ago57. Mitotic catastrophe is programmed cell death resulting from failed mitosis, including delayed mitosis and failure to repair DNA damaged by physical or chemical stresses1,58. CHK2 inhibition was shown to induce mitotic catastrophe and caspase-2-dependent cell death, and the inhibition/ablation of caspase-2 suppresses mitotic catastrophe, resulting in multinucleated and aneuploid cells57. Later, Sidi and colleagues showed that inhibition of CHK1, a cell cycle regulating kinase, causes ATM/ATR-mediated caspase-2 activation and results in nonapoptotic cell death program initiation, even in p53-deficient cells59. These reports suggest that inhibition of either CHK1 or CHK2 results in mitotic catastrophe involving caspase-2-mediated cell death.
Ongoing errors in chromosome segregation caused by dysfunctional cell cycle checkpoints, such as spindle assembly checkpoints (SACs) or cytokinesis failure during mitosis, can cause aneuploidy, which often leads to chromosomal instability (CIN), a hallmark of cancer60. This failure is often associated with defects in the activation of apoptotic pathways. For example, mutation of the p53 tumor suppressor or overexpression of the prosurvival BCL-2 family of proteins are common precursors to malignant transformation and therapy resistance61,62.
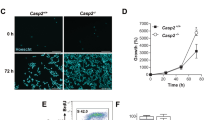
Caspase-2 deficiency is closely associated with enhanced aneuploidy in primary MEFs and tumors13,54,63,64,65. Similarly, bone marrow cells from aged but not young caspase-2-deficient mice also showed significantly higher aneuploidy than bone marrow cells from WT mice66. Recent studies showed that increased aneuploidy in caspase-2-deficient and caspase-2-catalytic mutant mouse cells reduced BID cleavage and cell death and clonogenic survival following polo-like kinase 1 (PLK1) inhibition in caspase-2-deficient cells (Fig. 3a, d)67. This finding implies a requirement for the enzymatic activity of caspase-2 in the apoptosis of aneuploid cells; however, it is unknown whether the PIDDosome is essential.

a Schematic showing that the cell cycle arrest and apoptotic functions of caspase are mediated via cleavage of MDM2 and BID following mitotic catastrophe, respectively. Figure created with BioRender.com. b An example of MDM2 cleavage by caspase-2 (Casp2) following treatment of WT or caspase-2-deficient (KO) U2OS cells with or without the AURKB inhibitor ZM447439 (ZM). c Panels showing examples of increased multinucleated (arrow) caspase-2-deficient U2OS cells following AURKB inhibition. Scale bar, 50 µm. d Examples of caspase-2-mediated BID cleavage (appearance of truncated BID, tBID) following treatment with a PLK1 inhibitor, BI2536 (BI), which induces mitotic arrest. fl.; full-length, cl.; cleaved, p33 and p18; cleaved caspase-2. The data and images in b–d were generated by Yoon Lim.
BUBR1 is a core protein of the mitotic checkpoint complex in mitosis control, and it has been shown to be an inhibitor of the PIDDosome in response to irradiation-mediated DNA damage36. Specifically, following DNA damage during mitosis, BUBR1 recruits ATM phosphorylated PIDD1 to the kinetochore, outcompeting RAIDD for PIDD1 binding, thus preventing PIDDosome formation. This mechanism helps prevent unplanned cell death during cell division and implicates the PIDDosome and caspase-2 as critical mitosis regulators36.
Caspase-2 stabilizes p53 following DNA damage
In addition to the findings discussed above, caspase-2 deficiency has often been linked to a defective DNA damage response. For example, caspase-2-deficient MEFs readily escape senescence in culture and exhibit increased micronuclei formation and sustained DNA damage during cell culture and following γ-irradiation54. A lack of caspase-2 is known to be associated with increased aneuploidy in both MEFs and in EμMyc lymphoma cells, and loss of caspase-2 leads to defective p53-mediated signaling, suggesting that caspase-2 is important in maintaining genomic integrity during cell proliferation and following DNA damage54,55. Other studies have implicated both PIDD1 and caspase-2 in DNA damage-dependent p53-mediated apoptosis68,69. Recent observations indicate that active caspase-2 stabilizes p53, resulting in cell cycle arrest and/or apoptosis (Fig. 3). The levels of p53 are regulated by the ubiquitin ligase MDM2, which is critical for the ubiquitination and degradation of p5370,71,72. In a seminal study published in 2011, Oliver and colleagues found that caspase-2 directly cleaves MDM2 at Asp367, resulting in the removal of the C-terminal RING domain that is critical for p53 ubiquitination68. N-terminally truncated MDM2 then binds p53 and promotes its stability68. Thus, this finding suggested for the first time that, following genotoxic stress and DNA damage, p53-dependent induction of the PIDDosome and caspase-2 activation generates a positive feedback loop that inactivates MDM2 and promotes p53 stability.
As MDM2 is also a transcriptional target of p53, increased p53 levels drive MDM2 expression, which in turn results in p53 degradation via the ubiquitin-proteasome system73,74. Thus, an MDM2-p53 negative feedback loop results in oscillatory p53 pulses following double-stand DNA breaks and ATM activation75,76,77. On the other hand, in response to UV-induced DNA damage that activates ATR, p53 is expressed as a broad pulse78. A study published in 2020 suggests that several days after ionizing radiation-induced DNA damage, some cells switch from oscillating to sustained p53 dynamics79. Using a Venus fluorescent reporter to monitor p53 levels in single cells, the authors found that following 10 Gy irradiation, 15–20% of cells exhibit stably increased p53 levels79. Interestingly, the fraction of cells switching from pulsatile to sustained p53 levels was maximal at 5 Gy but reduced with increasing radiation dose of 10 or 20 Gy dose. The cells with sustained p53 signaling showed chromosomal damage. The authors found that late phase p53 levels were reduced in irradiated PIDD1-deficient cells and in cells following caspase-2 inhibition79. Overall, their data suggest that PIDD1 and caspase-2 are required for cells that evade DNA damage-induced cell cycle arrest to switch from pulsating to sustained p53 signaling. Thus, the presence of damaged DNA activates caspase-2 via the PIDDosome to stabilize p53 signaling and thus limits the proliferation of cells carrying extensive DNA damage by inducing apoptosis (Fig. 4).

A proposed scheme showing that PIDDosome-mediated caspase-2 activation, MDM2 cleavage and p53 stabilization serves as a checkpoint under conditions of sustained DNA damage to induce cell cycle arrest or apoptosis. The figure was created with BioRender.com.
Caspase-2 in ploidy control
A study by Fava and coworkers demonstrated that extra centrosomes resulting from cytokinesis failure trigger PIDDosome-dependent caspase-2 activation37. Activated caspase-2 then cleaves MDM2, leading to p53 stabilization and p21-mediated cell cycle arrest of polyploid cells (Fig. 3a–c). The study showed that PIDD1 can localize to extra mother centrioles and that caspase-2 activation following AURKB inhibition is dependent on PIDD1 and RAIDD37. Importantly, the PIDDosome was essential for caspase-2 activation in this context and was required to protect cells against polyploidization and malignant transformation37. A more recent study showed p53 stabilization and effective cell cycle arrest following treatment with the same AURKB inhibitors in caspase-2-deficient U2OS cells80, suggesting that caspase-2-mediated p53 stabilization may be cell-type specific. Caspase-2 and PIDD1 have also been shown to be involved in the control of ploidy during postnatal development and regeneration in the liver81. Two very recent papers demonstrated how the PIDDosome may control centrosome amplification and aneuploidy82,83. The authors found that PIDD1 is directly recruited to mature centrosomes by the centriolar distal appendage protein ANKRD26, and this interaction is required for PIDDosome and caspase-2 activation following centrosome amplification82,83. These reports suggest that ANKRD26-dependent PIDDosome activation acts as a centriolar signal to limit cell proliferation via the caspase-2-p53 axis in response to centrosome amplification and extra chromosomes.
Caspase-2 as a tumor suppressor
From the discussion above, it is apparent that caspase-2 plays an important checkpoint function by stabilizing p53 to prevent the accumulation of cells carrying damage, thus potentially acting to suppress tumorigenesis. Indeed, there are several lines of evidence to suggest that loss of caspase-2 is associated with increased susceptibility to certain types of tumors. The human CASP2 gene is in Ch7q3484. As Ch7q is often deleted in hematological malignancies85, reduced expression of caspase-2 (along with other genes in this region) is not uncommon. Reduced caspase-2 expression has been reported in some cases of Burkitt lymphoma, mantle cell lymphoma (MCL), chronic lymphocytic leukemia (CLL), and acute myeloid leukemia (AML)86,87. In AML, reduced caspase-2 levels are linked to poor prognosis88. CASP2 mutations are rare but sometimes associated with colon, gastric lung and breast cancer89,90. Interestingly, aneuploidy tolerance and chromosomal instability in colon cancer correlate with low CASP2 levels due to BCL9L gene aberrations91.
In mouse models employing caspase-2 deficiency, a tumor suppressive effect of caspase-2 has been reported by several laboratories (Table 1). For example, caspase-2 deficiency enhances lymphomagenesis in Eμ-Myc transgenic mice55,64,92 and ATM kinase-deficient animals63 and accelerates breast tumor formation in MMTV/c-neu mice65 and in a KrasG12D-driven lung adenocarcinoma model93. The tumor suppressor role of caspase-2 was also apparent in a diethylnitrosamine-induced mouse hepatocellular carcinoma (HCC) model, where at 10 months of age, all caspase-2-knockout animals developed malignant HCC, whereas the WT animals showed only adenomas and rarely HCC94. Surprisingly, a new study showed that caspase-2 loss had no effect on diethylnitrosamine-induced carcinogenesis95. The reason for this discrepancy is unclear, but as the severity and timing of diethylnitrosamine-induced tumor appearance is dependent on many factors, the rate of HCC development may have masked the effects of caspase-2 deficiency. On the other hand, and interestingly, Sladky et al.95 found that PIDDosome deficiency caused hyperpolyploidization that prevented HCC development. In addition, caspase-2 deficiency somewhat delayed neuroblastoma development driven by the TH-MYCN transgene in mice96. Consistent with these findings, an analysis of expression array datasets of primary human neuroblastoma tumor patients demonstrated a significant correlation between high levels of caspase-2 and poor prognosis96. Notably, this correlation was observed in neuroblastoma patients with nonamplified MYCN96. The tissue- and context-specific tumor suppressor function of caspase-2 was also apparent in the distinct transcription profiles of caspase-2-deficient Eμ-Myc and TH-MYCN mouse tumors97.
Perspective
The work summarized here indicates that both the PIDDosome and caspase-2 play critical roles in p53 stabilization and cell death to prevent polyploidy, which is required to prevent the accumulation of harmful cells with chromosomal abnormalities. Importantly, caspase-2 activation can also occur independent of the PIDDosome to prevent aneuploidy and tumorigenesis. These data suggest that caspase-2 activation serves as a checkpoint under specific conditions of sustained cellular or DNA damage, which may explain the tumor suppressive effects of caspase-2 seen in various mouse models. However, many questions remain. Perhaps the most puzzling finding is that caspase-2-deficient mice acquire only mild phenotypes and do not spontaneously develop tumors or other specific pathologies in old age. Mice lacking caspase-2 demonstrate increased ploidy in the liver, and aged caspase-2 mice show increased aneuploidy in the bone marrow compartment66. Interestingly, this study also showed a possible role of caspase-2 in regulating hematopoietic stem cells (HSCs) and in progenitor cell differentiation66. Caspase-2-deficient animals showed a significant increase in short-term HSCs and multipotent progenitors with skewed differentiation towards myeloid progenitors with age66. Whether this phenotype represents a premyeloid state is unclear. Nevertheless, from studies with caspase-2-deficient mouse models, it is clear that caspase-2 deficiency is not a primary driver of tumorigenesis. Furthermore, it has been demonstrated that loss of caspase-2 significantly improves energy metabolism and reduces weight gain in mice on a high-fat diet that is associated with reduced incidence of non-alcoholic fatty liver disease (NAFLD) progression98,99,100. However, while caspase-2 has been shown to regulate both metabolism and polyploidization in the liver81,98,99,100, there appear to be no dramatic consequences in normal liver pathophysiology in caspase-2-deficient mice. Finally, caspase-2 deficiency is linked to aging-related traits, including increased oxidative stress and reduced levels of antioxidant signaling101,102. These findings support the idea that caspase-2 may play specific roles during replication stress or metabolic stress in cells. However, whether these phenotypes are acquired because of an alteration in caspase-2-mediated p53 regulation remains to be determined.
References
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Shalini, S., Dorstyn, L., Dawar, S. & Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 22, 526–539 (2015).
Vigneswara, V. & Ahmed, Z. The role of caspase-2 in regulating cell fate. Cells https://doi.org/10.3390/cells9051259 (2020).
Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 14, 32–43 (2007).
Dorstyn, L., Akey, C. W. & Kumar, S. New insights into apoptosome structure and function. Cell Death Differ. 25, 1194–1208 (2018).
Kist, M. & Vucic, D. Cell death pathways: intricate connections and disease implications. EMBO J. https://doi.org/10.15252/embj.2020106700 (2021).
Strasser, A. & Vaux, D. L. Viewing BCL2 and cell death control from an evolutionary perspective. Cell Death Differ. 25, 13–20 (2018).
Wei, M. C. et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 (2001).
Tait, S. W. & Green, D. R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a008706 (2013).
Wilson, N. S., Dixit, V. & Ashkenazi, A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat. Immunol. 10, 348–355 (2009).
Stennicke, H. R. et al. Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 273, 27084–27090 (1998).
Li, H., Zhu, H., Xu, C. J. & Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501 (1998).
Puccini, J., Dorstyn, L. & Kumar, S. Caspase-2 as a tumour suppressor. Cell Death Differ. 20, 1133–1139 (2013).
Dorstyn, L., Colussi, P. A., Quinn, L. M., Richardson, H. & Kumar, S. DRONC, an ecdysone-inducible Drosophila caspase. Proc. Natl Acad. Sci. USA 96, 4307–4312 (1999).
Kumar, S., Kinoshita, M., Noda, M., Copeland, N. G. & Jenkins, N. A. Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1 beta-converting enzyme. Genes Dev. 8, 1613–1626 (1994).
Guo, Y., Srinivasula, S. M., Druilhe, A., Fernandes-Alnemri, T. & Alnemri, E. S. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J. Biol. Chem. 277, 13430–13437 (2002).
Wang, Q. E., Han, C., Zhang, B., Sabapathy, K. & Wani, A. A. Nucleotide excision repair factor XPC enhances DNA damage-induced apoptosis by downregulating the antiapoptotic short isoform of caspase-2. Cancer Res. 72, 666–675 (2012).
Wejda, M. et al. Degradomics reveals that cleavage specificity profiles of caspase-2 and effector caspases are alike. J. Biol. Chem. 287, 33983–33995 (2012).
Wagner, K. W., Engels, I. H. & Deveraux, Q. L. Caspase-2 can function upstream of bid cleavage in the TRAIL apoptosis pathway. J. Biol. Chem. 279, 35047–35052 (2004).
Tu, S. et al. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat. Cell Biol. 8, 72–77 (2006).
Tiwari, M., Herman, B. & Morgan, W. W. A knockout of the caspase 2 gene produces increased resistance of the nigrostriatal dopaminergic pathway to MPTP-induced toxicity. Exp. Neurol. 229, 421–428 (2011).
Stefanis, L., Troy, C. M., Qi, H., Shelanski, M. L. & Greene, L. A. Caspase-2 (Nedd-2) processing and death of trophic factor-deprived PC12 cells and sympathetic neurons occur independently of caspase-3 (CPP32)-like activity. J. Neurosci. 18, 9204–9215 (1998).
Troy, C. M. et al. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J. Neurosci. 20, 1386–1392 (2000).
Baliga, B. C., Read, S. H. & Kumar, S. The biochemical mechanism of caspase-2 activation. Cell Death Differ. 11, 1234–1241 (2004).
Read, S. H., Baliga, B. C., Ekert, P. G., Vaux, D. L. & Kumar, S. A novel Apaf-1-independent putative caspase-2 activation complex. J. Cell Biol. 159, 739–745 (2002).
Butt, A. J., Harvey, N. L., Parasivam, G. & Kumar, S. Dimerization and autoprocessing of the Nedd2 (caspase-2) precursor requires both the prodomain and the carboxyl-terminal regions. J. Biol. Chem. 273, 6763–6768 (1998).
Ho, P. K., Jabbour, A. M., Ekert, P. G. & Hawkins, C. J. Caspase-2 is resistant to inhibition by inhibitor of apoptosis proteins (IAPs) and can activate caspase-7. FEBS J. 272, 1401–1414 (2005).
Sladky, V. C. & Villunger, A. Uncovering the PIDDosome and caspase-2 as regulators of organogenesis and cellular differentiation. Cell Death Differ. 27, 2037–2047 (2020).
Baliga, B. C. et al. Role of prodomain in importin-mediated nuclear localization and activation of caspase-2. J. Biol. Chem. 278, 4899–4905 (2003).
Miles, M. A., Kitevska-Ilioski, T. & Hawkins, C. J. Old and novel functions of caspase-2. Int. Rev. Cell Mol. Biol. 332, 155–212 (2017).
Dorstyn, L. & Kumar, S. Caspase-2 protocols. Methods Mol. Biol. 1133, 71–87 (2014).
Tang, Y., Wells, J. A. & Arkin, M. R. Structural and enzymatic insights into caspase-2 protein substrate recognition and catalysis. J. Biol. Chem. 286, 34147–34154 (2011).
Duan, H. & Dixit, V. M. RAIDD is a new ‘death’ adaptor molecule. Nature 385, 86–89 (1997).
Tinel, A. & Tschopp, J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304, 843–846 (2004).
Ando, K. et al. PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol. Cell 47, 681–693 (2012).
Thompson, R. et al. An inhibitor of PIDDosome formation. Mol. Cell 58, 767–779 (2015).
Fava, L. L. et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 31, 34–45 (2017).
Manzl, C. et al. Caspase-2 activation in the absence of PIDDosome formation. J. Cell Biol. 185, 291–303 (2009).
Kim, I. R. et al. DNA damage- and stress-induced apoptosis occurs independently of PIDD. Apoptosis 14, 1039–1049 (2009).
Sohn, D., Budach, W. & Janicke, R. U. Caspase-2 is required for DNA damage-induced expression of the CDK inhibitor p21(WAF1/CIP1). Cell Death Differ. 18, 1664–1674 (2011).
Imre, G. et al. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J. 31, 2615–2628 (2012).
Park, H. H. et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546 (2007).
Ando, K. et al. NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J. Cell Biol. 216, 1795–1810 (2017).
Lavrik, I. N., Golks, A., Baumann, S. & Krammer, P. H. Caspase-2 is activated at the CD95 death-inducing signaling complex in the course of CD95-induced apoptosis. Blood 108, 559–565 (2006).
Thome, M. et al. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol. 8, 885–888 (1998).
Bronner, D. N. et al. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and caspase-2-driven mitochondrial damage. Immunity 43, 451–462 (2015).
Robeson, A. C., Lindblom, K. R., Wojton, J., Kornbluth, S. & Matsuura, K. Dimer-specific immunoprecipitation of active caspase-2 identifies TRAF proteins as novel activators. EMBO J. https://doi.org/10.15252/embj.201797072 (2018).
Brown-Suedel, A. N. & Bouchier-Hayes, L. Caspase-2 substrates: to apoptosis, cell cycle control, and beyond. Front. Cell Dev. Biol. 8, 610022 (2020).
Nutt, L. K. et al. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 123, 89–103 (2005).
Shin, S. et al. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 24, 3532–3542 (2005).
Pham, D. D. et al. Caspase-2 and p75 neurotrophin receptor (p75NTR) are involved in the regulation of SREBP and lipid genes in hepatocyte cells. Cell Death Dis. 10, 537 (2019).
Andersen, J. L. et al. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J. 28, 3216–3227 (2009).
Lim, Y. et al. Phosphorylation by Aurora B kinase regulates caspase-2 activity and function. Cell Death Differ. 28, 349–366 (2021).
Dorstyn, L. et al. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 19, 1288–1298 (2012).
Ho, L. H. et al. A tumor suppressor function for caspase-2. Proc. Natl Acad. Sci. USA 106, 5336–5341 (2009).
Mendelsohn, A. R., Hamer, J. D., Wang, Z. B. & Brent, R. Cyclin D3 activates Caspase 2, connecting cell proliferation with cell death. Proc. Natl Acad. Sci. USA 99, 6871–6876 (2002).
Castedo, M. et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 23, 4362–4370 (2004).
Vitale, I., Galluzzi, L., Castedo, M. & Kroemer, G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 12, 385–392 (2011).
Sidi, S. et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 133, 864–877 (2008).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Fulda, S. Therapeutic opportunities based on caspase modulation. Semin. Cell Dev. Biol. 82, 150–157 (2018).
Schimmer, A. D. et al. Functional blocks in caspase activation pathways are common in leukemia and predict patient response to induction chemotherapy. Cancer Res. 63, 1242–1248 (2003).
Puccini, J. et al. Loss of caspase-2 augments lymphomagenesis and enhances genomic instability in Atm-deficient mice. Proc. Natl Acad. Sci. USA 110, 19920–19925 (2013).
Peintner, L. et al. The tumor-modulatory effects of Caspase-2 and Pidd1 do not require the scaffold protein Raidd. Cell Death Differ. 22, 1803–1811 (2015).
Parsons, M. J. et al. Genetic deletion of caspase-2 accelerates MMTV/c-neu-driven mammary carcinogenesis in mice. Cell Death Differ. 20, 1174–1182 (2013).
Dawar, S. et al. Impaired haematopoietic stem cell differentiation and enhanced skewing towards myeloid progenitors in aged caspase-2-deficient mice. Cell Death Dis. 7, e2509 (2016).
Dawar, S. et al. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 36, 2704–2714 (2017).
Oliver, T. G. et al. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol. Cell 43, 57–71 (2011).
Baptiste-Okoh, N., Barsotti, A. M. & Prives, C. A role for caspase 2 and PIDD in the process of p53-mediated apoptosis. Proc. Natl Acad. Sci. USA 105, 1937–1942 (2008).
Haupt, Y., Maya, R., Kazaz, A. & Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 (1997).
Kubbutat, M. H., Jones, S. N. & Vousden, K. H. Regulation of p53 stability by Mdm2. Nature 387, 299–303 (1997).
Momand, J., Zambetti, G. P., Olson, D. C., George, D. & Levine, A. J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245 (1992).
Juven, T., Barak, Y., Zauberman, A., George, D. L. & Oren, M. Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene. Oncogene 8, 3411–3416 (1993).
Wu, X., Bayle, J. H., Olson, D. & Levine, A. J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 7, 1126–1132 (1993).
Bakkenist, C. J. & Kastan, M. B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 (2003).
Purvis, J. E. et al. p53 dynamics control cell fate. Science 336, 1440–1444 (2012).
Lahav, G. et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 36, 147–150 (2004).
Batchelor, E., Mock, C. S., Bhan, I., Loewer, A. & Lahav, G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 30, 277–289 (2008).
Tsabar, M. et al. A switch in p53 dynamics marks cells that escape from DSB-induced cell cycle arrest. Cell Rep. 32, 107995 (2020).
Lim, Y., De Bellis, D., Dorstyn, L. & Kumar, S. p53 accumulation following cytokinesis failure in the absence of caspase-2. Cell Death Differ. 25, 2050–2052 (2018).
Sladky, V. C. et al. E2F-family members engage the PIDDosome to limit hepatocyte ploidy in liver development and regeneration. Dev. Cell 52, 335–349.e337 (2020).
Evans, L. T. et al. ANKRD26 recruits PIDD1 to centriolar distal appendages to activate the PIDDosome following centrosome amplification. EMBO J. https://doi.org/10.15252/embj.2020105106 (2020).
Burigotto, M. et al. Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. EMBO J. https://doi.org/10.15252/embj.2020104844 (2020).
Kumar, S. et al. Apoptosis regulatory gene NEDD2 maps to human chromosome segment 7q34-35, a region frequently affected in haematological neoplasms. Hum. Genet. 95, 641–644 (1995).
Honda, H., Nagamachi, A. & Inaba, T. -7/7q- syndrome in myeloid-lineage hematopoietic malignancies: attempts to understand this complex disease entity. Oncogene 34, 2413–2425 (2015).
Holleman, A. et al. Decreased PARP and procaspase-2 protein levels are associated with cellular drug resistance in childhood acute lymphoblastic leukemia. Blood 106, 1817–1823 (2005).
Hofmann, W. K. et al. Altered apoptosis pathways in mantle cell lymphoma detected by oligonucleotide microarray. Blood 98, 787–794 (2001).
Estrov, Z. et al. Caspase 2 and caspase 3 protein levels as predictors of survival in acute myelogenous leukemia. Blood 92, 3090–3097 (1998).
Kim, M. S. et al. Somatic mutations of caspase-2 gene in gastric and colorectal cancers. Pathol. Res. Pract. 207, 640–644 (2011).
Kim, M. S., Chung, N. G., Yoo, N. J. & Lee, S. H. Somatic mutation of proapoptotic caspase-2 gene is rare in acute leukemias and common solid cancers. Eur. J. Haematol. 86, 449–450 (2011).
Lopez-Garcia, C. et al. BCL9L Dysfunction Impairs Caspase-2 Expression Permitting aneuploidy tolerance in colorectal cancer. Cancer Cell 31, 79–93 (2017).
Manzl, C. et al. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ. 19, 1722–1732 (2012).
Terry, M. R. et al. Caspase-2 impacts lung tumorigenesis and chemotherapy response in vivo. Cell Death Differ. 22, 719–730 (2015).
Shalini, S. et al. Caspase-2 deficiency accelerates chemically induced liver cancer in mice. Cell Death Differ. 23, 1727–1736 (2016).
Sladky, V. C. et al. PIDDosome-induced p53-dependent ploidy restriction facilitates hepatocarcinogenesis. EMBO Rep. https://doi.org/10.15252/embr.202050893 (2020).
Dorstyn, L. et al. An unexpected role for caspase-2 in neuroblastoma. Cell Death Dis. 5, e1383 (2014).
Dorstyn, L. et al. Transcriptome profiling of caspase-2 deficient EmuMyc and Th-MYCN mouse tumors identifies distinct putative roles for caspase-2 in neuronal differentiation and immune signaling. Cell Death Dis. 10, 56 (2019).
Machado, M. V. et al. Caspase-2 promotes obesity, the metabolic syndrome and nonalcoholic fatty liver disease. Cell Death Dis. 7, e2096 (2016).
Machado, M. V. et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 64, 1148–1157 (2015).
Kim, J. Y. et al. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell 175, 133–145.e115 (2018).
Shalini, S. et al. Caspase-2 protects against oxidative stress in vivo. Oncogene 34, 4995–5002 (2015).
Tiwari, M. et al. A nonapoptotic role for CASP2/caspase 2: modulation of autophagy. Autophagy 10, 1054–1070 (2014).
Acknowledgements
The work in our laboratory was supported by the National Health and Medical Research Council (NHMRC) of Australia project grants 1043057 and 1156601, a NHMRC Senior Principal Research Fellowship (1103006) and a University of South Australia support package.
Author information
Authors and Affiliations
Contributions
Y.L. and S.K. conceptualized the review, Y.L. drafted the figures, and Y.L., L.D., and S.K. wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lim, Y., Dorstyn, L. & Kumar, S. The p53-caspase-2 axis in the cell cycle and DNA damage response. Exp Mol Med 53, 517–527 (2021). https://doi.org/10.1038/s12276-021-00590-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00590-2
This article is cited by
-
Targeting tumor suppressor p53 for organ fibrosis therapy
Cell Death & Disease (2024)
-
Effect of combined blue light and 5-ALA on mitochondrial functions and cellular responses in B16F1 melanoma and HaCaT cells
Cytotechnology (2024)
-
A dual role of RBM42 in modulating splicing and translation of CDKN1A/p21 during DNA damage response
Nature Communications (2023)
-
Permission to pass: on the role of p53 as a gatekeeper for aneuploidy
Chromosome Research (2023)
-
Identification of novel candidate genes and predicted miRNAs in atopic dermatitis patients by bioinformatic methods
Scientific Reports (2022)
