
Abstract
The stomach has been thought to host few commensal bacteria because of the existence of barriers, such as gastric acid. However, recent culture-independent, sequencing-based microbial analysis has shown that the stomach also harbors a wide diversity of microbiota. Although the stomach immune system, especially innate lymphoid cells (ILCs), has not been well elucidated, recent studies have shown that group 2 ILCs (ILC2s) are the dominant subtype in the stomach of both humans and mice. Stomach ILC2s are unique in that their existence is dependent on stomach microbiota, in sharp contrast to the lack of an impact of commensal microbiota on ILC2s in other tissues. The microbiota dependency of stomach ILC2s is partly explained by their responsiveness to interleukin (IL)-7. Stomach ILC2s express significantly higher IL-7 receptor protein levels on their surface and proliferate more in response to IL-7 stimulation in vitro than small intestinal ILC2s. Consistently, the stomach expresses much higher IL-7 protein levels than the small intestine. IL-5 secreted from stomach ILC2s promotes immunoglobulin (Ig) A production by plasma B cells. In a murine model, stomach ILC2s are important in containing Helicobacter pylori infection, especially in the early phase of infection, by promoting IgA production.
Similar content being viewed by others

Introduction
The surfaces of an animal’s body, such as the skin and mucosa, provide a boundary between the body and the surrounding environment and are colonized by numerous commensal bacteria, called microbiota. In particular, the intestinal mucosa hosts the largest microbial community, of 40 trillion bacteria, in the body1. Gut microbiota contribute to gut maturation2,3, host nutrition, and pathogen resistance4. By contrast, it has been thought that there are fewer commensal bacteria in the stomach due to the characteristic feature of the gastric barrier, such as the highly acidic conditions of the lumen due to acid-secreting parietal cells and the presence of proteolytic enzymes (pepsins) from pepsinogen-secreting zymogenic chief cells5,6. The impact of gastric microbiota on host pathophysiology is not well understood compared to the microbiota of the intestine. Compared to the intestine, where evidence has accumulated that it has evolved the unique gut-associated immune system to deal with gut microbiota and orally ingested pathogens7,8, studies on the stomach immune system are less common. However, recent studies have shown that wide bacterial diversity can be found in the human stomach as well5,9,10,11.
Here, we will review the recently identified characteristics of stomach group 2 innate lymphoid cells (ILC2s), especially their relationship with gastric microbiota and Helicobacter pylori infection.
Stomach and microbiota
Recent culture-independent analyses of microbial composition with 16 S rRNA sequencing have shown wide bacterial diversity in the human stomach5,9,10,11. In individuals who have neither of the two major gastric microbiota-perturbing events, H. pylori infection nor proton pump inhibitor (PPI) usage5, it has been shown that the microbiota in the stomach (and the duodenum) possesses a significantly lower number of bacteria than that of the lower gastrointestinal tract, which is probably correlated with the lower luminal pH in these organs6. The major phyla are Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria throughout the human gastrointestinal tract5,11. In contrast to the lower intestinal tract (and feces), where Firmicutes are dominant, followed by Bacteroidetes11,12, Firmicutes, and Proteobacteria are dominant in the stomach and the duodenum5,9,10. At the genus level, Streptococcus and Prevotella are the two major genera detected in the stomach9,10, which are minor genera in human feces, considered to reflect their decreased presence in the lower gastrointestinal tract12.
Gastric-microbiota-modulating factors: H. pylori and PPIs
As mentioned above, H. pylori infection perturbs the gastric microbiota5. H. pylori is a Gram-negative bacterium categorized as a pathobiont bacteria that colonizes the gastric mucosa of half or more of the human population in the world, ranging from 20% in developed countries to >90% in developing countries5,13,14. H. pylori colonizes the gastric mucosa using virulence properties, such as vacuolating cytotoxin, VacA and cytotoxin-associated gene A, cagA, and causes acute and chronic gastritis, eventually progressing to more severe disorders, including peptic ulcer disease and gastric cancer15.
H. pylori needs to overcome harsh acidic conditions in the stomach lumen to colonize and establish infection in the stomach5,13. It has been well documented that H. pylori infection occurs in early childhood, but rarely in adults13. H. pylori infection is basically acquired in childhood and persists throughout life without eradication therapy16,17. One of the possible explanations is that the ontogenetic development of the stomach in early childhood life is thought to be associated with the alleviation of gastric hostility, including achlorhydria, compared to adulthood18. Gastric hostility in adults sufficiently inhibits H. pylori infection. Therefore, H. pylori infection rarely occurs in adulthood, and when it does, it likely causes acute gastritis and is then eradicated19,20. H. pylori possesses urase and other enzymes that produce ammonia, which aid its infection by increasing the pH13,19.
Chronic H. pylori infection causes atrophic gastritis with a loss of parietal cells5,13. This leads to increased gastric pH and could modulate the gastric microbiota. The impact of H. pylori infection on the gastric microbiota has been studied. Although the majority of the gastric microbiota is occupied by H. pylori, its presence does not largely affect the composition of the gastric community9. H. pylori infection-associated gastric microbiota dysbiosis has been implicated in the pathogenesis of gastric diseases21,22, but the understanding of the complex interaction between the microbiota and H. pylori still needs to be further addressed in the development of gastric diseases.
PPIs are another gastric microbiota modifier5. PPIs suppress gastric acid secretion, are among the most commonly prescribed drugs and are widely used to treat gastric acid-related disorders, such as gastroesophageal reflux disease and peptic ulcers23,24. PPIs reduce bactericidal gastric acid secretion and lead to a significant change in the overall microbiota composition11,23. In the stomach, with PPI treatment, a greater diversity of bacterial species is observed, and the relative abundance of the phyla Firmicutes and Fusobacteria (whose abundance is very small without PPIs) is increased11.
ILCs and gut microbiota
ILCs are largely classified into three groups based on the characteristic expression of transcription factors and cytokine production (Fig. 1)25,26. ILC1s used to include all ILCs producing interferon-γ; however, it is now common to say that ILC1s are distinct from NK cells, whose differentiation is regulated by E4BP4 and Eomes25,26. ILC3s are known to produce interleukin (IL)-17 and IL-22 upon stimulation with IL-1β or IL-23; they are subdivided into (i) lymphoid tissue-inducer cells, which express CCR6 and possess lymphoid tissue-inducing ability; (ii) NKp46-ILC3s, which express NKp46 (also called natural cytotoxicity receptor 1); and (iii) CCR6−NKp46−ILC3s, which do not belong to (i) and (ii)27. ILC3s are basically distributed in tissues facing the external environment, especially the gastrointestinal tract, and play a role in defense mechanisms against pathogens27. Although the relationship between ILCs and gut microbiota has not been well understood until recently, recent advances in ILC biology, including single-cell analysis, have unveiled the details of the relationship28.

Outline of ILC differentiation pathways with the respective transcription factors required for their differentiation and cytokines produced by ILC subsets. CLP common lymphoid progenitor, NKP NK cell progenitor, CHILP common helper-like innate lymphoid precursor, E4BP4 E4 promoter-binding protein 4 (also known as NFIL3), ROR RAR-related orphan receptor, AHR aryl hydrocarbon receptor, cNK conventional NK cell, LTiP lymphoid tissue inducer projenitors, LTi lymphoid tissue inducer.
ILC2s in physiology and pathology
ILC2s were independently identified in 2010 by three groups29,30,31. ILC2s produce type 2 cytokines, such as IL-5, IL-9, and IL-13, upon stimulation with IL-25, IL-33 or TSLP, and are involved in the eradication of parasite infection29,30,31. Recent studies have attracted attention on ILC2s by showing that these cells are involved in the pathogenesis of diseases, including fibrosis, asthma, and obesity32,33,34,35,36. In these diseases, the common scheme of pathogenesis is that an alarmin, IL-33, released upon epithelial cell damage by allergens, inflammation, or stress responses, triggers the activation of ILC2s. IL-33-activated ILC2s secrete IL-4, IL-5, and IL-9, which, in turn, induce eosinophils and activate mast cells, while IL-13 from ILC2s induces fibrosis37,38,39. In addition, it has been reported that ILC2s produce not only cytokines, but also opioid methionine-enkephalin peptides to regulate adipose function and metabolic homeostasis by acting directly on adipocytes35. In contrast to ILC3s, commensal microbiota do not seem to be involved in the induction or differentiation of ILC2s40,41. These observations were made with ILC2s in adipose tissues, the intestine, and the lung, which is in sharp contrast to the microbiota dependency observed with stomach ILC2s as described below.
ILC2s and stomach microbiota
Studies on ILCs have mostly been performed in the intestine and lung, but information on the distribution and function of ILCs in the stomach is still lacking. We have recently examined ILC subsets in the murine stomach42. In the stomach, ILC2s are dominant, with a few ILC1s, and the stomach is almost devoid of ILC3s. This distribution is in sharp contrast to that observed in the intestine, where comparable numbers of the three ILC subsets exist. ILC2 dominance in the stomach has also been reported in humans43.
More importantly, ILC2s existing in the stomach are dependent on stomach microbiota42, unlike ILC2s in the other tissues/organs. In germ-free (GF) mice, the number of stomach ILC2s was significantly decreased (Fig. 2). This observation is also in sharp contrast to the intestine and the lung, where the number of ILC2s is not affected by a lack of a microbiota, as seen in GF mice40,41.

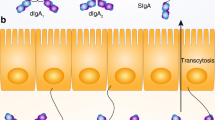
In germ-free (GF) mice, there are few ILC2s. Under GF conditions, IL-7 expression in stomach tissue is hardly detectable, and IL-7 receptor expression on stomach ILC2s is lower than that under SPF conditions. In contrast, the expression of the IL-33 receptor on stomach ILC2s is similar and that of IL-33 in the stomach tissue is slightly decreased in GF compared to SPF conditions. Under SPF conditions, the combination of IL-33 and IL-7 signaling leads to IL-5 secretion by ILC2s, which then promotes IgA production from plasma B cells.
Our findings indicate that stomach ILC2s possess unique characteristics compared to their counterparts in other tissues42. In fact, stomach ILC2s express significantly higher IL-7R levels on their surface and proliferate more in response to IL-7 stimulation than SI ILC2s in vitro. Consistently, the stomach expresses much higher IL-7 protein levels than the small intestine. Interestingly, the expression of IL-7 in stomach tissue is hardly detected under GF conditions. In addition, IL-7R expression on stomach ILC2s is also dependent on the existence of stomach microbiota, at least partly explaining the microbiota dependency of stomach ILC2s. In contrast, the expression of the IL-33 receptor on stomach ILC2s is comparable, and that of IL-33 in the stomach tissue is slightly decreased in GF compared to specific pathogen-free (SPF) conditions (Fig. 2)42.
Immunoglobulin (Ig) A coats the surface of commensal bacteria to prevent them from attaching to and invading the intestinal epithelium44. With the help of cytokines, B cells undergo class switch recombination to become different classes of Ig-producing B cells. In the case of IgA, IL-4 regulates class switching of B cells from IgM to IgA, and IL-5 further promotes the differentiation of IgA-expressing B cells to IgA-secreting plasma cells45. Although T helper (Th) 2 cells are the main source of IL-5, ILC2s have recently been established as another source of robust IL-5 secretion29,30,31,46. We have shown that microbiota-induced ILC2s are required as a major source of IL-5 for the induction of IgA+ plasma B cells, as well as efficient IgA secretion from these cells in the stomach (Fig. 2)42. The secreted IgA could modulate stomach microbiota composition by coating its subset.
Among stomach microbiota, members of the S24-7 family within the order Bacteroidales are suggested to be candidate bacteria for ILC2 induction42. Our preliminary results indicate that there seems to be differential ILC2-inducing ability among several S24-7 clones, and further studies should clarify the molecular mechanisms underlying bacterial induction of stomach ILC2s.
PPIs suppress gastric acid secretion and are among the most commonly prescribed drugs, widely used to treat gastric acid-related disorders, such as gastroesophageal reflux disease and peptic ulcer and, sometimes, H. pylori infection23,24. PPIs reduce bactericidal gastric acid secretion and lead to a significant change in the overall microbiota composition11,23. In the stomach, with PPI treatment, a greater diversity of bacterial species is observed, and the relative abundance of the phyla Firmicutes and Fusobacteria (whose abundance is very small without PPIs) is increased11. This change in gastric microflora composition might affect the number of ILC2-inducible bacteria and, thus, the number of ILC2s in the stomach, which, in turn, can alter the composition of the gastric microbiota.
Stomach ILC2s and H. pylori infection
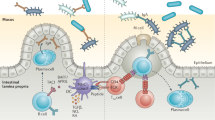
H. pylori infection, especially in the acute phase, is accompanied by a mucosal IgA response, although the T-cell response is predominantly Th1 type16,47,48. In a murine H. pylori infection model, we recently identified that stomach ILC2s are the source of IL-5 for IgA production in the early phase of H. pylori infection (Fig. 3)42. The expansion of IL-5-receptor-expressing plasmablasts is initiated by ILC2 activation, which is indispensable to restrain H. pylori infection. This is also supported by the fact that a significant reduction in plasma B cells and plasmablasts, as well as a reduction in H. pylori coated with IgA, was observed in mice with ILC2-specific deficiency. IgA-mediated H. pylori elimination has a strong correlation with the restraint of gastric inflammation, which is prominently diminished in mice lacking ILC2s42. It has also been shown that the Th2 cell-dependent IgA response is induced for mucosal protection in the late phase of infection (Fig. 3)42.

In the early phase of H. pylori infection, the induction of IL-7 and IL-33 in the stomach tissue increases and activates ILC2s to secrete IL-5, which promotes plasma B cells differentiation and IgA production. At this stage, there is no apparent increase in CD4+T cells (T-cell-independent pathway). In the later phase of the infection, IL-5 derived from Th2-type CD4+ T cells also promotes IgA production (T-cell dependent pathway).
As mentioned above, H. pylori infection is often acquired in childhood and persists throughout life without eradication therapy44,46, while H. pylori infection rarely occurs in adulthood, and when it does, it causes acute gastritis and is then eradicated47,48. Since early childhood is a particularly vulnerable period as the immune system is still developing, one bold hypothesis is that not enough ILC2 development occurs to eradicate H. pylori infection in childhood, although there is no evidence to support this notion at this time; further studies are awaited to clarify whether this hypothesis is true.
Conclusion
It is now clear that the stomach is not a totally sterile and immunologically silent organ, but instead harbors a wide diversity of commensal microbiota with unique composition and a unique immune system within the gastrointestinal tract. In particular, it possesses ILC2s with characteristics that are distinct from their counterparts in other organs/tissues. The stomach ILC2s are dependent on the presence of stomach microbiota, and they can modulate the composition of the microbiota by promoting IgA production. Stomach ILC2s are also important in H. pylori infection. H. pylori can evade or disturb innate and adaptive immune responses via multiple mechanisms to establish chronic infection, leading to clinical gastroduodenal disorders, including chronic gastritis, peptic ulcers, and two malignancies—gastric adenocarcinoma and mucosa-associated lymphoid tissue lymphoma. Therefore, it is important to understand the stomach immune system, including ILC2s, in more detail to develop novel treatment strategies to contain its infection.
References
Sender, R., Fuchs, S. & Milo, R. Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340 (2016).
Palmer, C., Bik, E. M., DiGiulio, D. B., Relman, D. A. & Brown, P. O. Development of the human infant intestinal microbiota. PLoS Biol. 5, e177 (2007).
Xu, J. & Gordon, J. I. Honor thy symbionts. Proc. Natl Acad. Sci. USA 100, 10452–10459 (2003).
Savage, D. C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 31, 107–133 (1977).
Yang, I., Nell, S. & Suerbaum, S. Survival in hostile territory: the microbiota of the stomach. FEMS Microbiol. Rev. 37, 736–761 (2013).
Wu, W. M., Yang, Y. S. & Peng, L. H. Microbiota in the stomach: new insights. J. Dig. Dis. 15, 54–61 (2014).
Brandtzaeg, P., Kiyono, H., Pabst, R. & Russell, M. W. Terminology: nomenclature of mucosa-associated lymphoid tissue. Mucosal Immunol. 1, 31–37 (2008).
Kanaya, T., Williams, I. R. & Ohno, H. Intestinal M cells: tireless samplers of enteric microbiota. Traffic 21, 34–44 (2020).
Bik, E. M. et al. Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl Acad. Sci. USA 103, 732–737 (2006).
Li, X. X. et al. Bacterial microbiota profiling in gastritis without Helicobacter pylori infection or non-steroidal anti-inflammatory drug use. PLoS ONE 4, e7985 (2009).
Mailhe, M. et al. Repertoire of the gut microbiota from stomach to colon using culturomics and next-generation sequencing. BMC Microbiol. 18, 157 (2018).
Nishijima, S. et al. The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res. 23, 125–133 (2016).
Rhee, K. H., Park, J. S. & Cho, M. J. Helicobacter pylori: bacterial strategy for incipient stage and persistent colonization in human gastric niches. Yonsei Med. J. 55, 1453–1466 (2014).
Fox, J. G. & Wang, T. C. Inflammation, atrophy, and gastric cancer. J. Clin. Invest 117, 60–69 (2007).
Camilo, V., Sugiyama, T. & Touati, E. Pathogenesis of Helicobacter pylori infection. Helicobacter 22, 1 (2017).
Reyes, V. E. & Peniche, A. G. Helicobacter pylori deregulates T and B cell signaling to trigger immune evasion. Curr. Top. Microbiol. Immunol. 421, 229–265 (2019).
Banatvala, N. et al. The cohort effect and Helicobacter pylori. J. Infect. Dis. 168, 219–221 (1993).
Koda, Y. K., Laudanna, A. A. & Barbieri, D. Variation and physiological significance of basal gastrinemia in normal children. Arq. Gastroenterol. 29, 66–70 (1992).
Kuipers, E. J. et al. Long-term sequelae of Helicobacter pylori gastritis. Lancet 345, 1525–1528 (1995).
Marshall, B. Helicobacter pylori: 20 years on. Clin. Med. 2, 147–152 (2002).
Coker, O. O. et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 67, 1024–1032 (2018).
Ferreira, R. M. et al. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 67, 226–236 (2018).
Eusebi, L. H. et al. Proton pump inhibitors: risks of long-term use. J. Gastroenterol. Hepatol. 32, 1295–1302 (2017).
Haastrup, P. F., Thompson, W., Sondergaard, J. & Jarbol, D. E. Side effects of long-term proton pump inhibitor use: a review. Basic Clin. Pharm. Toxicol. 123, 114–121 (2018).
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Trabanelli, S. et al. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping. Cytom. B Clin. Cytom. 94, 392–399 (2018).
Domingues, R. G. & Hepworth, M. R. Immunoregulatory sensory circuits in group 3 innate lymphoid cell (ILC3) function and tissue homeostasis. Front. Immunol. 11, 116 (2020).
Gury-BenAri, M. et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 166, 1231–1246 e1213 (2016).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Price, A. E. et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl Acad. Sci. USA 107, 11489–11494 (2010).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Hams, E. et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl Acad. Sci. USA 111, 367–372 (2014).
Moretti, S. et al. A mast cell-ILC2-Th9 pathway promotes lung inflammation in cystic fibrosis. Nat. Commun. 8, 14017 (2017).
Ealey, K. N., Moro, K. & Koyasu, S. Are ILC2s Jekyll and Hyde in airway inflammation? Immunol. Rev. 278, 207–218 (2017).
Brestoff, J. R. et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246 (2015).
Sasaki, T. et al. Innate lymphoid cells in the induction of obesity. Cell Rep. 28, 202–217.e207 (2019).
Halim, T. Y. et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014).
Noval Rivas, M., Burton, O. T., Oettgen, H. C. & Chatila, T. IL-4 production by group 2 innate lymphoid cells promotes food allergy by blocking regulatory T-cell function. J. Allergy Clin. Immunol. 138, 801–811.e809 (2016).
Pelly, V. S. et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol. 9, 1407–1417 (2016).
Monticelli, L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054 (2011).
Ricardo-Gonzalez, R. R. et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 19, 1093–1099 (2018).
Satoh-Takayama, N. et al. Bacteria-induced group 2 innate lymphoid cells in the stomach provide immune protection through induction of IgA. Immunity 52, 635–649.e634 (2020).
Li, R. et al. Group 2 innate lymphoid cells are involved in skewed type 2 immunity of gastric diseases induced by Helicobacter pylori infection. Mediators Inflamm. 2017, 4927964 (2017).
Bunker, J. J. et al. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 358, eaan6619 (2017).
Kunimoto, D. Y., Harriman, G. R. & Strober, W. Regulation of IgA differentiation in CH12LX B cells by lymphokines. IL-4 induces membrane IgM-positive CH12LX cells to express membrane IgA and IL-5 induces membrane IgA-positive CH12LX cells to secrete IgA. J. Immunol. 141, 713–720 (1988).
Gurram, R. K. & Zhu, J. Orchestration between ILC2s and Th2 cells in shaping type 2 immune responses. Cell Mol. Immunol. 16, 225–235 (2019).
Sobala, G. M. et al. Acute Helicobacter pylori infection: clinical features, local and systemic immune response, gastric mucosal histology, and gastric juice ascorbic acid concentrations. Gut 32, 1415–1418 (1991).
Mattapallil, J. J., Dandekar, S., Canfield, D. R. & Solnick, J. V. A predominant Th1 type of immune response is induced early during acute Helicobacter pylori infection in rhesus macaques. Gastroenterology 118, 307–315 (2000).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ohno, H., Satoh-Takayama, N. Stomach microbiota, Helicobacter pylori, and group 2 innate lymphoid cells. Exp Mol Med 52, 1377–1382 (2020). https://doi.org/10.1038/s12276-020-00485-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-020-00485-8
This article is cited by
-
Unveiling the gastric microbiota: implications for gastric carcinogenesis, immune responses, and clinical prospects
Journal of Experimental & Clinical Cancer Research (2024)
-
The Association Between the Gut Microbiome, Nutritional Habits, Antibiotics, and Gastric Cancer: a Scoping Review
Current Nutrition Reports (2022)
-
Interdisciplinary insights into the link between gut microbiome and gastric carcinogenesis—what is currently known?
Gastric Cancer (2022)
-
Impact of Helicobacter pylori eradication on the gastric microbiome
Gut Pathogens (2021)
-
Special issue on the human microbiome: from symbiosis to therapy
Experimental & Molecular Medicine (2020)
