
Abstract
Recent studies on mutations in cancer genomes have distinguished driver mutations from passenger mutations, which occur as byproducts of cancer development. The cancer genome atlas (TCGA) project identified 299 genes and 24 pathways/biological processes that drive tumor progression (Cell 173: 371-385 e318, 2018). Of the 299 driver genes, 12 genes are involved in histones, histone methylation, and demethylation. Among these 12 genes, those encoding the histone demethylases JARID1C/KDM5C and UTX/KDM6A were identified as cancer driver genes. Furthermore, gain-of-function mutations in genes encoding metabolic enzymes, such as isocitrate dehydrogenases (IDH)1/2, drive tumor progression by producing an oncometabolite, D-2-hydroxyglutarate (D-2HG), which is a competitive inhibitor of α-ketoglutarate, O2-dependent dioxygenases such as Jumonji domain-containing histone demethylases, and DNA demethylases. Studies on oncometabolites suggest that histone demethylases mediate metabolic changes in chromatin structure. We have reviewed the most recent findings regarding cancer-specific metabolic reprogramming and the tumor-suppressive roles of JARID1C/KDM5C and UTX/KDM6A. We have also discussed mutations in other isoforms such as the JARID1A, 1B, 1D of KDM5 subfamilies and the JMJD3/KDM6B of KDM6 subfamilies, which play opposing roles in tumor progression as oncogenes or tumor suppressors depending on the cancer cell type.
Similar content being viewed by others

Introduction
The three billion base pairs of DNA in the human genome contain genetic information that is inherited across generations; however, all genes are not accessible to the transcription machinery. Long DNA strands are packaged into chromatin structure, whose basic unit is the nucleosome, composed of the histone octamer (two copies each of H2A, H2B, H3, and H4) wrapped by two turns (147 bp) of DNA. These beads (nucleosomes)-on-a-string (DNA) structures are further organized into 30-nm-thick, compact heterochromatin structures. The three-dimensional structure of chromatin can be modulated by nucleosome positioning, modifications, and histone composition. Differences in chromatin structure can affect all events related to DNA, including transcription, replication, DNA repair, and splicing. Posttranslational modifications (PTMs) of histones critically affect interactions between histones and DNA and the recruitment of “reader” proteins, which recognize special histone modifications. For example, heterochromatin protein (HP)-1 specifically interacts with the trimethylated ninth lysine of histone 3 (H3K9me3) and triggers chromatin compaction via self-oligomerization. H3K9me3 and HP-1 are often detected in inaccessible heterochromatic regions. H3K27me3 also occurs in heterochromatic regions and is involved in transcriptional repression. In contrast, H3K4me3 occurs at the transcription start sites of active genes in euchromatic regions. Genome-wide analyses using RNA-seq and histone chromatin immunoprecipitation (ChIP)-seq elucidated the relationship between transcription and histone modification, which is more extensive than other molecular events on nucleosomes. Compact regions of the genome are less accessible for both the transcription and replication machineries1. Histone PTMs are determined by the balance between “writer” and “eraser” enzymes. These enzymes use cellular metabolites and coenzymes such as α-ketoglutarate (α-KG), S-adenosyl-methionine (SAM), flavin adenine dinucleotide (FAD), ATP, oxygen, Fe(II), acetyl-CoA, and vitamin C for their catalytic reactions2,3. For example, H3 lysine 4 (H3K4) can be methylated by MLL3/KMT2C or MLL4/KMT2D, members of the COMPASS family of histone H3K4 methyltransferases (writers), using SAM as the methyl donor, whereas H3K4me3 and H3K4me2 are demethylated by Jumonji domain-containing histone demethylases (erasers) (Table 1). The JARID1/KDM5 isoforms use oxygen, α-KG, vitamin C, and Fe(II) as cosubstrates (Fig. 1a).

a Substrates for O2- and α-KG-dependent histone demethylases. JHDMs (JmjC domain-containing histone demethylases) remove methyl (CH3-) groups from methylated lysines. JHDMs require O2, α-ketoglutarate, Fe(II), and vitamin C (vit C) for their catalytic activities and release CO2, succinate, and Fe(III). b Schematic illustration of the protein domains of histone demethylases. The domain architecture from the UniProt or NCBI gene (KDM1A, B) information was transformed using https://prosite.expasy.org/mydomains/. The numbers written in each functional domain column indicate the number of amino acids. ARID AT-rich interacting domain, amino oxidase amine oxidase domain, C5HC2 C5HC2 zinc-finger domain, CW CW-type zinc-finger domain, CXXC CXXC zinc-finger domain, C6 C6 zinc-finger domain, JMJC Jumonji C domain, LRR Leu-rich repeat domain, PHD plant homeodomain, SWIRM Swi3p, Rsc8p, and Moira domain, TPR tetratricopeptide domain, TUDOR Tudor domain
More than 20 different Jumoni domain-containing histone demethylases have been identified (Fig. 1b), and their specific substrates and acronyms have been summarized in Table 2. Although these demethylases share common cosubstrates, their Km values for each substrate vary. We have summarized the Km values for O2 and α-KG for each histone demethylase isoform in Table 3. The O2 Km values of the H3K9me2 and me1 demethylase JMJD1A/KDM3A are lower than those of other isoforms, such as JMJD2A; thus, the catalytic activity of JMJD1A persists even under severe hypoxia4,5. Therefore, the catalytic activities of histone demethylases are differentially regulated in response to changes in the concentrations of their cosubstrates. The sensitivities of each histone demethylase to oncometabolites such as D(R)-2-hydroxyglutarate (HG) and L(S)-2HG vary, suggesting that histone methylation can be differently altered in response to IDH mutations and the presence of oncometabolites (Table 3)6.
Although histones are the most abundant molecules in the nucleus, each is fixed at different positions and is hence immobile inside the nucleus. Each histone is unique, as they are wrapped by different DNA sequences. Histone modification enzymes, both writers and erasers, cannot recognize specific DNA sequences in their targets (except for JARID1) but can be recruited in the vicinity of specific histones via interactions with transcription factors (TF) or long noncoding RNAs (lncRNA), which can recognize and interact with specific DNA sequences. Therefore, only a certain subset of histones can be modified when the enzymes are sufficiently close to their substrate for a chemical reaction to occur. In the “signal control” mechanism, certain signals select specific histone substrates and alter their methylation status by activating specific TFs or lncRNAs, which recognize DNA sequences to recruit specific histone methyltransferases and/or demethylases in the vicinity of the selected histones (Fig. 2). In contrast, “metabolic control” mechanisms instantly change histone methylation without activation of additional TFs. Changes in the metabolic substrates or inhibitors can instantly alter the activities of enzymes that are already recruited on specific regions of the genome, resulting in altered levels of histone methylation. Therefore, metabolic control of histone modification depends on (i) the availability of substrates and inhibitors and (ii) the genomic regions where the writers and erasers already reside. As the designated histone molecules where the writers and erasers reside are cell-specific, the effects of metabolic control of histone modification vary within cells, thereby increasing cellular heterogeneity (Fig. 2).

Metabolic control and signaling control interpret how the levels of O2, α-ketoglutarate (α-KG), and 2-hydroxyglutarate (2-HG) modulate gene expression by altering histone methylation. DNA-bound proteins (DBP) or DNA-bound long noncoding RNAs recruit histone demethylases (KDM), such as JARID and JMJD. In low oxygen or α-KG concentrations and a 2-HG high concentration, the activities of JARID or JMJD are suppressed at different loci for each cell, resulting in a high methylation level on the corresponding loci. Transcription factors (TF) such as hypoxia-inducible factor (HIF) are activated under low oxygen conditions or high 2-HG conditions. Activated TFs recruit histone methyltransferase (HMT), which forms a complex with KDM. In most cells, although KDM and HMT are recruited together, the histone methylation level was upregulated by HMT, as the KDM activity is suppressed under hypoxic or high 2-HG conditions. Dynamic histone modification can regulate transcription, replication, and DNA repair
Hypoxic tumor microenvironments have been considered the main cause for increased chemo- and radiotherapy resistance. Recent studies revealed that hypoxic conditions limit the effective concentration of α-KG and increase the 2-HG level via metabolic reprogramming. Although several studies showed that hypoxia itself, hypoxia-induced metabolic reprogramming, and IDH mutations increase the total abundance of methylated histones in different cancer cells, the mechanism through which the increased level of histone methylation is related to cellular heterogeneity, cancer resistance, and progression remains unknown. This review introduces recent observations regarding (i) metabolic reprogramming of the α-KG balance by IDH mutation and hypoxia in cancers, (ii) the tumor-suppressive functions of the cancer driver genes JARID1C/KDM5C and UTX/KDM6A, and (iii) mutations in genes encoding other isoforms in various cancers, namely, JARID1A, 1B, 1D, and JMJD3/KDM6B of the KDM5 and KDM6 subfamilies.
Hypoxic reprogramming of the α-KG balance in cancer metabolism
Production of an oncometabolite, D(R)-2HG
Supra-physiological concentrations of the D(R) form of 2-hydroxyoxoglutartate (D-2HG or R-2HG), a stereo-specific isoform of the natural and endogenous L-2HG (or S-2HG) molecule, was detected in the sera of patients with acute myeloid leukemia (AML) and glioma7,8. Cancer genomic analyses have identified that an abnormal increase in the D-2HG level is associated with mutations in either IDH1 or IDH2. Wild-type IDH1 and IDH2 catalyze the oxidative decarboxylation of isocitrate to generate α-KG and CO2 (Fig. 3). IDH1 localizes to the peroxisomes and cytosol, whereas IDH2 localizes to the mitochondria. In several tumors, including glioma, AML, chondrosarcoma, intrahepatic cholangiocarcinoma, and thyroid carcinoma (Table 1), missense mutations of arginine residues in the active sites of both IDH1 and IDH2 (IDH1R123H, IDH2R140Q, or IDH2R172K) result in new activities that further convert α-KG to D-2HG9,10,11,12,13,14,15,16,17. The observation that D-2HG is sufficient to promote transformation of hematopoietic cells suggested that R-2HG is an oncometabolite18. Specific inhibitors of these IDH mutants have been developed and some are in clinical trials19,20. Nonetheless, the molecular mechanisms through which D-2HG promotes tumorigenesis remain poorly understood. Since D-2HG is a competitive inhibitor of α-KG-dependent dioxygenases, such as histone-, DNA- and RNA demethylases, D-2HG may induce dysregulation of histone, DNA, and RNA methylation in various cancer cell lines. In addition to mutated IDHs, D-3-phosphoglycerate dehydrogenase (PHGDH) also catalyzes the reduction of α-KG to D-2HG using NADH (Fig. 3)21. Accordingly, PHGDH is frequently amplified in breast cancer, and a subset of breast malignancies have been shown to accumulate 2-HG22,23. Under normal conditions, the D-2HG level remains low because of the catalytic activity of endogenous D-2HG dehydrogenase (D2HGDH), which catalyzes the conversion of D-2HG to α-KG; however, mutant IDHs produce excess D-2HG, which accumulates in patients with glioma and AML18,24,25.

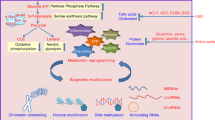
Glucose and glutamine are transported inside cells using individual transporters (circles with arrows). Glucose is converted to pyruvate via glycolysis, which is then converted into acetyl CoA by PDH and used in the TCA cycle. Intermediates in the TCA cycle, from citrate to oxaloacetate, are converted by individual enzymes. IDH1 and IDH2 are localized in the cytosol and mitochondria, respectively. Both enzymes convert isocitrate to α-KG. Glutamine is converted to glutamate by GLS, which can be converted to α-KG by GDH or AT. α-KG is used as a substrate by α-KG-dependent dioxygenase together with oxygen and iron ions. The types of α-KG-dependent dioxygenases include enzymes that induce metabolic epigenetic changes in histones, DNA, and RNA, and enzymes that induce changes in signaling. As these enzymes are α-KG- and O2-dependent, they are inhibited via deprivation of these factors. Regarding α-KG, (i) mutated IDH1/2 and PHGDH produce D-2HG, and (ii) LDHA and MDH1/2, whose levels are increased under hypoxic conditions, produce L-2HG. Both types of 2-HG compete with α-KG to inhibit α-KG-dependent dioxygenases. Both D-2HG and L-2HG can be converted to α-KG by D2HGDH and L2HGDH, respectively. Under hypoxic conditions, the activity of α-KG-dependent dioxygenases is inhibited via the regulation of glucose and glutamine pathways as well as by the O2 concentration. Hypoxia increases glycolysis by upregulating glucose transporters and enzymes related to glycolysis. Hypoxia-inducible factor 1 alpha (HIF1α) activates PDK, an enzyme that inactivates PDH (which converts pyruvate to acetyl-CoA); therefore, hypoxia inhibits the entry of pyruvate into the TCA cycle. Glutamine uptake and GDH (the enzyme that converts glutamate to α-KG) are upregulated under hypoxic conditions. Glutamine is converted to α-KG to provide intermediates for the TCA cycle. Details are provided in the text and the enzymes are shown in green. AT aminotransferases, D2HGDH D-2HG dehydrogenase, FH fumarate hydratase, FTO fat mass and obesity-associated protein, GDH glutamate dehydrogenases, GLS glutaminase, IDH1/2 isocitrate dehydrogenases 1/2, JHDM JmjC domain-containing histone demethylases, L2HGDH L-2HG dehydrogenase, LDHA lactate dehydrogenase A, MDH 1/2 malate dehydrogenase 1/2, PDH pyruvate dehydrogenase, PDK1 pyruvate dehydrogenase kinase 1, PHGDH D-3-phosphoglycerate dehydrogenase, SDH succinate dehydrogenase, TET ten-eleven translocation, TCA tricarboxylic acid
Excess endogenous L(S)-2HG in hypoxic cells
Similar to D-2HG (or R-2HG), excessive L-2HG (or S-2HG), an enantiomer of D-2HG, can also inhibit numerous α-KG-dependent enzymes26,27. Recent studies showed that hypoxia dramatically induces L-2HG production in both normal and malignant cells28,29. Under hypoxic or acidic conditions, L-2HG production was increased via promiscuous catalytic activities of lactate dehydrogenase A (LDHA) and malate dehydrogenase (MDH1 and 2) (Fig. 3). Purified LDH and MDH enzymes stereo-specifically reduced α-KG to L-2HG using NADH. Acidic conditions increased the levels of the protonated α-KG form, which more stably binds to the catalytic region of LDH-A, thereby increasing L-2HG production30. Hypoxic condition in the core region of many solid tumors enhanced L-2HG production via HIF-1α-dependent induction of LDH-A expression. Similar to D-2HG, L-2HG inhibits the Jumonji family histone lysine demethylase KDM4C, resulting in aberrant accumulation of trimethylated histone 3 lysine 9 (H3K9me3)28.
D- and L-2HG accumulation due to defects in 2-HG dehydrogenases
L-2HG is oxidized and converted to α-KG by L-2HG dehydrogenase in the mitochondria. Similarly, D-2HG is also oxidized by D2HGDH. These 2-HG oxidation reactions are coupled with the reduction of FAD or NAD+31. Oxidation of 2-HG increases the NADH and FADH levels in cells; thus, 2-HG may act as a reservoir of reducing equivalents32,33,34. Consistent with this idea, the NADH/NAD ratio increases when mitochondrial respiration is impaired; limited 2-HG oxidation in hematopoietic stem cells increases the levels of 2-HG, which is accompanied by inhibition of DNA and histone demethylation, leading to increased hypermethylation of both DNA and histones34. Homozygous germline loss-of-function mutations in D2HGDH or L-2HG dehydrogenase (L2HGDH) increase the D-2HG or L-2HG levels, respectively, in both urine and blood35. Systemic L-2HG elevations arising from inherited L2HGDH mutations have been associated with brain tumors36. L2hgdh knockout mice display an increased L-2HG level in the brain with progressive leukoencephalopathy and neurodegeneration37.
Glutamine deprivation and reduction of a-KG in cancer
Hypoxic environments in solid tumors reduce pyruvate production by inducing a less active M2 form of pyruvate kinase (PKM2) in an HIF-1α-dependent manner, such that hypoxia enhances glycolysis but limits oxidative phosphorylation38,39,40. Under hypoxic conditions, glutamine (the most abundant amino acid in blood) is used as the major precursor that can be converted into intermediates of the tricarboxylic acid cycle to support cancer cell survival and proliferation by generating nucleotides, amino acids, and fatty acids. Glutamine is transported into the cytoplasm by transporters such as solute carrier family 1 (neutral amino acid transporter) member 5 (SLC1A5), followed by conversion to glutamate by glutaminase41. Glutamate can be converted to α-KG either by glutamate dehydrogenases, or by aminotransferases42. Therefore, in hypoxic tumor environments, α-KG production depends on glutamine supplied by the blood. The increased glutamine catabolism in tumors may deplete the local supply, leading to glutamine deprivation. This possibility is supported by in vivo findings that the glutamine level decreases to almost undetectable levels in numerous tumors, including hepatomas and sarcomas43,44,45. A recent study using metabolomic analysis comparing paired pancreatic tumor patient samples with benign adjacent tissue specimens revealed that glutamine is one of the most strongly depleted metabolites in tumors44. Glutamine is further depleted in the hypoxic core regions of tumors due to poor blood supply and increased consumption by multiple anaerobic metabolic processes46. Using patient-derived melanoma, Pan et al. showed that glutamine deficiency also contributed to drug resistance and tumor heterogeneity45. They showed that glutamine depletion increased the abundance of methylated histone via α-KG reduction, a substrate of KDMs. Knockdown of KDM6B/JMJD3 (H3K27me3 demethylase) reproduced the effects of low glutamine, suggesting that O2- and α-KG-dependent histone demethylases mediate signals from tumor microenvironments and metabolic status to chromatin.
Several studies showed that hypoxia in tumors contributes to inhibition of histone demethylases via multiple processes: (i) by limiting their substrate O2, and (ii) by reprogramming anaerobic metabolism to deplete α-KG and increase 2-HG, an inhibitor of α-KG. Furthermore, mutations in IDH1 and 2 in several cancers result in high D-2HG levels. Many studies have shown that hypoxia and the oncometabolite increase the total amount of methylated histones in various cancer cells. However, there are many unsolved questions: (i) does this metabolic control of histone methylation vary with single cell status? (ii) Is this metabolic regulation related to tumor-suppressive functions of the cancer driver genes JARID1C/KDM5C and UTX/KDM6A? (iii) What is the molecular mechanism through which changes in histone methylation influence tumor progression? Several studies estimated the Km values for either O2 and α-KG and the IC50 for 2-HG of several histone demethylases and other dioxygenases, demonstrating that each dioxygenase has different sensitivities for substrates and inhibitors (Table 3). Next, we will discuss the tumor-suppressive and oncogenic functions of the KDM5 and KDM6 subfamilies, among which the JARID1C/KDM5C and UTX/KDM6A isoforms have been identified as cancer drivers via TCGA analyses (Table 1)47.
JARID1C/KDM5C, a cancer driver gene, and other isoforms of the KDM5 (H3K4me3, me2, and me1 demethylases) subfamily
TCGA predicted JARID1C/KDM5C as a tumor suppressor, mutations in which drive cancer progression (Table 1). JARID1C/KDM5C is one of the four different isoforms of the JARID1/KDM5 subfamily, which catalyzes demethylation of H3K4me3, me2, and me1 (Tables 1 and 2). Similar to other histone demethylases, JARID1 isoforms have different names and cell type-specific expression patterns. Several studies have revealed the diverse roles of JARID 1A, 1B, and 1D both as tumor suppressors and oncogenes depending on the type of cancer cells. JARID1 proteins contain multiple conserved domains (Fig. 1b). Their catalytic domains are located in two cores of the JmjN and JmjC domains, which are separated by two different domains, namely, an AT-rich interacting domain (ARID) and a plant homeodomain 1 (PHD1) finger48. The interaction between the JmjN and JmjC domains causes a conformational change in the catalytic core of JmjC, thereby activating the enzyme. The ARID domain in JARID1B recognizes the GCACA/C consensus motif, whereas the PHD1 domain binds only unmethylated H3K4; thus, JARID1B persists after demethylation and is involved in repressing target genes49,50,51. This function of PHD1 is unique for JARID1B but not for its other isoforms. Studies on yeast Jhd2 (a JARID1 homolog) showed that the C5HC2 domains and the other C-terminal PHD domain are dispensable for its catalytic activity, suggesting that the C-terminal domains of JARIDs participate in diverse interactions with other proteins to cell type-specific and nonenzymatic functions52,53.
JARID1C/KDM5C as a tumor suppressor
JARID1C was identified as an X-linked mental retardation-related gene that escapes X inactivation during embryogenesis. Thus, females harbor two active copies of JARID1C, whereas males possess a single copy54. Loss of JARID1C in males contributes to sex bias phenotypes55,56. Mutations in JARID1C are associated with short stature, hyperreflexia, and autism57,58. Mutations in JARID1C have been identified in many cancers, such as clear cell renal cell carcinoma (ccRCC), pancreatic cancer, and human papillomavirus (HPV)-associated cancer (Table 1)59,61,62,63,63. In ccRCC, von Hippel Lindau (VHL), a tumor suppressor, is dominantly inactivated. As VHL is an HIF-α subunit-specific E3 ubiquitin ligase, HIF-α is constitutively activated in VHL-inactive ccRCC. Whole-exome sequencing analyses of ccRCC revealed that in addition to VHL, JARID1C/KDM5C, UTX/KDM6A, and SETD2 (H3K36 methyltransferase) were new cancer genes in ccRCC61. In 786-O VHL−/− ccRCC cells, JARID1C knockdown significantly enhanced tumor growth in a xenograft mouse model, showing that JARID1C is a tumor suppressor and that its inactivating mutations in ccRCC promote tumors60. Whole-exome sequencing of human pancreatic cancers identified truncating insertions and deletion mutations in JARID1C63. Genome-wide siRNA screening has identified SMCX (another name of JARID1C) as an E2-dependent regulator of the HPV oncogenes E6 and E762. The HPV protein E2 represses the expression of oncogenic E6 and E7 genes by inactivating p53 and Rb, respectively. JARID1C was shown to repress E6 and E7 by interacting with E2. JARID1C knockdown increased the expression levels of the E6 and E7 oncogenes, suggesting that JARID1C can function as a tumor suppressor in HPV-associated cancers.
The molecular mechanism through which JARID1C suppresses tumor growth remains unclear. One explanation is that JARID1C represses important oncogenic target genes by reducing the active H3K4me3 marks at several promoters, where it is recruited via interactions with DNA-binding proteins64; furthermore, JARID1C is recruited to the promoters of IGFBP1, DNAJC12, COL6A1, and GDF15 by interacting with HIF in 786-O VHL−/− ccRCC cells60, and at the long control region promoter of the viral E6 and E7 genes via interaction with E262. Rondinelli et al. suggested that in addition to transcriptional regulation, the H3K4me3 demethylase activity of JARID1C is also associated with heterochromatin assembly and genomic instability after replication in ccRCC cells65. JARID1C binds to heterochromatic regions where transcription is tightly suppressed. Demethylation of H3K4me3 by JARID1C is required for Suv39H1-mediated methylation of H3K9 for postreplication heterochromatin reconstruction. In ccRCC cells, JARID1C inactivation failed to reduce H3K4me3 at heterochromatic regions, preventing Suv38H1 H3K9 methyl transferase and HP1a from binding to heterochromatin. Under this condition, heterochromatin assembly is hindered, leading to unrestrained expression of aberrant noncoding RNAs, which trigger genomic rearrangement. Therefore, JARID1C prevents tumor progression by guarding genomic stability via maintenance of heterochromatic regions. JARID1C is also required for proper DNA replication by removing tri-methyl groups from H3K4 at replication origins, which drives origin binding by the preinitiation proteins CDC45 and PCNA66. Whether promotion of DNA replication is related to the tumor suppression function of JARID1C is unclear.
Other KDM5 isoforms, JARID1A, 1B, and 1D, as tumor suppressors
JARID1A and 1B are not cancer drivers; however, they are the most intensively studied isoforms. Several studies provided evidence that they are involved in tumor suppression. JARID1A/KDM5A/RBP2/RBBP2 was identified as an Rb-interacting protein, while JARID1B/KDM5B/PLU-1 was identified as a gene that is upregulated by overexpression of c-ErbB267,68. The H3K4 demethylation activities of both JARID1A and 1B mediate Rb’s function of repressing E2F target genes, suggesting that JARID1A and 1B may be involved in tumor suppression by enhancing cellular senescence69. Consistent with this idea, Nijwening et al. demonstrated that JARID1B depletion led to phenotypes similar to those observed after Rb1 knockdown and that JARID1B associates with E2F target genes during cellular senescence70. The genome-wide epigenetic state of leukemic stem cells in MLL-associated leukemia revealed that leukemic stem cells maintained a high level of H3K4me3 and a low level of H3K79me2. JARID1B negatively regulates self-renewal and the oncogenic potential of leukemic stem cells by reducing H3K4me3 abundance71. A genome-wide transcriptional analysis revealed that the JARID1B-LSD1-NuRD1 complex targeted the cell migration- and angiogenesis-related CCL14 chemokine pathway52. Experiments using MDA-MB-231 and MCF7 breast cancer cells showed that JARID1B suppressed cell invasion and angiogenesis by repressing CCL14 expression, suggesting that JARID1B suppresses tumor progression. JARID1D was originally reported as a minor histocompatibility antigen on the Y chromosome72. JARID1D was considered a tumor suppressor because of its downregulation, mutation, or loss in prostate cancer and ccRCC73,74. JARID1D directly interacts with the androgen receptor (AR) to attenuate the transcriptional activation of AR target genes in hormone-sensitive prostate cancers, whereas JARID1D expression is downregulated during the progression to hormone-resistant tumors.
These diverse roles for the JARID1 isoforms in multiple cancer types highlight the roles of JARID1A, 1B, and 1D as tumor suppressers; however, unlike JARID1C, the functions of JARID1A, 1B, and 1D depend on the cellular context, which underscores their potential as cancer biomarkers and drug targets. In contrast, several studies showed that JARID1A and 1B are involved in tumor progression rather than in suppression.
Oncogenic function of JARID1A and 1B
JARID1A is significantly amplified and overexpressed in various human tumors, including breast cancer and head and neck squamous cell carcinoma75,76. Unlike JARID1B, JARID1A knockdown altered H3K4 methylation, resulting in inhibition of proliferation and reduced drug resistance, which is suggestive of the oncogenic roles of JARID1A in breast cancer75. Wang et al. (2009) reported that fusion of the C-terminal PHD domain of JARID1A/PHF23 to nucleoporin-98 generated potent oncoproteins in human leukemia. In the NUP98-PHD fusion protein, the ability of the PHD domain, which recognizes the H3K4me3/2 mark, was essential for leukemogenesis77. Lin et al. showed that loss of JARID1A/RBP2 promotes senescence and inhibits proliferation in a histone demethylase activity- and Rb-dependent manner in mouse embryonic fibroblasts (MEFs). Furthermore, genetic ablation of JARID1A/RBP2 decreased tumor formation in the pancreatic islets of Men1 (a tumor suppressor)-defective mice and in the pituitary glands of Rb(+/−) mice, thereby improving the survival of these mice. These results suggested that JARID1A activates tumorigenesis78. Similarly, JARID1A depletion inhibits proliferation, migration, invasion, and metastasis of lung cancer, suggesting oncogenic roles of JARID1A in lung tumorigenesis and progression79.
Roesch et al. have recently demonstrated that even within highly proliferative melanomas, there is a slow-cycling cell subpopulation with a doubling time of >4 weeks80,81. They observed that the expression level of the RBP2-homolog 1/JARID1B/KDM5B/PLU-1 is particularly high in the slow-cycling cell subpopulation. Melanomas are highly heterogeneous tumors, but the biological significance of their different subpopulations is not clear. The same authors characterized the slow-cycling subpopulation by isolating JARID1Bhigh melanoma cells. JARID1B knockdown leads to an initial acceleration of tumor growth, followed by exhaustion. The JARID1Bhigh subpopulation has cancer stem cell-like molecular and functional traits, which are essential for continuous tumor growth. The JARID1Bhigh slow-cycling melanoma cells were characterized by high expression levels of enzymes related to mitochondrial bioenergetics and increased drug resistance, suggesting that JARID1B contributes to the maintenance of cancer stem cell traits. Although the H3K4me3 demethylation activity of JARID1B is required for maintaining cancer stem cell traits, the mechanisms through which the JARID1 enzymatic activity is involved in the cell cycle, drug tolerance, and oxidative metabolism are unclear82,83. Similar to the heterogenic cell populations in melanoma, other types of cancers, such as head and neck cancers, also contain slow-cycling cell populations that show cancer stem cell traits and drug resistance84. Sharma et al. also isolated a drug-tolerant cancer cell population with cancer stem cell properties from an EGFR-mutant non-small cell lung carcinoma (NSCLC)-derived cell line (PC9). They observed that JARID1A is required for maintaining the drug-tolerant cancer cell population85. These studies showed that JARID1A and 1B contribute to intratumor heterogeneity and cancer stem cell traits, which are required for tumor progression.
UTX/KDM6A, a cancer driver gene, and other isoforms in the KDM6 (H3K27me3, me2, and me1 demethylase) subfamily
Two functional isoforms, ubiquitously transcribed tetratricopeptide repeats on the X chromosome (UTX)/KDM6A and JMJD3/KDM6B, have been identified as H3K27me3-, me2- and, me1-specific histone demethylases. Similar to JARID1C, UTX has been identified as an X-linked gene that escapes X chromosome inactivation and is ubiquitously expressed. This escape from X-inactivation contributes to sex bias in many tumors, as a single mutation in UTX is sufficient for its loss of function in males but not in females56. The UTX paralog, Y chromosome-linked UTY, is inactive due to a mutation in the JmjC domain86,87. Both UTX and JMJD3 have C-terminal JmjC domains, which possess catalytic activities. UTX/KDM6A, but not JMJD3/KDM6B, harbors N-terminal six tetratricopeptide repeat (TPR) domains that mediate interactions with other proteins (Fig. 1b). UTX/KDM6A has been identified as a cancer driver gene via TCGA, as its loss or inactivation promotes several malignancies. JMJD3/KDM6B plays important roles in development, tissue regeneration, stem cell biology, inflammation, cellular senescence, and aging. The role of JMJD3 in cancer is poorly understood. Despite sequence similarity in the catalytic domains of JMJD3 and UTX, they have contrasting roles in various cancers, particularly leukemia.
UTX/KDM6A as a tumor suppressor
Genomic analyses of several tumors revealed that loss of UTX occurs in various cancers, including B-cell lymphoma, bladder urothelial carcinoma, head and neck squamous cell carcinoma, pancreatic adenocarcinoma, lung squamous cell carcinoma, and kidney renal papillary cell carcinoma (Table 1)88,89,90,91,92,93. However, the molecular mechanism through which UTX suppresses tumor progression remains unclear. Depending on the cancer type, UTX is involved in tumor suppression, not only via its H3K27me3 demethylase activity but also via interactions with other epigenetic complexes.
Due to its H3K27me3, me2, and me1 demethylase activities, loss of UTX increases the H3K27me3 level at its target genes. Several studies showed that loss of UTX represses a certain subset of genes by increasing the levels of the repressive H3K27me3 mark, ultimately promoting proliferation, clonogenicity, adhesion, and tumorigenicity in myeloid, bladder, and lung transformation94,96,97,97. Upon loss of the UTX demethylases, H3K27 methyl transferases, such as PRC2/EZH2, determine the levels of H3K27me3 on the UTX-EZH2 target genes. Consistently, loss of UTX increases cellular sensitivity to EZH2 inhibition. In UTX-mutated multiple myeloma cells, such altered sensitivity was related to changes in gene expression triggered by a rebalancing of the H3K27me3 levels at specific genes, such as IRF4 and c-MYC94. In UTX/KDM6A-mutated urothelial bladder carcinoma, PRC2/EZH2 target genes, such as IGFBP3, are deregulated due to H3K27me3 enrichment. EZH2 inhibition upregulated UTX-EZH2 target genes and contributed to the EZH2-dependent growth suppression in KDM6A-null bladder tumors in both patient-derived and cell line xenograft models95. PRC2/EZH2 inhibition has emerged as a potential precision therapeutic strategy for genetically defined UTX/KDM6A null cancers that acts by rebalancing the H3K27me3 levels at specific genes98,99,100. EZH2 inhibition also restored the normal gene expression patterns and impaired the proliferation of tumor cells harboring mutations in an H3K4 methyltransferase, MLL3/KMT2C, or a tumor suppressor, BAP1, by rebalancing the H3K27me3 levels at MLL3/BAP1 target genes101. In a range of human tumor types, a cancer-associated mutational hotspot was detected in the PHD domain of MLL3, which mediates association with BAP1. Cancer cells that harbored mutations in the PHD domain of MLL3 or lacked BAP1 showed reduced recruitment of UTX/KDM6A to gene enhancers, with no reduction in the levels of the repressive H3K27me3 mark. Thus, in cancer cells with MLL3 or BAP1 mutations, reduction of H3K27me3 via EZH2 inhibitors can restore the balance of H3K27me3 at enhancers where UTX is not properly recruited. Thus, UTX and PRC2/EZH2 inhibition contribute to enhancer restoration by preventing the deposition of excess H3K27me3102. Several studies showed that PRC2/EZH2 inhibition compensates for the loss of UTX function, suggesting that the catalytic activity of UTX plays an important role in tumor suppression. However, other studies showed that in certain types of cancer, an inactive UTX paralog is required for tumor development in males concomitant with loss of UTY and that the UTX-UTY double-knockout cells exhibited higher proliferation than the single-knockout cells, suggesting demethylase-independent tumor suppressor functions of UTX/KDM6A and UTY103.
Several studies showed that UTX is essential for the establishment of the active enhancer histone marks H3K4me1 and H3K27ac in a demethylase activity-independent manner via recruitment and coupling of an H3K4 methyltransferase complex (named COMPASS) and the histone acetyl transferase p300. In embryonic stem cells, loss of UTX reduced the levels of H3K4me1/H3K27ac at enhancers and transcription. An interaction between UTX and MLL4 (a COMPASS component) enhances p300-dependent H3K27 acetylation not only via UTX-dependent recruitment of p300 but also via MLL4-mediated H3K4 monomethylation. The UTX-MLL4-p300 crosstalk coordinately establishes an active enhancer for transcription104. Similarly, in many cancers and other diseases, enhancer-associated chromatin-modifying components, such as UTX and members of H3K4 methyltransferase complexes, are frequently mutated, leading to enhancer malfunction. In pancreatic cancer, loss of UTX causes aberrant activation of superenhancers that regulate oncogenes (such as Delta-Np63, MYC, and RUNX3) to drive an aggressive subtype of squamous-like pancreatic cancer103. To investigate the tumor-suppressive function of UTX in vivo, Gozdecka et al. developed myeloid-specific Utx knockout mice, most of which (63%) developed AML105. To mimic human leukemia, they expressed the AML1-ETO fusion gene in Utx−/− hematopoietic stem and progenitor cells. Global genomic and proteomic analyses using an Utx-null mouse leukemia model revealed that UTX suppresses myeloid leukemogenesis via noncatalytic functions. Loss of UTX/KDM6A leads to only minor changes in the level H3K27me3 modification, but significant changes in the levels H3K4me1/H3K27ac modifications and alterations in the binding of pioneering enhancer transcription factors, ETS and GATA. These findings suggest that loss of UTX contributes to drive tumor progression via repositioning of histone modification enzymes.
JMJD3, a KDM6 isoform, as a tumor suppressor
JMJD3 can be considered a tumor suppressor based on the finding that it increases the expression levels of the tumor suppressors p16INK4a, p14ARF, and p15INK4B and the activities of the tumor suppressors p53 and Rb. During oncogene-induced senescence (OIS) in human fibroblasts, JMJD3 demethylates the repressive H3K27me3 mark in the INK locus, which contains the genes encoding p16INK4a, p14ARF, and p15INK4B 106. Oncogenes such as B-RAF or RAS induce JMJD3 expression. JMJD3 is recruited by a long noncoding-RNA, ANRIL, to the INK locus107. Cyclin-dependent kinase inhibitors, p16INK4a and p15INK4B, maintain the activity of the Rb protein by inhibiting CDK4/6 (which normally phosphorylates and inactivates the Rb tumor suppressor), whereas p14ARF increases p53 stability by inhibiting the p53-specific E3 ligase, MDM2108. JMJD3 also directly regulates Rb function during cellular senescence. Specific methylation of the K810 residue of Rb facilitates its interaction with CDK4 protein kinase. JMJD3 demethylates the K810 residue of Rb, which prevents CDK4 from phosphorylating the S807 and S811 residues of Rb in senescent cells109. Both JMJD3 and Rb are together linked to the formation of senescence-associated heterochromatin foci (SAHF). SAHF is essential for silencing genes involved in cell cycle progression. JMJD3 and p53 mutually increase their activities. p53 increases the nuclear localization of JMJD3 via protein−protein interaction and recruits JMJD3 to its target genes, such as that encoding p21, where it demethylates the H3K27me3 repressive mark110,111. The demethylase activity of JMJD3 eventually increases the activities of potent tumor suppressors, such as Rb and p53, leading to cellular senescence, a process that blocks tumor initiation, thereby supporting the role of JMJD3 as a tumor suppressor.
Several studies have shown potential tumor-suppressive roles for JMJD3 in different types of cancers. In glioblastoma, JMJD3 overexpression reduced the growth of glioma stem cells by increasing the p16 level and stabilizing p53 in the nucleus112. JMJD3 expression correlates with the p15INK4B expression level in clinical colorectal cancer samples. Low JMJD3 expression correlates with poor prognosis in patients with surgically resected colorectal cancer113. Loss of JMJD3 heterozygosity at chromosome 17p13.1 increased the aggressiveness of pancreatic ductal adenocarcinoma. JMJD3 expression is lower in patient samples of hepatocellular carcinoma, lung adenocarcinoma, non-Hodgkin’s lymphoma, and hematopoietic malignancies compared with that in normal tissues106.
Oncogenic function of JMJD3/KDM6B
JMJD3 is upregulated in various cancers, such as glioblastomas, breast carcinoma, melanoma, renal cell carcinoma, Hodgkin’s lymphoma, and myelodysplastic syndrome114,115,116,117,118,119,119. Furthermore, JMJD3 induces epithelial−mesenchymal transition in clear renal cell carcinoma120. Thus, JMJD3 plays contradictory roles as both a tumor suppressor and an oncogene. Ezh2, a H3K27 methyltransferase that counteracts JMJD3, has been reported to function as a tumor suppressor in myeloid tumors, whereas it acts as an oncogene in other tumors121,122. One simple but convincing explanation for the opposite roles of JMJD3 is that the effects of JMJD3 are cancer cell type-specific. For example, the absence of JMJD3 is permissive for cell division under senescence stimuli in tumors that express senescence effectors such as p16, p53, and Rb, indicating that JMJD3 functions as a tumor suppressor. However, in tumors with dysfunctional senescence or apoptotic effectors, increased JMJD3 activity is beneficial for tumor progression.
Independent of the activation of the INK locus, senescence is associated with changes in lysosomal activities and secretion of inflammatory cytokines, which are collectively termed the “senescence-associated secretory phenotype (SASP)”123,124,125. The SASP stimulates inflammation, aging, proliferation, and recruitment of immune and stem cells. JMJD3 has been considered a key regulator of cytokine production, as it activates NFκB and Stat1/3, which are the main transcription factors required for the induction of inflammatory cytokine expression126,127,128,129. Therefore, an increase in the robustness of the SASP due to JMJD3 reactivation might trigger both cellular senescence and immune responses. Conversely, JMJD3 reactivation might facilitate the recruitment of stem cells to tumors to replace old damaged cells, leading to tumor progression. Thus, JMJD3 can be considered an oncogene as it promotes development of the SASP, which enhances tumor progression.
Conclusion
Among the several isoforms of histone demethylases, JARID1C/KDM5C and UTX/KDM6A have been identified as cancer drivers. Several studies have consistently shown that JARID1C and UTX function as tumor suppressors in various cancers, whereas the other isoforms, JARID1A, 1B, and 1D of the KDM5 subfamily and JMJD3 of the KDM6 subfamily, function as tumor suppressors and oncogenes, depending on the cancer type. The molecular mechanisms through which JARID1C and UTX are involved in tumor suppression still remain unclear. Depending on cancer type, they suppress tumor progression either through their catalytic or noncatalytic function. The observation that the Km values for O2 and α-KG and the IC50 value for 2-HG differ among demethylases suggests that heterogeneous microenvironments and the metabolic status of individual cancer cells within solid tumors affect the heterogeneous epigenetic landscapes among single cells in a population. The localization of histone demethylases in the genome may vary with individual cell status, and metabolic control of histone demethylases may elicit epigenetic changes in a cellular context-dependent manner, which can increase cellular heterogeneity (shown in Fig. 2). Although the catalytic activities of these histone demethylases are reversibly inhibited, the consequences of temporal impairment of their activities are irreversible, as histone demethylases are involved in regulating gene expression, genome stability, and replication. A better understanding of the molecular mechanisms through which mutations in histone demethylases promote epigenetic plasticity to drive cancer progression will guide precision therapeutic strategies for the selection of histone demethylase and methyl-transferase inhibitors.
References
Wu, R., Wang, Z., Zhang, H., Gan, H. & Zhang, Z. H3K9me3 demethylase Kdm4d facilitates the formation of pre-initiative complex and regulates DNA replication. Nucleic Acids Res. 45, 169–180 (2017).
Myllyla, R., Kuutti-Savolainen, E. R. & Kivirikko, K. I. The role of ascorbate in the prolyl hydroxylase reaction. Biochem. Biophys. Res. Commun. 83, 441–448 (1978).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004).
Krieg, A. J. et al. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 30, 344–353 (2010).
Lee, H. Y., Yang, E. G. & Park, H. Hypoxia enhances the expression of prostate-specific antigen by modifying the quantity and catalytic activity of Jumonji C domain-containing histone demethylases. Carcinogenesis 34, 2706–2715 (2013).
Laukka, T., Myllykoski, M., Looper, R. E. & Koivunen, P. Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J. Mol. Biol. 430, 3081–3092 (2018).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
DiNardo, C. D. et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 121, 4917–4924 (2013).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008).
Mardis, E. R. et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 (2009).
Yan, H. et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009).
Ward, P. S. et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 (2010).
Borger, D. R. et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17, 72–79 (2012).
Waitkus, M. S., Diplas, B. H. & Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro. Oncol. 18, 16–26 (2016).
Medeiros, B. C. et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 31, 272–281 (2017).
Yang, H., Ye, D., Guan, K. L. & Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin. Cancer Res. 18, 5562–5571 (2012).
Kang, M. R. et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 125, 353–355 (2009).
Losman, J. A. et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 (2013).
Dang, L. & Su, S. M. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Annu. Rev. Biochem. 86, 305–331 (2017).
Gagné, L. M., Boulay, K., Topisirovic, I., Huot, M. E. & Mallette, F. A. Oncogenic activities of IDH1/2 mutations: from epigenetics to cellular signaling. Trends Cell Biol. 27, 738–752 (2017).
Fan, J. et al. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. Acs Chem. Biol. 10, 510–516 (2015).
Possemato, R. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011).
Terunuma, A. et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 124, 398–412 (2014).
Gross, S. et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 207, 339–344 (2010).
Weller, M., Wick, W. & von Deimling, A. Isocitrate dehydrogenase mutations: a challenge to traditional views on the genesis and malignant progression of gliomas. Glia 59, 1200–1204 (2011).
Chowdhury, R. et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 (2011).
Xu, W. et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011).
Intlekofer, A. M. et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell. Metab. 22, 304–311 (2015).
Oldham, W. M., Clish, C. B., Yang, Y. & Loscalzo, J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 22, 291–303 (2015).
Intlekofer, A. M. et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat. Chem. Biol. 13, 494–500 (2017).
Linster, C. L., Van Schaftingen, E. & Hanson, A. D. Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 9, 72–80 (2013).
Rzem, R., Van Schaftingen, E. & Veiga-da-Cunha, M. The gene mutated in l-2-hydroxyglutaric aciduria encodes l-2-hydroxyglutarate dehydrogenase. Biochimie 88, 113–116 (2006).
Engqvist, M. K., Esser, C., Maier, A., Lercher, M. J. & Maurino, V. G. Mitochondrial 2-hydroxyglutarate metabolism. Mitochondrion 19 Pt B, 275–281 (2014).
Anso, E. et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 19, 614–625 (2017).
Kranendijk, M. et al. IDH2 mutations in patients with D-2-hydroxyglutaric aciduria. Science 330, 336 (2010).
Moroni, I. et al. L-2-hydroxyglutaric aciduria and brain malignant tumors: a predisposing condition? Neurology 62, 1882–1884 (2004).
Ma, S. et al. L2hgdh deficiency accumulates l-2-hydroxyglutarate with progressive leukoencephalopathy and neurodegeneration. Mol Cell Biol 37, https://doi.org/10.1128/MCB.00492-16 (2017).
Luo, W. et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744 (2011).
Wong, N., Ojo, D., Yan, J. & Tang, D. PKM2 contributes to cancer metabolism. Cancer Lett. 356, 184–191 (2015).
Xie, H. & Simon, M. C. Oxygen availability and metabolic reprogramming in cancer. J. Biol. Chem. 292, 16825–16832 (2017).
Altman, B. J., Stine, Z. E. & Dang, C. V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634 (2016).
Moreadith, R. W. & Lehninger, A. L. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+ -dependent malic enzyme. J. Biol. Chem. 259, 6215–6221 (1984).
R, E. et al. Amino acids in epidermal carcinogenesis in mice. Cancer Res. 9, 350–353 (1949).
Kamphorst, J. J. et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015).
Pan, M. et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 18, 1090–1101 (2016).
Wilson, W. R. & Hay, M. P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410 (2011).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385 e318 (2018).
Horton, J. R. et al. Characterization of a linked Jumonji domain of the KDM5/JARID1 family of histone H3 lysine 4 demethylases. J. Biol. Chem. 291, 2631–2646 (2016).
Scibetta, A. G. et al. Functional analysis of the transcription repressor PLU-1/JARID1B. Mol. Cell. Biol. 27, 7220–7235 (2007).
Klein, B. J. et al. The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 6, 325–335 (2014).
Zhang, Y. et al. The PHD1 finger of KDM5B recognizes unmodified H3K4 during the demethylation of histone H3K4me2/3 by KDM5B. Protein Cell 5, 837–850 (2014).
Li, Q. et al. Binding of the JmjC demethylase JARID1B to LSD1/NuRD suppresses angiogenesis and metastasis in breast cancer cells by repressing chemokine CCL14. Cancer Res. 71, 6899–6908 (2011).
Wong, P. P. et al. Histone demethylase KDM5B collaborates with TFAP2C and Myc to repress the cell cycle inhibitorp21(cip) (CDKN1A). Mol. Cell. Biol. 32, 1633–1644 (2012).
Jensen, L. R. et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 76, 227–236 (2005).
Ricketts, C. J. & Linehan, W. M. Gender specific mutation incidence and survival associations in clear cell renal cell carcinoma (CCRCC). PLoS ONE 10, e0140257 (2015).
Dunford, A. et al. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet. 49, 10–16 (2017).
Abidi, F. E. et al. Mutations in JARID1C are associated with X-linked mental retardation, short stature and hyperreflexia. J. Med. Genet. 45, 787–793 (2008).
Vallianatos, C. N. et al. Altered gene-regulatory function of KDM5C by a novel mutation associated with autism and intellectual disability. Front. Mol. Neurosci. 11, 104 (2018).
Dalgliesh, G. L. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463, 360–363 (2010).
Niu, X. et al. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 31, 776–786 (2012).
Varela, I. et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542 (2011).
Smith, J. A. et al. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 107, 3752–3757 (2010).
Wang, L. et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 22, 208–219 (2012).
Outchkourov, N. S. et al. Balancing of histone H3K4 methylation states by the Kdm5c/SMCX histone demethylase modulates promoter and enhancer function. Cell Rep. 3, 1071–1079 (2013).
Rondinelli, B. et al. Histone demethylase JARID1C inactivation triggers genomic instability in sporadic renal cancer. J. Clin. Invest. 125, 4625–4637 (2015).
Rondinelli, B. et al. H3K4me3 demethylation by the histone demethylase KDM5C/JARID1C promotes DNA replication origin firing. Nucleic Acids Res. 43, 2560–2574 (2015).
Defeo-Jones, D. et al. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature 352, 251–254 (1991).
Lu, P. J. et al. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J. Biol. Chem. 274, 15633–15645 (1999).
Chicas, A. et al. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc. Natl. Acad. Sci. USA 109, 8971–8976 (2012).
Nijwening, J. H., Geutjes, E. J., Bernards, R. & Beijersbergen, R. L. The histone demethylase Jarid1b (Kdm5b) is a novel component of the Rb pathway and associates with E2f-target genes in MEFs during senescence. PLoS ONE 6, e25235 (2011).
Wong, S. H. et al. The H3K4-methyl epigenome regulates leukemia stem cell oncogenic potential. Cancer Cell. 28, 198–209 (2015).
Wang, W. et al. Human H-Y: a male-specific histocompatibility antigen derived from the SMCY protein. Science 269, 1588–1590 (1995).
Arseneault, M. et al. Loss of chromosome Y leads to down regulation of KDM5D and KDM6C epigenetic modifiers in clear cell renal cell carcinoma. Sci. Rep. 7, 44876 (2017).
Li, N. et al. JARID1D is a suppressor and prognostic marker of prostate cancer invasion and metastasis. Cancer Res. 76, 831–843 (2016).
Hou, J. et al. Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am. J. Transl. Res. 4, 247–256 (2012).
Li, H. et al. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: a rational approach to preclinical model selection. Mol. Cancer Res. 12, 571–582 (2014).
Wang, G. G. et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 459, 847–851 (2009).
Lin, W. et al. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc. Natl. Acad. Sci. USA 108, 13379–13386 (2011).
Teng, Y. C. et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 73, 4711–4721 (2013).
Roesch, A. et al. Retinoblastoma-binding protein 2-homolog 1: a retinoblastoma-binding protein downregulated in malignant melanomas. Mod. Pathol. 18, 1249–1257 (2005).
Roesch, A. et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 141, 583–594 (2010).
Scott, D. A. et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J. Biol. Chem. 286, 42626–42634 (2011).
Roesch, A. et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23, 811–825 (2013).
Facompre, N. D. et al. JARID1B enables transit between distinct states of the stem-like cell population in oral cancers. Cancer Res. 76, 5538–5549 (2016).
Sharma, S. V. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010).
Lan, F. et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449, 689–694 (2007).
Shpargel, K. B., Sengoku, T., Yokoyama, S. & Magnuson, T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 8, e1002964 (2012).
Liu, J. et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 22, 2315–2327 (2012).
Liao, L., Testa, J. R. & Yang, H. The roles of chromatin-remodelers and epigenetic modifiers in kidney cancer. Cancer Genet 208, 206–214 (2015).
Li, X. et al. UTX is an escape from X-inactivation tumor-suppressor in B cell lymphoma. Nat. Commun. 9, 2720 (2018).
Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322 (2014).
Cancer Genome Atlas Research, N. et al. . Comprehensive molecular characterization of papillary renal-cell carcinoma. N. Engl. J. Med. 374, 135–145 (2016).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Ezponda, T. et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 21, 628–640 (2017).
Ler, L. D. et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 9, eaai8312 (2017).
Wu, Q. et al. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proc. Natl. Acad. Sci. USA 115, E3978–E3986 (2018).
Zheng, L. et al. Utx loss causes myeloid transformation. Leukemia 32, 1458–1465 (2018).
Van der Meulen, J. et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood 125, 13–21 (2015).
Xu, B., Konze, K. D., Jin, J. & Wang, G. G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 43, 698–712 (2015).
Yan, N. et al. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp. Cell Res. 359, 405–414 (2017).
Wang, L. et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat. Med. 24, 758–769 (2018).
Saxena, M. et al. Transcription factor-dependent ‘anti-repressive’ mammalian enhancers exclude H3K27me3 from extended genomic domains. Genes Dev. 31, 2391–2404 (2017).
Andricovich, J. et al. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell. 33, 512–526 e518 (2018).
Wang, S. P. et al. A UTX-MLL4-p300 transcriptional regulatory network coordinately shapes active enhancer landscapes for eliciting transcription. Mol. Cell 67, 308–321 e306 (2017).
Gozdecka, M. et al. UTX-mediated enhancer and chromatin remodeling suppresses myeloid leukemogenesis through noncatalytic inverse regulation of ETS and GATA programs. Nat. Genet. 50, 883–894 (2018).
Agger, K. et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 (2009).
Aguilo, F., Zhou, M. M. & Walsh, M. J. Long noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a expression. Cancer Res. 71, 5365–5369 (2011).
Sherr, C. J. Divorcing ARF andp53: an unsettled case. Nat. Rev. Cancer 6, 663–673 (2006).
Zhao, L. et al. JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death Differ. 22, 1630–1640 (2015).
Akdemir, K. C. et al. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 42, 205–223 (2014).
Williams, K. et al. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS ONE 9, e96545 (2014).
Ene, C. I. et al. Histone demethylase Jumonji D3 (JMJD3) as a tumor suppressor by regulating p53 protein nuclear stabilization. PLoS ONE 7, e51407 (2012).
Pereira, F. et al. KDM6B/JMJD3 histone demethylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 20, 4655–4665 (2011).
Anderton, J. A. et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein−Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 30, 2037–2043 (2011).
Ramadoss, S., Chen, X. & Wang, C. Y. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J. Biol. Chem. 287, 44508–44517 (2012).
Shen, Y. et al. Expression and significance of histone H3K27 demethylases in renal cell carcinoma. Bmc Cancer 12, 470 (2012).
Wei, Y. et al. Global H3K4me3 genome mapping reveals alterations of innate immunity signaling and overexpression of JMJD3 in human myelodysplastic syndrome CD34+ cells. Leukemia 27, 2177–2186 (2013).
Perrigue, P. M. et al. The histone demethylase jumonji coordinates cellular senescence including secretion of neural stem cell-attracting cytokines. Mol. Cancer Res. 13, 636–650 (2015).
Park, W. Y., Hong, B. J., Lee, J., Choi, C. & Kim, M. Y. H3K27 Demethylase JMJD3 employs the NF-kappaB and BMP signaling pathways to modulate the tumor microenvironment and promote melanoma progression and metastasis. Cancer Res. 76, 161–170 (2016).
Li, Q. et al. KDM6B induces epithelial−mesenchymal transition and enhances clear cell renal cell carcinoma metastasis through the activation of SLUG. Int. J. Clin. Exp. Pathol. 8, 6334–6344 (2015).
Ernst, T. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 42, 722–726 (2010).
Hock, H. A complex Polycomb issue: the two faces of EZH2 in cancer. Genes Dev. 26, 751–755 (2012).
Acosta, J. C. et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 (2008).
Coppe, J. P. et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 (2008).
Kuilman, T. et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 (2008).
De Santa, F. et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130, 1083–1094 (2007).
Das, N. D., Jung, K. H. & Chai, Y. G. The role of NF-kappaB and H3K27me3 demethylase, Jmjd3, on the anthrax lethal toxin tolerance of RAW 264.7 cells. PLoS ONE 5, e9913 (2010).
Przanowski, P. et al. The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia. J. Mol. Med. (Berl.) 92, 239–254 (2014).
Na, J. et al. Histone H3K27 demethylase JMJD3 in cooperation with NF-kappaB regulates keratinocyte wound healing. J. Invest. Dermatol. 136, 847–858 (2016).
Markolovic, S. et al. Structure−function relationships of human JmjC oxygenases-demethylases versus hydroxylases. Curr. Opin. Struct. Biol. 41, 62–72 (2016).
Cascella, B. & Mirica, L. M. Kinetic analysis of iron-dependent histone demethylases: alpha-ketoglutarate substrate inhibition and potential relevance to the regulation of histone demethylation in cancer cells. Biochemistry 51, 8699–8701 (2012).
Carbonneau, M. et al. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat. Commun. 7, 12700 (2016).
Lu, C. et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012).
Upadhyay, A. K. et al. An analog of BIX-01294 selectively inhibits a family of histone H3 lysine 9 Jumonji demethylases. J. Mol. Biol. 416, 319–327 (2012).
Walport, L. J. et al. Human UTY(KDM6C) is a male-specific N-methyl lysyl demethylase. J. Biol. Chem. 289, 18302–18313 (2014).
Laukka, T. et al. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 291, 4256–4265 (2016).
Elkashef, S. M. et al. IDH mutation, competitive inhibition of FTO, and RNA methylation. Cancer Cell. 31, 619–620 (2017).
Su, R. et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172, 90–105 e123 (2018).
Koivunen, P., Hirsila, M., Gunzler, V., Kivirikko, K. I. & Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 279, 9899–9904 (2004).
Ehrismann, D. et al. Studies on the activity of the hypoxia-inducible-factor hydroxylases using an oxygen consumption assay. Biochem. J. 401, 227–234 (2007).
Koivunen, P. et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 (2012).
Hirsila, M., Koivunen, P., Gunzler, V., Kivirikko, K. I. & Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 278, 30772–30780 (2003).
Myllyharju, J. & Kivirikko, K. I. Characterization of the iron- and 2-oxoglutarate-binding sites of human prolyl 4-hydroxylase. EMBO J. 16, 1173–1180 (1997).
Wang, P. et al. Oncometabolite D-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 13, 2353–2361 (2015).
Fu, X. et al. 2-Hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell. Metab. 22, 508–515 (2015).
Acknowledgements
We thank Prof. Sung Soo Kim (School of Medicine, Kyung Hee University) for his critical comments and Ramhee Moon for assistance in organizing the references. This work was supported by grants NRF-2016R1A2B4012840 and NRF-2018R1A4A1025985 from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT). Sujin Yim was supported by NRF and the Center for Women In Science, Engineering and Technology (WISET) Grant funded by the Ministry of Science and ICT under the Program for Returners into R&D.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, S., Yim, S. & Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp Mol Med 51, 1–17 (2019). https://doi.org/10.1038/s12276-019-0230-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0230-6
