
Abstract
We identified principal genetic alterations in 97.1% (99/102) of patients with T-acute lymphoblastic leukemia (T-ALL) using integrative genetic analyses, including massive parallel sequencing and multiplex ligation-dependent probe amplification (MLPA). A total of 133 mutations were identified in the following genes in descending order: NOTCH1 (66.7%), FBXW7 (19.6%), PHF6 (15.7%), RUNX1 (12.7%), NRAS (10.8%), and DNMT3A (9.8%). Copy number alterations were most frequently detected in CDKN2B, CDKN2A, and genes on 9p21.3 in T-ALL (45.1%). Gene expression data demonstrated the downregulation of CDKN2B in most cases of T-ALL, whereas CDKN2A downregulation was mainly restricted to deletions. Additional quantitative methylation analysis demonstrated that CDKN2B downregulation stemmed from deletion and hypermethylation. Analysis of 64 patients with CDKN2B hypermethylation indicated an association with an older age of onset and early T cell precursor ALL, which involved very early arrest of T cell differentiation. Genes associated with methylation and myeloid neoplasms, including DNMT3A and NRAS, were more commonly mutated in T-ALL with CDKN2B hypermethylation. In particular, a CDKN2B biallelic deletion or high methylation level (≥45%), the age of onset, and the GATA3 and SH2B3 mutations were factors associated with a poor prognosis. This study clarifies that one of the most important genetic events in T-ALL, namely, CDKN2B downregulation, occurs mechanistically via deletion and hypermethylation. Different susceptible genetic backgrounds exist based on the CDKN2B downregulation mechanism.
Similar content being viewed by others

Introduction
T-acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic malignancy that accounts for approximately 20% of all cases of ALL. T-ALL tends to be more common in adults than in children1,2. Efforts to develop a differential diagnosis of various leukemias have focused on morphology, immunophenotype, and molecular and/or cytogenetic factors. The genetic approach is hindered by the lack of information on T-ALL. Large-scale sequencing studies of the T-ALL genome have identified driver mutations involved in the loss of transcription factors, epigenetic tumor suppressors, cell cycle inhibitors, gain of oncogenes, and chromosomal rearrangements that can result in fusion products3,4,5,6. There is broad consensus that an aberrantly activated NOTCH1 pathway due to NOTCH1 and FBXW7 gene mutations is predominant in both pediatric and adult T-ALL. NOTCH1 and FBXW7 mutations have been reported in approximately 60% and 8–30% of T-ALL patients, respectively3,4. Deletion and hypermethylation of promoter sequences of the 9p21 region, which includes the CDKN2A and CDKN2B genes, are present in >70% of T-ALL cases3,7,8. In addition, alterations in PHF6 (20%), PTEN (20%), WT1 (15%), RUNX1 (10–20%), LEF1 (10–15%), and ETV6 (5–15%) genes are present at a slightly lower frequency3,4,5. However, the mutational impact of these genes on survival is controversial based on different reports, even in the most extensively investigated cases affecting the NOTCH1 gene1,3,4,9. This knowledge is insufficient to enable the genetic classification of T-ALL.
A further hindrance is the paucity of data on the role of genetic changes on risk stratification and prognosis in T-ALL patients compared with B cell ALL patients. A study involving patients with early T cell precursor (ETP)-ALL reported one subset with unique biology that might represent a provisional entity with a characteristic immunophenotypic and genetic profile (myeloid-associated gene mutations, such as mutations in DNMT3A, ETV6, FLT3, GATA3, IDH1, IDH2, JAK3, NRAS/KRAS, and RUNX1); the prognostic significance of the profile remains debated10,11,12.
Nevertheless, understanding the disease pathogenesis and expected biological behavior of T-ALL on the basis of genetic profiles is essential for the successful treatment of T-ALL. Thus we explored the association of the pathogenesis and biology of T-ALL based on genetic aberrations by integrative genetic analyses using massive parallel sequencing and copy number analysis of T-ALL patients. The goal was to identify commonly altered genetic aberrations. The identified genetic alterations were combined with gene expression and methylation data to understand the underlying mechanisms associated with the biological behaviors of T-ALL, such as surface antigen expression, and their prognostic impact.
Materials and methods
Patients
The study included 102 T-ALL patients (69 males and 33 females comprising 42 children and 60 adults; median age, 22 years (range, 2–77 years)) diagnosed between October 2004 and August 2015. The diagnosis of T-ALL was based on morphology, cytochemistry, and immunophenotyping characteristics according to the 2008 World Health Organization classifications. The median follow-up was 20.8 months from initial presentation (range, 0.5–134.7 months). Cytogenetic G-banding analysis was performed by standard methods. We also examined 28 leukemic fusion transcripts using a HemaVision multiplex RT-PCR system (DNA Diagnostic, Risskov, Denmark). ETP-ALL was defined based on the immunophenotype [CD1a(−), CD8(−) and CD5(−/dim)], stem cell expression phenotype (CD34, HLA-DR, CD117), and/or coexpression of myeloid antigens (CD13, CD33, CD11b, CD65)13. All patients were treated with remission induction chemotherapy according to our institutional protocol. Allogeneic hematopoietic stem cell transplantation (HSCT) was performed after consolidation. Autologous HSCT is used when an allogeneic donor is not available. Details of the treatment protocols were previously described14,15,16. Following the Declaration of Helsinki, all patients provided written informed consent for genetic analyses. The study protocol was approved by the Institutional Review Board of Seoul St. Mary’s Hospital, The Catholic University of Korea.
Targeted capture and massive parallel sequencing of commonly mutated genes in T-ALL
To comprehensively characterize the patterns of somatic mutations in T-ALL, massive parallel sequencing of all coding exons of 11 genes (NOTCH1, DNMT3A, FBXW7, RUNX1, PHF6, PTEN, GATA3, KRAS, EZH, NRAS, and SH2B3) previously found to be associated with T-ALL was performed using semiconductor sequencing technology (IonTorrent PGM, Thermo Fisher Scientific, Carlsbad, CA, USA). Custom primers were designed online using the Ion AmpliSeq™ designer software (http://www.ampliseq.com; Thermo Fisher Scientific) to generate a total of 178 primer pairs in a two-pool design with predicted target coverage of 100%. Libraries were templated and enriched using Ion OneTouch™ 2 system and Ion OneTouch™ ES system (Thermo Fisher Scientific), respectively. Sequencing was performed on an Ion 318™ chip (Thermo Fisher Scientific) using the Sequencing Kit 200 (Thermo Fisher Scientific) per the manufacturer’s instructions.
Sequence alignment to the reference human genome (GRCh37/hg19) and base calling were performed using the Torrent Suite software version 4.2 (Thermo Fisher Scientific). Torrent Variant Caller v.4.2-r88446 (Thermo Fisher Scientific) was used for calling variants from PGM mapped reads, and the called variants were annotated using the Ion Reporter software version 4.4 (Thermo Fisher Scientific). Integrative Genomics Viewer facilitated the identification of variants. To minimize the risk of false positives, we required “novel” mutations to be detected at >10% of reads. Variants with a quality score >20, sequencing read depth >100, and variant allele frequency >4% were selected. Variants reported in dbSNP version 138, the 1000 Genomes Project, or the Korean Reference Genome databases and scored as tolerated or benign by Sorting Intolerant From Tolerant or polymorphism phenotyping methods were excluded. Variants were described according to the recommendations of the Human Genome Variation Society. Variant descriptions were assessed by Mutalyzer Name Checker (http://mutalyzer.nl). The mutant allele burden was calculated as the ratio of mutant reads to total reads for the affected base.
Multiplex ligation-dependent probe amplification (MLPA) for the detection of commonly deleted genes in T-ALL
MLPA analysis was performed to detect commonly deleted and/or amplified genes in T-ALL using a SALSA MLPA Kit (P383-A1 T-ALL; MRC Holland, Amsterdam, The Netherlands) according to the manufacturer’s protocol. Amplification products were quantified and identified by capillary electrophoresis on an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Data were analyzed using the GeneMarker software v1.91 (SoftGenetics, State College, PA, USA). Intensity ratios of 0.6–1.4, 0.3–0.6, and 0.3 were considered to represent normal copy number (wild type), monoallelic deletion, and biallelic deletion, respectively.
Measurement of CDKN2A and CDKN2B gene expression
To quantify the CDKN2A and CDKN2B mRNA expression, reverse transcription–quantitative polymerase chain reaction (RT-qPCR) was performed using the TaqMan® gene expression assay (Applied Biosystems) according to the manufacturer’s instructions. RNA was isolated from bone marrow (BM) aspirates of 49 patients and 6 normal controls using the High Pure RNA Isolation Kit (Roche Diagnostics, Mannheim, Germany). Taqman probes for the CDKN2A gene (HS00923894_m1) with transcript p16, CDKN2B gene (HS00793225_m1) with transcript p15, and the endogenous control (glyceraldehyde-3-phosphate dehydrogenase (GAPDH); Hs99999905_m1) were used. All reactions were performed using an ABI 7500 Real-Time PCR system (Applied Biosystems). Gene expression levels were calculated based on the 2−ΔΔCt × 1000 method after normalization to the transcript levels of the GAPDH housekeeping gene.
CDKN2A and CDKN2B promoter methylation analysis
CpG methylation in the promoter regions of the CDKN2A and CDKN2B genes was quantified using pyrosequencing. Bisulfite conversion of genomic DNA was performed using an Epitect Bisulfite Kit (Qiagen). Pyrosequencing was performed using the PyroMark® Gold Q96 Reagent Kit (Qiagen) according to the manufacturer’s instructions. All reactions were performed using the PyroMarkTM Q96 ID (Biotage AB, Uppsala, Sweden). A total of 12 CpG sites (CDKN2A, 5 sites; CDKN2B, 7 sites) were analyzed per sample. For CDKN2A, all assays were performed with commercially available gene-specific PyroMark CpG assays (Qiagen). The primers for the analysis of CDKN2B-specific CpG regions were designed using the Pyrosequencing Assay Design Software (Biotage AB, Supplementary Table S1).
Statistical analyses
Differences in clinical variables according to mutation status were investigated using Fisher’s exact test for categorical variables and Mann–Whitney U test for continuous variables. Correlations were measured by Spearman’s Rho (ρ). Survival functions were calculated using a Kaplan–Meier survival analysis, and the differences in survival curves were compared using two-sided log-rank test. Cox proportional hazard models were used to estimate hazard ratios (HRs) for univariate and multivariate analyses for differences in survival. All statistical analyses were performed using SPSS 12.0.1 for Windows (SPSS, Chicago, IL, USA). A two-sided P value <0.05 indicated statistical significance.
Results
Genetic profile of T-ALL
We performed targeted NGS of 11 genes that were recurrently mutated in T-ALL. This gene panel includes NOTCH13,4, DNMT3A8,17, FBXW73,4, RUNX13,4,8,18, PHF63,4, PTEN3,4, GATA318, KRAS18, EZH23,4,18, NRAS3,4, and SH2B318. These genes are mutated in T-ALL at relatively high frequencies, and six genes (DNMT3A, RUNX1, GATA3, KRAS, EZH2, and SH2B3) are associated with ETP-ALL based on previous reports.
Among all enrolled T-ALL patients, 97.1% (99/102) possessed at least one somatic mutation and/or copy number alteration (Fig. 1a). A total of 133 non-synonymous somatic mutations were identified in 89 patients (87.3%). The number of mutations in each patient varied, ranging from 1 to 5 (median, 2). The majority of the mutations detected were missense mutations (76/133, 57.1%), followed by frameshift (25/133, 18.8%), nonsense (18/133, 13.5%), and in-frame (14/133, 10.5%) mutations. The median mutant allele burden was 40.0% (range, 3.5–98.4%).

a Single-nucleotide variations, small indels, and copy number alterations are presented for the 31 most frequently altered genes. Cases have been grouped by the presence or absence of a CDKN2B deletion, followed by cases with copy number alterations of genes on 9p21.3 (CDKN2A, MTAP, and MLLT3 genes). The patient cohort is further annotated according to the ETP status, gender, and age groups. Each column represents one patient, and each colored box indicates a mutation. b Each colored box represents each genetic alteration (copy number alterations, single-nucleotide variations, and small indels) per gene. Each column corresponds to one sample
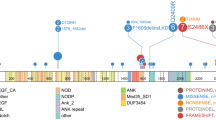
NOTCH1 mutations were the most frequent pathogenetic events in T-ALL patients (68/102, 66.7%). The 48 types of mutations observed comprised 23 missense, 12 in-frame, 8 nonsense, and 5 frameshift mutations. The mutations most commonly occurred in the heterodimerization (HD) domain (34/48, 71%) followed by the proline–glutamate–serine–threonine-rich (PEST) domain (11/48, 23%). Of the 19 patients who had ≥2 NOTCH1 mutations, 10 had mutations in both the PEST and HD domains. The mutant allele burden was widely distributed (range, 3.5–98.4%; median, 36.3%) with two peaks at 10–20% and 40–50% (Fig. 2).

The frequency of the obtained mutations is illustrated by the number of colored circles, which also indicate the effect at the amino acid level. The histogram presents the distribution of the NOTCH1 mutant allele level detected in our T-ALL cases. The schematics are based on the following NCBI protein reference sequences: NOTCH1, NP_060087.3; FBXW7, NP_361014.1; PTEN, NP_000305.3; EZH2, NP_004447.2; DNMT3A, NP_072046.2; KRAS, NP_ 203524.1; NRAS, NP_002515.1; SH2B3, NP_005466.1; GATA3, NP_001002295.1; RUNX1, NP_001745.2; and PHF6, NP_115834.1
The mutational incidence in the other genes was as follows: FBXW7 (20/102, 19.6%), PHF6 (n = 16, 15.7%), RUNX1 (n = 13, 12.7%), NRAS (n = 11, 10.8%), DNMT3A (n = 10, 9.8%), GATA3 (n = 8, 7.8%), PTEN (n = 6, 5.9%), KRAS (n = 6, 5.9%), EZH2 (n = 5, 4.9%), and SH2B3 (n = 3, 2.9%). In total, 11 of the 16 PHF6 mutations and 4 of the 6 PTEN mutations that were detected were frameshift or nonsense mutations, respectively, which could result in a loss-of-function mutation. By contrast, missense substitutions were predominantly observed in the other genes (Fig. 1a).
We used MLPA to identify copy number alterations in genes commonly altered in T-ALL19,20. The P383-A1 probe mix contains 56 probes for 13 different chromosomal regions: 1p33 (SIL-TAL1), 4q25 (LEF1), 6q15 (CASP8AP2), 6q23.3 (MYB), 7q36.1 (EZH2), 9p21.3 (CDKN2A, CDKN2B, MTAP, MLLT3), 9q34.1 (NUP214-ABL1), 10q23.31 (PTEN), 11p15.4 (LMO1), 11p13 (LMO2, CD44, SLC1A2, RAG2), 17q11.2 (NF1, SUZ12), 18p11.21 (PTPN2), and Xq26.2 (SH2D1A, PHF6, ARHGEF6). Copy number alterations were detected in 80/102 (78.4%) T-ALL patients and occurred in 23 genes. CDKN2A or CDKN2B deletions were observed in 46 of the 102 patients (45.1%). Among 43 patients with CDKN2A deletions, 41 (95.3%) harbored biallelic deletions. Monoallelic deletions were only detected in two patients. CDKN2B deletions were observed in 38 (37.3%) patients, including 76.3% biallelic (n = 29) and 23.7% monoallelic (n = 9) deletions. Both CDKN2A and CDKN2B deletions were noted in 35 (34.3%) of the T-ALL patients. Copy number alterations (deletion or gain) in other genes were as follows: MTAP (26/102, 25.5%), LEF1 (n = 25, 24.5%), MLLT3 (n = 20, 19.6%), and TAL1 (n = 20, 19.6%). The MTAP and MLLT3 genes on the 9p21.3 locus were co-deleted with CDKN2A/B in 25 and 20 patients, respectively. Interestingly, the number of copy number alterations in a patient was generally inversely correlated with the number of somatic mutations in a patient (Fig. 1b).
Some of the identified genetic lesions closely correlated with the age of onset. Copy number alterations in CDKN2A (66.7% vs. 28.3%, P < 0.001), CDKN2B (54.8% vs. 25.0%, P = 0.003), and MLLT3 (33.3% vs. 10.0%, P = 0.005) were more prevalent among pediatric patients. DNMT3A and KRAS alterations were exclusively found in adults (DNMT3A, 16.7%, P = 0.005; KRAS, 10.0%, P = 0.041; Supplementary Figure S1).
Characteristics of ETP-ALL
A total of 26 (25.5%) patients were classified as having ETP-ALL, which more commonly occurred in adult (≥18 years of age) patients (21/60, 35.0%) compared with pediatric (<18 years of age) patients (5/42, 11.9%) (P = 0.011). The characteristics of ETP-ALL are presented in Table 1. Some genetic profiles differed between ETP and non-ETP-ALL. DNMT3A mutations were more frequently detected in ETP-ALL (23.1% vs. 5.3%, P = 0.016), whereas deletions of the CDKN2A, CDKN2B, and MTAP genes located on 9p21 were more commonly noted in non-ETP-ALL (11.5% vs. 55.3%, 7.7% vs. 47.4%, and 7.7% vs. 31.6%, respectively; P < 0.001, P < 0.001, and P = 0.016, respectively). Three of the four SET-NUP214 T-ALL were included in ETP-ALL (P = 0.036; Supplementary Figure S2).
Downregulation of CDKN2B gene expression in T-ALL
To understand whether these changes corresponded to changes in gene expression, CDKN2A and CDKN2B gene expression was measured by RT-qPCR in 49 available T-ALL samples, and the values were compared with the value attained from normal BM. CDKN2A expression did not differ between T-ALL and normal BM (median, 7.137; range, 0.048–238.943 vs. 31.523; 4.445–56.243, P = 0.307). Among those with T-ALL, patients with a CDKN2A deletion revealed significantly reduced CDKN2A expression compared with those without a deletion (0.258; 0.048–3.176 vs. 41.288; 1.912–238.943, P < 0.001; Fig. 3a, c). CDKN2B expression was significantly reduced in patients with T-ALL (median, 0.221; range, 0.000–4.833) compared with those with normal BM (median, 4.067; range, 2.043–8.721, P < 0.001). Of note, CDKN2B expression was reduced in patients with a CDKN2B deletion (median, 0.208; range, 0.010–4.833) and those without a deletion (median, 0.221; range, 0.000–1.303, P = 0.406) (Fig. 3a, c). The results indicate a downregulation of CDKN2B in most cases of T-ALL. However, CDKN2A downregulation is mainly restricted to cases with a CDKN2A deletion.

a Expression level of CDKN2A and CDKN2B genes in T-ALL cases and normal bone marrow. Comparison among biallelic deletion (DelBi), monoallelic deletion (DelMono), no deletion (No del), and normal bone marrow. b Comparison of methylation levels of CDKN2A and CDKN2B genes between groups in T-ALL cases. c Three-dimensional (3D) scatter plot of the relationships among gene expression level, methylation level, and gene deletion. CDKN2A downregulation is mainly confined to the cases with CDKN2A deletion. CDKN2B gene downregulation is observed in almost all cases of T-ALL. CDKN2B deletion and hypermethylation appear to be mutually exclusive in our cohort. d Frequency and distribution of recurring genetic alterations according to the CDKN2B status (deletion vs. hypermethylation). **P < 0.001; * P < 0.05; CNA copy number alteration
CDKN2B hypermethylation in T-ALL
Given that promoter hypermethylation is an alternative method to inactivate tumor-suppressor genes in a variety of malignancies, we evaluated the methylation status of 93 CDKN2A and CDKN2B genes. The median methylation level of CDKN2B CpG sites was 50.6% (range, 2.2–87.6%), which was increased compared with CDKN2A (median, 3.8%; range, 0.5–44.4%, P < 0.001). The methylation levels of the CDKN2B and CDKN2A genes were positively correlated (Spearman rho = 0.55, P < 0.001). CDKN2A and CDKN2B methylation was dependent on the deletion status of each gene. Patients without a CDKN2A deletion exhibited increased CDKN2A methylation compared to those with a CDKN2A deletion (median, 7.1%; range, 1.1–44.4% vs. 2.6%; 0.5–16.3%, P < 0.001). CDKN2B methylation in patients without CDKN2B deletion was significantly increased compared with those with a deletion (median, 66.3%; range, 5.1–87.6% vs. 13.3%; 2.2–81.5%, P < 0.001). All patients without CDKN2B deletion exhibited a >5% CDKN2B methylation level. The methylation level was highest in patients without CDKN2B deletion, followed by monoallelic and biallelic deletions (median, 27.5%; range, 2.4–81.5% and 11.0%; 2.2–39.3%, respectively; Fig. 3b, c). These methylation and gene expression data indicate that hypermethylation is the main downregulation mechanism of CDKN2B expression, especially in patients without CDKN2B deletion.
Comparison of CDKN2B deletion and hypermethylation
Based on the above results, we grouped patients according to the CDKN2B downregulation mechanism. As expected, no significant difference in CDKN2B expression was noted between the two groups: deletion and hypermethylation (median, 0.2; range, 0.01–4.8 vs. 0.2; 0.0–7.3, P = 0.436, respectively). The clinical characteristics and genetic profiles were influenced by the downregulation mechanism (Table 2). T-ALL with CDKN2B deletion was diagnosed at a younger age than in patients harboring hypermethylation (median, 15.7 vs. 29.5 years, P = 0.001) and was associated with lower platelet counts (median, 52.0 × 1000/μL vs. 79.0 × 1000/μL, P = 0.018) and higher BM blast values (median, 95.0% vs. 91.0%, P = 0.043). Interestingly, most of the ETP-ALL cases (92.3%) were included in the CDKN2B hypermethylation group. DNMT3A (P = 0.012) and NRAS (P = 0.006) mutations exclusively occurred in the CDKN2B hypermethylation group. RUNX1, EZH2, KRAS, SH2B3, and PHF6 mutations tended to occur more frequently in the CDKN2B hypermethylation group. However, the results did not reach statistical significance, perhaps due to the limited number of patients (Fig. 3d, Supplementary Figure S3 and S4).
Prognosis
To assess the clinical relevance of genetic alterations, their prognostic significance was examined in a cohort of 102 T-ALL cases. The estimated mean overall survival (OS) and event-free survival (EFS) were 72.1 months (95% confidence interval (CI), 59.5–84.7) and 58.9 months (46.7–71.0), respectively, among all patients. ETP-ALL did not exhibit different OS and EFS values compared with non-ETP-ALL in our cohort. Among the genetic alterations, RUNX1 mutation (HR, 2.059; CI, 0.995–4.259; P = 0.051) and CDKN2A methylation (HR, 1.038; CI, 1.005–1.072; P = 0.022) were associated with reduced OS by univariate analysis. However, these factors did not reach statistical significance by multivariate analysis (Fig. 4a, b, g). Small cohorts of 8 and 3 patients with GATA3 and SH2B3 mutations, respectively, exhibited shorter OS compared with wild-type patients (GATA3 mutated vs. wild-type: median OS, 13.4 months vs. not reached, P = 0.008; SH2B3 mutated vs. wild-type: median OS, 1.2 months vs. 50.8 months, P < 0.001). NOTCH1 mutations including the involved domain and mutant allele burden did not influence disease prognosis in our cohort. In multivariate analyses, age of onset (HR, 1.019; CI, 1.003–1.036; P = 0.020), GATA3 mutation (HR, 4.658; CI, 1.827–11.875; P = 0.001), and SH2B3 mutation (HR, 5.926; CI, 1.337–26.270; P = 0.019) were identified as independent markers of a poor prognosis (Fig. 4c, d, g). No significant differences in survival were noted between patients with and without CDKN2A gene deletion. No statistically significant difference in OS was observed between patients with CDKN2B gene deletion and hypermethylation.

RUNX1 mutation (a) and CDKN2A hypermethylation (b) were associated with reduced OS in T-ALL patients. Met<15% methylation level <15%, Met≥15% methylation level ≥15%. GATA3 (c) and SH2B3 (d) mutations were independent markers of a poor prognosis for the OS in T-ALL patients. Kaplan–Meier analyses for OS (e) and EFS (f) for patients with T-ALL according to CDKN2B status (deletion and methylation). DelMono monoallelic deletion, DelBi biallelic deletion, Met<45% methylation level <45%, Met≥45% methylation level ≥45%. g Univariate and multivariate analyses of factors associated with overall survival in T-ALL patients
In addition, we designed genetic indices predictive of a poor outcome based on the combination of CDKN2B methylation and deletion status, which are common mechanisms of downregulation. Patients with either biallelic deletion or high methylation level (≥45%) were considered to exhibit a poor prognosis given that their estimated 3-year OS and EFS were shorter than those with monoallelic deletion or low (<45%) methylation (OS: 43.0% vs. 85.2%, P = 0.005; EFS: 35.9% vs. 59.1%, P = 0.042). The majority of patients in the former group were more likely to die during follow-up (44/79, 55.7%) compared with the latter (2/16, 12.5%, P = 0.002). CDKN2B biallelic deletion or high methylation remained an important independent significant prognostic factor after multivariate analysis (HR, 6.358; CI, 1.503–26.888; P = 0.012; Fig. 4e–g). Given that age was a significant prognostic factor, survival analyses were conducted for each of the two age groups. GATA3 and SH2B3 mutations were associated with poor OS in adult patients (P = 0.008 and P < 0.001, respectively). GATA3, SH2B3, NRAS, and FBXW7 mutations were associated with poor EFS in adult patients (P = 0.022, P = 0.001, P = 0.033, and P = 0.018). In pediatric patients, CDKN2A gene deletion was associated with an inferior EFS (P = 0.033).
Discussion
The CDKN2B gene encoding the tumor-suppressor p15INK4B is closely chromosomally linked to CDKN2A and is involved in the cell cycle and senescence. This gene is frequently inactivated by deletion, methylation, or mutation in a wide variety of tumors21,22. In acute leukemias, homozygous deletion of CDKN2B and CDKN2A occurs in approximately 30% of ALL patients23. In T-ALL, deletion occurred in approximately 50% of childhood T-ALL patients, whereas mutation is rarely observed24. Methylation studies demonstrated that the CDKN2B gene but not CDKN2A is frequently inactivated by hypermethylation of the promoter in acute leukemias25,26,27. However, few T-ALL cases were included in these studies, and the functional implications of CDKN2B in T-ALL pathogenesis and prognostic significance remain uncharacterized.
To our knowledge, this study is the first study to demonstrate that CDKN2B downregulation mechanisms are activated in most T-ALL cases. Reduced CDKN2B gene expression was clearly validated by RT-qPCR. One downregulation mechanism involves gene deletion. CDKN2B deletion was observed in 37.3% of our T-ALL cohort. Previous studies have demonstrated that CDKN2B and CDKN2A are inactivated by monoallelic or biallelic deletion in a number of ALL types6,24,28,29. In the current and prior studies28,29, most CDKN2B deletions (35/38, 92.1%) occurred along with CDKN2A deletion. MTAP, MLLT3, and CDKN2A genes were also frequently deleted with CDKN2B. Therefore, these codeletions are thought to result from the genetic instability of chromosome 9p21.3 in T-ALL. However, because the previous study design involved mixed B-ALL with T-ALL23, and the techniques used did not enable discrimination of the gene(s) that were actually deleted28,30,31, it is difficult to gauge the significance of individual gene deletion and to identify which factor contributes more to the pathogenesis and biological behavior. CDKN2A deletion is generally considered to arise as a secondary event cooperatively with initiating driver mutations24,32. Few studies have addressed CKDN2B downregulation in T-ALL. Reduced CDKN2B expression in individuals with the rs77728904 polymorphism has been reported to be associated with an increased risk of B cell precursor ALL33. We used MLPA to easily discriminate the deleted gene. CDKN2A gene expression was reduced along with the deletion, whereas CDKN2B gene expression was low, even in cases with no deletion. This consistent downregulation indicates that CDKN2B plays a more important role in T-ALL pathogenesis compared with CDKN2A.
The current results indicate that another CDKN2B downregulation mechanism is hypermethylation. Hypermethylation of the CpG island in promoter regions is critical for the regulation of gene expression. The comprehensive analysis accounts for different influences of two genes on T-ALL even though they are located close together. Unlike CDKN2A, CDKN2B was hypermethylated in >5% of patients without a CDKN2B deletion. It was of particular interest to compare the characteristics between the two mechanisms. We identified several differences between the hypermethylation and deletion groups. CDKN2B hypermethylation was related to older age and ETP-ALL. Genes associated with the pathogenesis of acute myeloid leukemia, such as DNMT3A, NRAS, RUNX1, EZH2, and KRAS, were more commonly mutated in the hypermethylation group. These distinctive genetic differences are consistent with the findings in ETP-ALL involving very early arrest in T cell differentiation, which is related to hematopoietic stem cells and myeloid progenitors8,18,34,35. DNMT3A mutations are frequently observed in hematological malignancies and occur in a comparable range of lymphoid and myeloid disorders. Furthermore, the mutations are common in individuals with clonal hematopoiesis associated with aging36. DNMT3A mutations lead to localized hypermethylation affecting tumor-suppressor genes, including CDKN2B37; inhibited hematopoietic stem cell differentiation; and obstructed differentiation and leukemic transformation38. Thus the current results support the view that DNMT3A mutation is one of the CDKN2B hypermethylation-susceptible conditions in T-ALL.
We used targeted gene sequencing given that it is an effective method to detect a large number of actionable mutations in driver genes in T-ALL. Non-synonymous somatic mutations were identified in 87.3% of T-ALL cases with a median of two mutations per patient. A total of 133 mutations in 11 genes were identified. The mutations most frequently found were in NOTCH1, FBXW7, PHF6, RUNX1, NRAS, DNMT3A, and GATA3 (reported in descending order of frequency). Of these, NOTCH1 mutations were detected in 66.7% of T-ALL patients; 27.9% of patients had >2 mutations. Although most mutations were clustered in the HD domain (69%) and PEST domain (22%), each mutation was unique. Massive parallel sequencing was used in the study; the approach detects individual mutations and allows for measurement of the mutant burden, which represents the percentage of leukemic cells that harbor a specific mutation. The NOTCH1 mutant burden ranged from 3.5% to 98.4%, and 23/93 (24.7%) mutations revealed a burden of <20%. We investigated the impact of mutant burden of NOTCH1 mutations on disease phenotype and prognosis. The finding was consistent with prior descriptions that some NOTCH1 mutations at diagnosis were present in minor subclones and were lost at relapse, indicating that such mutations are secondary events30. Although we did not identify any different characteristics according to the mutant burden and the location and type of mutations, the knowledge gained about mutations using massive parallel sequencing cannot be underestimated. This and further collaborative efforts will help definitively characterize the impact of mutant burden in T-ALL and clarify the characteristics of NOTCH1 target therapy that will enhance its success.
From the perspective of prognosis, age of onset and mutations in RUNX1, GATA3, and SH2B3 were associated with poorer outcome. CDKN2A methylation also exhibited prognostic significance. Multivariate analyses identified age of onset, GATA3 mutation, and SH2B3 mutation as independent markers of a poor prognosis. A recent study revealed that RUNX1 and GATA3 were recurrent targets for mutation in ETP-ALL, and GATA3 and SH2B3 mutations were associated with poor prognosis in evaluating the relapse risk in T-ALL18. In another study, GATA3 silencing occurred in approximately one third of adult ETP-ALL patients and was associated with GATA3 DNA hypermethylation39. In addition, Perez-Garcia et al. reported that western blot analysis with a SH2B3 C-terminal antibody revealed the complete loss of SH2B3 protein expression in SH2B3 mutant cells compared with cells derived from a SH2B3 wild-type control40. Whether deletion or hypermethylation caused CDKN2B downregulation did not influence the clinical outcome. In most previous studies4,41,42, CDKN2B deletion did not play a prognostic role in T-ALL. Although it has been reported that hypermethylation of CDKN2B was associated with inferior EFS in ALL27, the prognostic impact of the methylation of CDKN2B on T-ALL remains inconclusive. This finding is a predictable result because the CDKN2B downregulation itself is important to T-ALL pathogenesis regardless of the mechanism of downregulation. However, the clinical outcome depended upon the indices affecting the level of downregulation. CDKN2B biallelic deletion or high methylation level (≥45%) was associated with a poorer outcome compared to monoallelic deletion or low methylation (<45%) (OS: 43.0% vs. 85.2%, P = 0.005). Particularly, the newly designed indices remained an important independent significant prognostic factor after multivariate analysis (HR, 6.358, P = 0.012).
This study identified principal genetic alterations in most of the T-ALL patients. In particular, we identified one of the most important genetic events in T-ALL, namely, CDKN2B downregulation, which results from two mechanisms: deletion and hypermethylation. Specific genetic backgrounds are susceptible to each mechanism, such as genetic instability on 9p21.3 and DNMT3A mutation, respectively. An integrative evaluation demonstrated that CDKN2B biallelic deletion or high methylation (≥45%) are factors associated with a poor prognosis, paving the way for trials of hypomethylating agents.
References
Mullighan, C. G. Mutations of NOTCH1, FBXW7, and prognosis in T-lineage acute lymphoblastic leukemia. Haematologica 94, 1338–1340 (2009).
Pui, C. H. & Evans, W. E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 354, 166–178 (2006).
Van Vlierberghe, P. & Ferrando, A. The molecular basis of T cell acute lymphoblastic leukemia. J. Clin. Invest. 122, 3398–3406 (2012).
Karrman, K. & Johansson, B. Pediatric T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 56, 89–116 (2017).
Neumann, M. et al. Mutational spectrum of adult T-ALL. Oncotarget 6, 2754–2766 (2015).
Batova, A. et al. Frequent and selective methylation of p15 and deletion of both p15 and p16 in T-cell acute lymphoblastic leukemia. Cancer Res. 57, 832–836 (1997).
Belver, L. & Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 16, 494–507 (2016).
Grossmann, V. et al. The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer 52, 410–422 (2013).
Trinquand, A. et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-cell acute lymphoblastic leukemia: a Group for Research in Adult Acute Lymphoblastic Leukemia study. J. Clin. Oncol. 31, 4333–4342 (2013).
Arber, D. A. et al. The2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Coustan-Smith, E. et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 10, 147–156 (2009).
Patrick, K. et al. Outcome for children and young people with early T-cell precursor acute lymphoblastic leukaemia treated on a contemporary protocol, UKALL 2003. Br. J. Haematol. 166, 421–424 (2014).
Chopra, A. et al. Immunophenotypic analysis of T-acute lymphoblastic leukemia. A CD5-based ETP-ALL perspective of non-ETP T-ALL. Eur. J. Haematol. 92, 211–218 (2014).
Park, J. et al. Chromosome abnormalities in T-cell acute lymphoblastic leukemia in Korea. Int. J. Hematol. 99, 279–287 (2014).
Lee, S. et al. Donor-specific differences in long-term outcomes of myeloablative transplantation in adults with Philadelphia-negative acute lymphoblastic leukemia. Leukemia 24, 2110–2119 (2010).
Lee, J. W. et al. Treatment of children with acute lymphoblastic leukemia with risk group based intensification and omission of cranial irradiation: A Korean study of 295 patients. Pediatr. Blood Cancer 63, 1966–1973 (2016).
Neumann, M. et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 121, 4749–4752 (2013).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
Kunz, J. B. et al. Pediatric T-cell lymphoblastic leukemia evolves into relapse by clonal selection, acquisition of mutations and promoter hypomethylation. Haematologica 100, 1442–1450 (2015).
Richter-Pechanska, P. et al. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood Cancer J. 7, e523 (2017).
Nobori, T. et al. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 368, 753–756 (1994).
Herman, J. G. et al. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res. 57, 837–841 (1997).
Bertin, R. et al. CDKN2A, CDKN2B, and MTAP gene dosage permits precise characterization of mono- and bi-allelic 9p21 deletions in childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 37, 44–57 (2003).
Sulong, S. et al. A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood 113, 100–107 (2009).
Wong, I. H., Ng, M. H., Huang, D. P. & Lee, J. C. Aberrant p15 promoter methylation in adult and childhood acute leukemias of nearly all morphologic subtypes: potential prognostic implications. Blood 95, 1942–1949 (2000).
Chim, C. S., Tam, C. Y., Liang, R. & Kwong, Y. L. Methylation of p15 and p16 genes in adult acute leukemia: lack of prognostic significance. Cancer 91, 2222–2229 (2001).
Mai, H. et al. Hypermethylation of p15 gene associated with an inferior poor long-term outcome in childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 142, 497–504 (2016).
Kim, M. et al. Homozygous deletion of CDKN2A (p16, p14) and CDKN2B (p15) genes is a poor prognostic factor in adult but not in childhood B-lineage acute lymphoblastic leukemia: a comparative deletion and hypermethylation study. Cancer Genet. Cytogenet. 195, 59–65 (2009).
Mirebeau, D. et al. The prognostic significance of CDKN2A, CDKN2B and MTAP inactivation in B-lineage acute lymphoblastic leukemia of childhood. Results of the EORTC studies 58881 and 58951. Haematologica 91, 881–885 (2006).
Karrman, K. et al. Deep sequencing and SNP array analyses of pediatric T-cell acute lymphoblastic leukemia reveal NOTCH1 mutations in minor subclones and a high incidence of uniparental isodisomies affecting CDKN2A. J. Hematol. Oncol. 8, 42 (2015).
Yu, L. et al. Microarray detection of multiple recurring submicroscopic chromosomal aberrations in pediatric T-cell acute lymphoblastic leukemia. Leukemia 25, 1042–1046 (2011).
Schwab, C. J. et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica 98, 1081–1088 (2013).
Hungate, E. A. et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat. Commun. 7, 10635 (2016).
Schafer, V. et al. EZH2 mutations and promoter hypermethylation in childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 142, 1641–1650 (2016).
Danis, E. et al. Ezh2 controls an early hematopoietic program and growth and survival signaling in early T cell precursor acute lymphoblastic leukemia. Cell Rep. 14, 1953–1965 (2016).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Scourzic, L. et al. DNMT3A(R882H) mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia 30, 1388–1398 (2016).
Zhang, X. et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 48, 1014–1023 (2016).
Gao, J., Chen, Y. H. & Peterson, L. C. GATA family transcriptional factors: emerging suspects in hematologic disorders. Exp. Hematol. Oncol. 4, 28 (2015).
Perez-Garcia, A. et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood 122, 2425–2432 (2013).
Kuchinskaya, E. et al. Interphase fluorescent in situ hybridization deletion analysis of the 9p21 region and prognosis in childhood acute lymphoblastic leukaemia (ALL): results from a prospective analysis of 519 Nordic patients treated according to the NOPHO-ALL 2000 protocol. Br. J. Haematol. 152, 615–622 (2011).
Krieger, D. et al. Frequency and clinical relevance of DNA microsatellite alterations of the CDKN2A/B, ATM and p53 gene loci: a comparison between pediatric precursor T-cell lymphoblastic lymphoma and T-cell lymphoblastic leukemia. Haematologica 95, 158–162 (2010).
Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2015R1A2A2A04002756); a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C3076); and the Research Fund of Seoul St. Mary’s Hospital, The Catholic University of Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jang, W., Park, J., Kwon, A. et al. CDKN2B downregulation and other genetic characteristics in T-acute lymphoblastic leukemia. Exp Mol Med 51, 1–15 (2019). https://doi.org/10.1038/s12276-018-0195-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-018-0195-x
This article is cited by
-
Characteristics of DNMT3a mutation in acute myeloid leukemia and its prognostic implication
Egyptian Journal of Medical Human Genetics (2024)
-
Allogeneic haematopoietic stem cell transplantation might overcome the poor prognosis of adolescents and adult patients with T-lineage acute lymphoblastic leukaemia and CDKN2 deletion
Bone Marrow Transplantation (2024)
-
Analytical and Potential Clinical Performance of Oncomine Myeloid Research Assay for Myeloid Neoplasms
Molecular Diagnosis & Therapy (2020)
