
Abstract
Neural progenitor cell (NPC) transplantation has been shown to be beneficial in the ischemic brain. However, the low survival rate of transplanted NPCs in an ischemic microenvironment limits their therapeutic effects. Tumor necrosis factor-alpha (TNF-α) is one of the proinflammatory cytokines involved in the pathogenesis of various injuries. On the other hand, several studies have shown that TNF-α influences the proliferation, survival, and differentiation of NPCs. Our study investigated the effect of TNF-α pretreatment on human NPCs (hNPCs) under ischemia-related conditions in vitro. hNPCs harvested from fetal brain tissue were pretreated with TNF-α before being subjected to oxygen–glucose deprivation (OGD) to mimic ischemia in vitro. TNF-α pretreatment improved the viability and reduced the apoptosis of hNPCs after OGD. At the molecular level, TNF-α markedly increased the level of NF-κB signaling in hNPCs, and an NF-κB pathway inhibitor, BAY11-7082, completely reversed the protective effects of TNF-α on hNPCs. These results suggest that TNF-α improves hNPC survival by activating the NF-κB pathway. In addition, TNF-α significantly enhanced the expression of cellular inhibitor of apoptosis 2 (cIAP2). Use of a lentivirus-mediated short hairpin RNA targeting cIAP2 mRNA demonstrated that cIAP2 protected against OGD-induced cytotoxicity in hNPCs. Our study of intracellular NF-κB signaling revealed that inhibition of NF-κB activity abolished the TNF-α-mediated upregulation of cIAP2 in hNPCs and blocked TNF-α-induced cytoprotection against OGD. Therefore, this study suggests that TNF-α pretreatment, which protects hNPCs from OGD-induced apoptosis by activating the NF-κB pathway, provides a safe and simple approach to improve the viability of transplanted hNPCs in cerebral ischemia.
Similar content being viewed by others


Introduction
Self-renewing neural progenitor cells (NPCs) have the capacity to differentiate into multiple neural cell lineages, such as neurons, astrocytes, and oligodendrocytes; migrate toward damaged sites in the central nervous system (CNS); and provide various neurotrophic factors1,2,3,4. Recent studies have shown that transplantation of NPCs into the ischemic brain can restore neurological function and induce tissue regeneration via replacement of lost cells, enhancement of axonal plasticity, and promotion of angiogenesis, neurogenesis, and synaptogenesis5,6,7. Despite these promising results, NPC therapy is limited by poor cell survival due to increased apoptosis of transplanted cells in the ischemic microenvironment6,8,9. Therefore, protecting NPCs from apoptosis in the ischemic microenvironment is essential for improving cell therapy. Several strategies have been suggested to improve the efficacy of stem cell therapy, such as genetically modified stem cells overexpressing survival-related or paracrine factors10,11,12,13. However, although transplant outcomes have been improved by genetic modification of stem/progenitor cells, safer and simpler strategies might be needed for future clinical applications. Pretreatment of NPCs with pharmacological agents might elicit significant cytoprotective effects against ischemia14,15,16.
The proinflammatory cytokine tumor necrosis factor-alpha (TNF-α) is induced in response to various pathological processes. In the event of brain injury, TNF-α is released by macrophages and activated microglia within a few minutes and remains in the injured tissues over the following days. Furthermore, TNF-α is upregulated in various neurodegenerative disorders, including Huntington’s disease, Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and stroke, in which it is considered to play a central role in a proinflammatory function17,18. On the other hand, several studies have shown that TNF-α has a neuroprotective function against stroke19 and influences the proliferation, differentiation, and survival of neural stem/progenitor cells in the brain, thereby modulating tissue remodeling and repair after stroke, neurodegeneration, and inflammation20,21,22. At the molecular level, the effects of TNF-α on NPCs depend on binding to TNF receptor and subsequent intracellular signaling through nuclear factor-κB (NF-κB) and the mitogen-activated protein kinase (MAPK) pathways23. The three major types of MAPK, namely, extracellular signal-regulated kinase 1/2 (ERK1/2), p38 MAPK, and c-Jun N-terminal kinase (JNK), regulate gene expression by phosphorylating downstream kinases24. Therefore, we suggest that TNF-α pretreatment might decrease apoptosis and increase survival of NPCs through the NF-κB or MAPK pathways in ischemic conditions.
In this study, we used fetal brain-derived primary human NPCs (hNPCs) to investigate the effects of TNF-α on cell survival under ischemic conditions in vitro. Primary hNPCs are usually derived from fetal CNS tissue at approximately 5–22 weeks of gestation and are expanded as cell clusters in the presence of mitogenic cytokines25,26,27,28,29. The purpose of the present study was to determine whether TNF-α pretreatment improves hNPC survival in ischemic conditions. We also investigated the molecular mechanisms underlying the effects of TNF-α pretreatment on hNPCs.
Materials and methods
hNPC culture
Human fetal brain tissue was obtained from a cadaver at 13 weeks of gestation with full parental consent and the approval of the Research Ethics Committee of Yonsei University College of Medicine, Seoul, Korea (Permit Number: 4-2003-0078)28. All procedures conformed to the guidelines of the National Institutes of Health and the Korean Government. hNPCs isolated from the telencephalon were grown as neurospheres in serum-free growth medium, which consisted of a 1:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F12 (DMEM/F12; Gibco, Carlsbad, CA, USA) containing 1% penicillin/streptomycin (P/S; Gibco), 1% N2 formulation (Gibco), 20 ng/ml fibroblast growth factor-2 (R&D Systems, Minneapolis, MN, USA), 10 ng/ml leukemia inhibitory factor (Sigma, St. Louis, MO, USA) and 8 μg/ml heparin (Sigma). All cultures were maintained in a humidified incubator at 37 °C and 5% CO2 in air, and half of the growth medium was replaced every 3–4 days. Cells were passaged every 7–8 days by dissociation of bulk neurospheres with 0.05% trypsin/EDTA (Gibco) and cryopreserved at each passage in a Good Manufacturing Practice facility. For TNF-α (Peprotech, Rocky Hill, NJ, USA) pretreatment, TNF-α was added to the cell culture medium (final concentration: 0; 5; 10; 20; 50; or 100 ng/ml) for 24 h and then washed out prior to in vitro experiments.
Oxygen–glucose deprivation (OGD)
To mimic cerebral ischemia in vitro, we seeded hNPCs (106 cells/well) in six-well plates, grew them for 48 h in growth medium in a 5% CO2 atmosphere, and then treated them with TNF-α (0, 5, 10, 20, 50, or 100 ng/ml) for 24 h. To induce oxygen–glucose deprivation (OGD), we placed the cells in medium containing 116 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgSO4, 26.2 mM NaHCO3, and 1.0 mM NaH2PO4, and incubated them in an anaerobic chamber (DG250, Don Whitley Scientific Limited, Bridgend, UK). The chamber was flushed with a gas mixture containing 85% N2, 10% H2, and 5% CO230. OGD was carried out for 6 h.
Blockade of NF-κB and p38 MAPK signaling
hNPCs were plated at a density of 1 × 106 cells/well in six-well tissue culture plates and cultured for 48 h. Subsequently, the cells were treated with or without TNF-α, with or without pretreatment with inhibitors of various signaling pathways. An NF-κB-specific inhibitor (BAY11-7082, 10 µM; Calbiochem, San Diego, CA, USA) or p38 MAPK-specific inhibitor (SB203580, 20 µM; Sigma) was added to the hNPC culture medium 2 h before TNF-α treatment to block NF-κB and p38 MAPK signaling.
Cell viability assay
Cell viability was measured using a Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan), which detects cellular dehydrogenase activity in living cells. Cells treated with or without TNF-α and with or without signaling inhibitors were placed in oxygen-depleted, glucose-free medium and then incubated in an anaerobic chamber for 6 h. CCK-8 reagent was added to each well, and the plate was incubated for an additional 4 h at 37 °C. Then, the absorbance of each sample at 450 nm was immediately evaluated using an automatic microplate reader (BioTek Instruments; Winooski, VT, USA). All experiments were performed three times independently with three technical replicates each.
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) release was assayed using a Cytotoxicity Detection Kit PLUS (Roche, Mannheim, Germany), which evaluates cytotoxicity by measuring the amount of LDH released from the cytosol of damaged cells according to the manufacturer’s protocols. LDH activity was measured in the supernatant of cultures exposed to OGD for 6 h and of control cultures to assess cell damage. The data point represents the average value of three independent replicates.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from hNPCs using TRIzol reagent (Molecular Research Center, Cincinnati, OH, USA). RNA was quantified with a spectrophotometer, and 1 μg of isolated RNA was reverse-transcribed into cDNA using a first-strand cDNA synthesis kit (Promega Corp., Madison, WI, USA) following the manufacturer’s protocol. Synthesized cDNA was stored at −20 °C until use as a template in PCR. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed in 384-well plates, with 0.5 μl of cDNA in a 10 μl reaction volume per well, using LightCycler 480 SYBR Green I Master mix (Roche) on a LightCycler 480 System (Roche) as follows: 95 °C for 5 min and 45 cycles of 95 °C for 10 s, 60 °C for 20 s, and 72 °C for 15 s, followed by a melting curve program. Forward and reverse primers were designed to evaluate the expression levels of factors in hNPCs (GAPDH: sense, 5′-CCATGAGAAGTATGACAACAGCC-3′, and antisense, 5′-GGGTGCTAAGCAGTTGGTG-3′; cellular inhibitor of apoptosis 2 [cIAP2]: sense, 5′-CAACATGCCAAGTGGTTTCC-3′, and antisense, 5′-CTCCTGGGCTGTCTGATGTG-3′; and SOD2: sense, 5′-CTGGACAAACCTCAGCCCTA-3′, and antisense, 5′-TGATGGCTTCCAGCAACTC-3′). Relative gene expression was normalized to that of the housekeeping gene GAPDH and analyzed using advanced relative quantification based on the E-method provided by Roche Applied Science.
Western blotting
hNPCs were lysed in tissue protein extraction reagent (Pierce Biotechnology, Rockford, IL, USA) containing protease and phosphatase inhibitors (Sigma), and lysates were centrifuged (16 000 × g, 30 min, 4 °C). The supernatant was collected and stored at −70 °C. The protein concentrations of the lysates were determined using the Pierce BCA protein assay kit (Pierce Biotechnology). The protein was mixed with 2% β-mercaptoethanol in tricine sample buffer (Bio-Rad, Hercules, CA, USA) and denatured by heating to 95 °C. Then, the samples were electrophoresed in 10% Tris-glycine gels and 4–15% Mini-PROTEAN TGX precast gels (Bio-Rad) and transferred to nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin prepared in Tris-buffered saline (TBS) containing 0.1% TWEEN 20 (TBST) and then incubated overnight at 4 °C with the following antibodies: rabbit anti-NF-κB p65 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA); rabbit anti-phospho-NF-κB p65; rabbit anti-phospho-p38 MAPK; rabbit anti-p38 MAPK; rabbit anti-phospho-ERK1/2; rabbit anti-ERK1/2; rabbit anti-phospho-JNK; rabbit anti-JNK; rabbit anti-cleaved caspase-3; mouse anti-caspase-2 (all 1:1000; Cell Signaling Technology, Inc., Danvers, MA, USA); mouse anti-cIAP2 (1:500; BD Biosciences Pharmingen, San Diego, CA, USA); goat anti-lamin B (1:1,000; Santa Cruz Biotechnology); and mouse anti-β-actin (1:2500; Sigma). Next, the immunoblots were washed in TBST, incubated with a horseradish peroxidase-conjugated secondary antibody (1:20 000; Jackson Immunoresearch, West Grove, PA, USA) for 1 h at room temperature, and treated with SuperSignal West Pico or Dura Chemiluminescent substrate (Thermo Scientific, Suwanee, GA, USA). The images were observed with a LAS-4000 mini imager (GE Healthcare, Buckinghamshire, UK) and analyzed with MultiGauge software (Fujifilm, Tokyo, Japan).
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Cells were treated in various ways and then exposed to OGD. After the cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.3% Triton X-100 for 15 min, an In Situ Cell Death Detection Kit FITC (Roche) was used for immunocytochemical detection of apoptosis through the detection of single- and double-stranded DNA breaks by a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay according to the manufacturer’s instructions. Stained cells were analyzed by fluorescence microscopy to detect green fluorescence of apoptotic cells and blue fluorescence of cell nuclei. The proportion of TUNEL-positive cells was determined by analyzing three randomly selected fields per group.
Extraction of cytoplasmic and nuclear proteins
Cytoplasmic and nuclear proteins were extracted using the NE-PER Nuclear and Cytoplasmic Extraction Reagent kit (Pierce Biotechnology) according to the manufacturer’s instructions. Cells were grown in a six-well tissue culture dish. Briefly, the cells were washed with sterile phosphate-buffered saline (PBS) and centrifuged at 500 × g for 5 min at 4 °C. The cell pellet was resuspended in 400 µl of Cytoplasmic Extraction Reagent I solution containing protease inhibitors. The suspension was vortexed for 15 s, incubated on ice for 10 min, supplemented with 22 µl of Cytoplasmic Extraction Reagent II solution, placed on ice for 1 min, and then vortexed again. The suspensions were centrifuged at 16 000 × g for 5 min at 4 °C. The supernatant was collected as the cytoplasmic fraction. The remaining pellet was resuspended in 200 µl of Nuclear Extraction Reagent. The suspensions were vortexed and incubated on ice for 10 min. This process was repeated three times more for a total of 40 min. The resulting nuclear suspension was centrifuged at 16 000 × g for 10 min at 4 °C, and the supernatant was collected as the nuclear fraction.
Detection of nuclear NF-κB p65 in TNF-α-stimulated hNPCs
hNPCs were stimulated with 20 ng/ml TNF-α. After 3 h, cells were fixed with 4% paraformaldehyde for 30 min at room temperature. Blocking and permeabilization were carried out in 10% normal donkey serum (NDS; Jackson ImmunoResearch), 3% bovine serum albumin, and 0.3% Triton X-100 (Sigma) prepared in PBS. Samples were incubated at 4 °C overnight with a primary antibody against NF-κB p65 (1:50; Santa Cruz Biotechnology) diluted in PBS containing 3% NDS. After being washed with PBS, slides were labeled with an Alexa Fluor 594-conjugated donkey anti-rabbit secondary antibody (1:500; Invitrogen, Carlsbad, CA, USA) in PBS containing 3% NDS for 1 h at room temperature. Cells were counterstained using Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector, Burlingame, CA, USA) and visualized by fluorescence microscopy (BX51; Olympus, Center Valley, PA, USA).
Transfection of cIAP2-targeting short hairpin RNA
The pLKO-shcIAP2-A plasmid was a gift from William Hahn (Addgene, Cambridge, MA, USA), and the scrambled short hairpin RNA (shRNA) control plasmid was a gift from David Sabatini (Addgene). 293T cells were cultured in DMEM (Gibco) containing 10% fetal bovine serum (Gibco) and 1% P/S. Subconfluent 293T cells in DMEM were co-transfected with 20 µg of the lentiviral plasmid pLKO-shcIAP2-A or the control scrambled shRNA empty vector, 15 µg of the psPAX2 packaging plasmid, and 6 µg of the pMD2.G envelope plasmid using calcium phosphate. The lentiviral vectors were donated by the Trono laboratory (Addgene). After 6 h, the medium containing the transfection reagent was removed and replaced with fresh DMEM containing 10% fetal bovine serum and 1% P/S. After 2 days, the culture medium containing lentiviral particles was harvested, centrifuged at 3000 r.p.m. for 5 min to remove cell debris, filtered, and concentrated by ultracentrifugation at 26 000 r.p.m. on a sucrose cushion. hNPCs were transduced with lentiviral particles encoding shcIAP2 or scrambled shRNA, and then puromycin (1 μg/ml) was added to kill non-transfected cells. These cells were used for subsequent experiments.
Next-generation sequencing
hNPCs were stimulated with 20 ng/ml TNF-α or left untreated. After 24 h, total RNA was extracted from hNPCs using TRIzol reagent (Molecular Research Center). Preparation of an RNA library using a TruSeq RNA kit (Illumina, San Diego, CA, USA) and sequencing using a HiSeq 2000 (Illumina) according to the manufacturer’s instructions were carried out by Macrogen Co. (Seoul, Korea).
All primary analysis of sequence data was performed with the Bowtie2, TopHat2, and Cufflinks packages31,32,33. Bowtie2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. TopHat is a specially designed tool for fast splice-junction mapping of RNA-Seq reads. It also analyzes the mapping results to identify splice junctions between exons. In our study, sequence reads were mapped to the UCSC hg19 reference genome using Bowtie2 and TopHat. Once the sequence reads were successfully mapped to the reference genome, Cufflinks3 was used for transcript assembly and gene quantification, and then differentially expressed genes (DEGs) were identified using rigorous statistical analysis using Cuffdiff. DEGs were identified by comparing the expression profiles before and after TNF-α treatment.
Induction of experimental focal hypoxic-ischemic brain injury and cell transplantation
All procedures were approved by the Institutional Animal Care and Use Committee at Yonsei University College of Medicine. Unilateral hypoxic-ischemic (HI) brain injury was induced in ICR mice at postnatal day 7 by permanent right common carotid artery occlusion under isoflurane anesthesia, followed by exposure to hypoxia (8% oxygen/92% nitrogen) for 1.5 h. Body temperature was maintained at 37 °C on a warm plate. All mice received the same care and housing and were evenly distributed between control and treatment groups. On day 3 after HI injury (postnatal day 10), the pups were anesthetized, and an incision was made through the dorsal midline of the scalp to inject TNF-α-pretreated hNPCs, non-pretreated hNPCs (10 μl of cell suspension at 8 × 104 cells/μl), or vehicle into the HI-injured site of each mouse brain with a glass micropipette (0.3 mm diameter). Cyclosporine (10 mg/kg; Sandimmune, Novartis Korea, Seoul) was intraperitoneally administered to all groups of mice once per day from the day before transplantation until sacrifice.
Immunohistochemistry
Mice were anesthetized, transcardially perfused, and fixed with cold PBS followed by 4% paraformaldehyde. The brains were removed, cryoprotected in 30% sucrose, and frozen in O.C.T. compound (Sakura Finetek, Torrance, CA, USA). The brains were coronally sliced into 16 μm sections with a freezing cryostat (Leica, Wetzlar, Germany). The sections were mounted on slides and stained with antibodies specific for a human nucleus marker (STEM101; 1:100; Stem Cell Inc., Cambridge, UK) and a human cytoplasmic marker (STEM121; 1:500; Stem Cell Inc.) to trace transplanted hNPCs. Immunofluorescence images were captured using an Olympus BX51 microscope.
Statistical analyses
All statistical analyses were performed using SPSS version 23 (IBM Corp., Armonk, NY, USA). The two groups were compared by the Mann–Whitney U-test. Multiple groups were compared with a repeated measures analysis of variance, followed by Bonferroni post hoc analysis. All data are represented as the means ± standard error of the means. P < 0.05 was considered to indicate a statistically significant difference.
Results
TNF-α promotes survival of hNPCs upon OGD
The viability of hNPCs pretreated with various concentrations of TNF-α was evaluated by a CCK-8 assay after OGD exposure (Fig. 1a). hNPC viability was significantly decreased after OGD exposure (OGD alone, 46.5% ± 2.0%; n = 3), and TNF-α pretreatment mitigated the OGD-induced reduction of hNPC viability (5 ng/ml, 53.4% ± 0.2%; 10 ng/ml, 56.1% ± 0.6%; 20 ng/ml, 75.6% ± 2.9%; 50 ng/ml, 66.7% ± 1.7%; 100 ng/ml, 59.1% ± 0.8%; n = 3/group). Similarly, the LDH assay showed that hNPC cytotoxicity was significantly increased after OGD exposure, and TNF-α pretreatment diminished the OGD-induced cytotoxicity, as indicated by reduced LDH release compared with the untreated group (5 ng/ml, 91.5% ± 0.4%; 10 ng/ml, 84.5% ± 1.7%; 20 ng/ml, 80.8% ± 2.1%; 50 ng/ml, 85.3% ± 0.7%; 100 ng/ml, 91.3% ± 0.6%; n = 3/group; Fig. 1b). TNF-α treatment at concentrations up to 100 ng/ml did not affect hNPC cytotoxicity in normal culture conditions (Supplementary Figure S1). Death of hNPCs pretreated with various concentrations of TNF-α was evaluated by a TUNEL assay after OGD exposure (Fig. 1c, d). The number of apoptotic TUNEL-positive hNPCs was notably increased after OGD exposure. However, the number of apoptotic hNPCs (fluorescein isothiocyanate-positive cells) was significantly reduced in the TNF-α-pretreated group (0 ng/ml, 14.8% ± 2.9%; 5 ng/ml, 6.9% ± 1.9%; 10 ng/ml, 6.3% ± 1.5%; 20 ng/ml, 4.4% ± 0.5%; 50 ng/ml, 4.1% ± 0.4%; 100 ng/ml, 5.2% ± 0.7%; n = 4/group; Fig. 1c, d). The data showed that pretreatment of hNPCs with 20 ng/ml TNF-α markedly increased cell viability and decreased cell cytotoxicity compared with the values in the untreated group after OGD exposure. On the basis of these findings, we pretreated hNPCs with 20 ng/ml TNF-α in further experiments. The pro-survival effect of TNF-α was verified by western blotting with an anti-cleaved caspase-3 antibody (Fig. 1e, f). TNF-α-pretreated hNPCs showed a reduction of OGD-induced caspase-3 activation compared with untreated hNPCs. However, the levels of caspase-2, known as an apoptotic initiator, were not significantly different between TNF-α-pretreated and untreated hNPCs (Supplementary Figure S2). These results demonstrated that TNF-α protects against the OGD-induced reduction of hNPC viability by reducing apoptosis and cytotoxicity. In addition, to evaluate whether TNF-α pretreatment of hNPCs could enhance engraftment and survival of implanted cells within the HI-injured brain in vivo, we transplanted TNF-α-pretreated and untreated hNPCs into the severely HI-injured brain of a neonatal mouse model. Compared with the untreated control group, the TNF-α-pretreated group showed significantly increased numbers of implanted cells positive for the human-specific cell markers STEM121 and STEM101 within the peri-infarct region of the HI-injured brain 3 weeks following transplantation (Supplementary Figure S3). These findings demonstrate therapeutically beneficial effects of TNF-α pretreatment on the survival and engraftment of implanted hNPCs in HI brain injury; such effects would lead to functional improvement.

a–d hNPCs were pretreated with different doses of TNF-α (0, 5, 10, 20, 50, or 100 ng/ml) for 24 h before OGD exposure. a The viability of hNPCs was assessed by a CCK-8 assay after OGD exposure for 6 h. *P < 0.05 and **P < 0.01 vs. OGD-alone group. b Cytotoxicity was measured using LDH assays. **P < 0.01 vs. OGD-alone group. c TUNEL-positive cells were visualized by fluorescence microscopy (arrows). The data shown are representative images. Scale bar, 50 μm. d Quantitative analysis of the TUNEL assay. TUNEL-positive cells were counted in four images of randomly selected fields per group. *P < 0.05 and **P < 0.01 vs. non-treated group. (e) Protein levels of cleaved caspase-3 were determined by western blotting. Cells cultured in the presence or absence of 20 ng/ml TNF-α were incubated for 6 h under OGD conditions. β-Actin was used as an internal control. f Quantification of cleaved caspase-3 (n = 3). *P < 0.05 vs. control. Data are presented as the mean ± SEM
TNF-α activates the NF-κB and p38 MAPK pathways
To investigate the mechanisms by which TNF-α promotes hNPC survival, we assessed several signaling pathways associated with the regulation of cell proliferation, cell survival, and stress, specifically the most common kinases activated by TNF-α: NF-κB; p38 MAPK; ERK1/2; and JNK. Upon treatment of hNPCs with 20 ng/ml TNF-α for various durations, protein expression of p-NF-κB p65 and p-p38 MAPK increased substantially; however, the expression levels of NF-κB p65 and p38 MAPK remained relatively unaltered (Fig. 2a). Similarly, the protein levels of p-JNK, JNK, p-ERK1/2, and ERK1/2 were relatively stable in hNPCs treated with TNF-α for different durations (Fig. 2a, b). These results suggest that the TNF-α-induced increase in hNPC survival upon OGD exposure is mediated by NF-κB or p38 MAPK signaling activation.

a Cell lysates were analyzed by western blotting for p-NF-κB p65, phosphorylated MAPK signaling proteins (p-p38 MAPK, p-ERK1/2, and p-JNK), total NF-κB p65, and total MAPK (p38 MAPK, ERK1/2, and JNK) after TNF-α (20 ng/ml) administration. β-Actin was used as a loading control. b Densitometric quantification of NF-κB p65 and MAPK proteins (n = 3). **P < 0.01 vs. pretreatment group. Data are presented as the mean ± SEM
To study the TNF-α-induced activation of the NF-κB and p38 MAPK pathways, we evaluated the effects of an NF-κB inhibitor (BAY11-7082) and a p38 MAPK inhibitor (SB203580) on hNPCs. Western blot analysis indicated that B22Y11-7082 greatly inhibited TNF-α-induced nuclear translocation of NF-κB p65 (Fig. 3a–d) and that SB203580 significantly inhibited phosphorylation of p38 MAPK (Fig. 3f, g). To analyze the subcellular distribution of NF-κB p65 by immunocytochemistry, we fixed hNPCs and immunostained them with an anti-NF-κB p65 antibody (Fig. 3e). The level of nuclear NF-κB p65 was higher in TNF-α-treated hNPCs (TNF-α) than in untreated hNPCs (control). Moreover, the TNF-α-induced nuclear localization of NF-κB p65 was inhibited in hNPCs incubated with BAY11-7082 prior to TNF-α treatment (TNF-α + BAY). Thus, the subcellular localization of NF-κB p65 correlates with the western blot data.

a TNF-α induces nuclear translocation of NF-κB p65 in hNPCs. A representative western blot analysis of NF-κB p65 in cytosolic and nuclear extracts of hNPCs confirmed the TNF-α-related nuclear translocation of NF-κB. TNF-α treatment increased the nuclear translocation of NF-κB p65 in hNPCs. b Summarized data (n = 3) show the average protein level of NF-κB p65 (normalized to the protein level of β-actin or lamin B) corresponding to the bands shown in the western blots. *P < 0.05 vs. control. c Western blot analysis of cells treated with 20 ng/ml TNF-α for 6 h in the presence or absence of BAY11-7082 using an anti-NF-κB p65 antibody. d Quantification of NF-κB p65 protein (n = 3). e Representative immunofluorescence staining of NF-κB p65 in hNPCs. DAPI staining was performed to indicate cell nuclei. An overlay of the images (NF-κB p65 and DAPI) demonstrates the nuclear translocation of NF-κB p65 in hNPCs treated with (TNF-α) or without (control) TNF-α (20 ng/ml), with BAY11-7082 alone (BAY), or with TNF-α and BAY11-7082 (TNF-α + BAY). The data shown are representative images. Scale bar, 25 μm. f Western blot analysis of cells treated with 20 ng/ml TNF-α for 6 h in the presence or absence of SB203580 using anti-p38 MAPK and anti-p-p38 MAPK antibodies. g Quantification of p38 MAPK and p-p38 MAPK protein levels. **P < 0.01. Data are presented as the mean ± SEM
TNF-α improves the viability of hNPCs upon OGD by activating the NF-κB pathway
To further investigate whether the NF-κB or p38 MAPK pathways are responsible for the reduction in OGD-induced apoptosis, we treated hNPCs with an NF-κB inhibitor (BAY11-7082) or a p38 MAPK inhibitor (SB203580) for 2 h before TNF-α treatment. BAY11-7082 exacerbated OGD-induced apoptosis compared with that of hNPCs pretreated with TNF-α alone (18.2% ± 0.9% vs. 10.6% ± 0.3%, P < 0.01; Fig. 4a, b). Preincubation with BAY11-7082 before TNF-α treatment increased OGD-induced cytotoxicity compared with the level observed on TNF-α treatment alone, as measured by the LDH assay (109.4% ± 0.2% vs. 79.3% ± 0.2%, P < 0.01; n = 3/group; Fig. 4c). The LDH and TUNEL assays revealed that BAY11-7082 significantly reduced TNF-α-mediated cytoprotection after OGD exposure. On the other hand, preincubation with SB203580 before TNF-α treatment did not significantly alter hNPC death (5.5% ± 0.6% vs. 4.1% ± 0.1%; Fig. 4d, e) or cytotoxicity compared with the levels observed upon TNF-α treatment alone (83.4% ± 0.2% vs. 80.9% ± 0.5%; n = 3/group; Fig. 4f). hNPC cytotoxicity was significantly increased after OGD exposure but was not affected by treatment of cells with TNF-α or inhibitors under normal conditions (Fig. 4c, f). Our results reveal that TNF-α improved the viability of hNPCs upon OGD by activating the NF-κB pathway.

a Cells were pretreated with the following compounds before OGD exposure: 20 ng/ml TNF-α; 10 μM BAY11-7082; or vehicle (DMSO). TUNEL-positive cells were visualized by fluorescence microscopy (arrows). The data shown are representative images. Scale bars, 25 μm. b Quantitative analysis of the TUNEL assay. TUNEL-positive cells were counted from four images of randomly selected fields per group. **P < 0.01. c Cytotoxicity was measured using the LDH assay. **P < 0.01. NS not significant. d Cells were pretreated with the following compounds before OGD exposure: 20 ng/ml TNF-α; 20 μM SB203580; or vehicle (DMSO). TUNEL-positive cells were visualized by fluorescence microscopy (arrows). The data shown are representative images. Scale bars, 50 μm. e Quantitative analysis of the TUNEL assay. TUNEL-positive cells were counted from four images of randomly selected fields per group. *P < 0.05. f Cytotoxicity was measured using an LDH assay. **P < 0.01. NS, not significant. Data are presented as the mean ± SEM
TNF-α induces cIAP2 expression via the NF-κB pathway
To determine the influence of TNF-α on hNPC gene expression, we performed next-generation sequencing (NGS) to assess the expression of survival-related genes in stem/progenitor cells. NGS analysis was carried out using hNPCs incubated in the absence or presence of TNF-α (20 ng/ml) for 24 h. Expression of anti-apoptotic and antioxidant genes was higher in TNF-α-pretreated hNPCs than in untreated control cells. cIAP2 and SOD2, which are anti-apoptotic and antioxidant genes, respectively, were significantly upregulated only in TNF-α-pretreated hNPCs (Fig. 5a).

a Heatmap of NGS data analysis of transcripts encoding genes associated with anti-apoptotic and antioxidant biological pathways. FPKM-normalized values of each gene were used and converted to their log10 values to generate the graphs. Heatmaps were generated using the cummeRbund R package. Asterisks in heatmaps denote genes discussed in the text. b qRT-PCR analysis of anti-apoptotic and antioxidant genes showing key factors (such as cIAP2 and SOD2, respectively) whose expression was significantly increased by TNF-α treatment. TNF-α-induced cIAP2 and SOD2 mRNA expression was eliminated by the NF-κB inhibitor BAY11-7082. **P < 0.01 vs. control. The control group consisted of hNPCs not treated with TNF-α or BAY11-7082. c In hNPCs pretreated with the p38 MAPK inhibitor, TNF-α-induced SOD2 mRNA expression was moderately reduced, but TNF-α-induced cIAP2 mRNA expression was not. qRT-PCR data are representative of three independent experiments. *P < 0.05, **P < 0.01 vs. control and #P < 0.05 vs. TNF-α alone. The control group consisted of hNPCs not treated with TNF-α or SB203580. d Protein levels of cIAP2 by the NF-κB inhibitor BAY11-7082 were determined by western blotting. The results are representative of three independent experiments. e Quantification of cIAP2 protein. *P < 0.05, **P < 0.01. f Protein levels of cIAP2 under treatment with the p38 MAPK inhibitor SB203580 were determined by western blotting. The results are representative of three independent experiments. g Quantification of cIAP2 protein. **P < 0.01. Data are presented as the mean ± SEM
To confirm the data obtained by NGS, we analyzed the mRNA expression of cIAP2 and SOD2 by quantitative reverse-transcription polymerase chain reaction in hNPCs cultured in the presence and absence of TNF-α (20 ng/ml) for 24 h. The expression levels of cIAP2 and SOD2 were approximately 450-fold and 120-fold higher, respectively, in TNF-α-pretreated hNPCs than in untreated hNPCs (Fig. 5b, c). Thus, qRT-PCR analysis of hNPCs confirmed the NGS data, showing that TNF-α treatment upregulates the gene expression of cIAP2 and SOD2.
To determine whether the NF-κB and p38 MAPK pathways are involved in regulating TNF-α-induced expression of cIAP2 and SOD2 in hNPCs, we treated hNPCs with an NF-κB inhibitor (BAY11-7082) and a p38 MAPK inhibitor (SB203580) for 2 h before TNF-α treatment. TNF-α-induced cIAP2 and SOD2 mRNA expression was eliminated in hNPCs pretreated with the NF-κB inhibitor (Fig. 5b). By contrast, in hNPCs pretreated with the p38 MAPK inhibitor, TNF-α-induced SOD2 mRNA expression was moderately reduced but TNF-α-induced cIAP2 mRNA expression was not (Fig. 5c). Therefore, NF-κB improved the survival of TNF-α-pretreated hNPCs after OGD via overexpression of cIAP2, suggesting that TNF-α pretreatment enhanced hNPC survival upon OGD via the NF-κB pathway. We also confirmed that TNF-α-induced overexpression of cIAP2 was not induced through the p38 MAPK pathway, but rather through NF-κB signaling at the protein level (Fig. 5d–g). In addition, the significantly increased expression of cIAP2 protein was maintained in TNF-α-pretreated hNPCs under OGD conditions (Supplementary Figure S4).
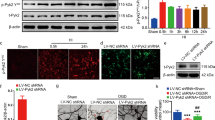
NF-κB-mediated cIAP2 expression is essential for TNF-α-induced protection of hNPCs against OGD
To investigate whether cIAP2, which was induced by TNF-α via NF-κB, affects the apoptosis of hNPCs upon OGD, we combined genetic knockdown of cIAP2 with an shRNA-mediated system to examine its effects in hNPCs. Using this approach, we demonstrated that TNF-α-induced cIAP2 supported TNF-α-mediated protection of hNPCs under OGD. The level of cIAP2 in TNF-α-pretreated lentivirus (LV)-shcIAP2-transfected hNPCs was 36% and 23% of that of TNF-α-pretreated LV-shScramble (shScr)-transfected hNPCs under normal (Supplementary Figure S5) and OGD conditions (Fig. 6a), respectively. Cytotoxicity was comparable between LV-shcIAP2-transfected cells and LV-shScr-transfected cells under normoxia. The level of cytotoxicity upon OGD, as measured by LDH release, was significantly lower in TNF-α-pretreated LV-shScr-transfected cells than in non-pretreated LV-shScr-transfected cells (82.8% ± 0.7% vs. 100% ± 0.7%, P < 0.01; n = 4/group; Fig. 6b). However, the level of cytotoxicity upon OGD was significantly higher in TNF-α-pretreated LV-shcIAP2-transfected cells than in TNF-α-pretreated LV-shScr-transfected cells (96.3% ± 0.7% vs. 82.8% ± 0.7%, P < 0.01; n = 4/group; Fig. 6b). TNF-α-pretreated LV-shScr-transfected cells reduced OGD-induced apoptosis compared with the level in untreated LV-shScr-transfected cells (5.6% ± 1.2% vs. 14.2% ± 1.8%, P < 0.01; Fig. 6c, d). On the other hand, cell death did not significantly differ between TNF-α-pretreated LV-shcIAP2-transfected cells and untreated LV-shcIAP2-transfected cells (16.5% ± 1.2% vs. 15.0% ± 0.6%; Fig. 6c, d). Thus, we demonstrated that TNF-α-mediated cIAP2 expression via the NF-κB pathway is essential to the protection of hNPCs against OGD.

a After TNF-α treatment, cIAP2 expression in LV-shcIAP2-transfected hNPCs under OGD was 23% of the level in LV-shScr-transfected cells. *P < 0.05 vs. TNF-α-pretreated LV-shScr. b After OGD, cytotoxicity, as measured by LDH release, was significantly higher in TNF-α-pretreated LV-shcIAP2-transfected hNPCs than in TNF-α-pretreated LV-shScr-transfected cells. **P < 0.01. NS not significant. c TUNEL-positive cells were visualized by fluorescence microscopy (arrows). The data shown are representative images. Scale bars, 50 μm. d Quantitative analysis of the TUNEL assay. TUNEL-positive cells were counted from four images of randomly selected fields per group. *P < 0.05. Data are presented as the mean ± SEM
Discussion
In the current study, we demonstrated that TNF-α pretreatment protected hNPCs against OGD, which was used to mimic ischemia in vitro; the pretreated group showed substantially increased cell viability and a reduced number of apoptotic cells upon OGD compared with the untreated control. We found that survival-related proteins such as NF-κB p65 and cIAP2 were markedly increased after TNF-α pretreatment, while an NF-κB inhibitor (BAY11-7082) and a cIAP2-targeting shRNA, both of which significantly reduced cIAP2 expression after TNF-α treatment, abolished TNF-α-induced cytoprotection against OGD. Taken together, these data suggest that TNF-α pretreatment protects hNPCs from ischemic damage by upregulating cIAP2 via NF-κB. In addition, the preliminary in vivo experiment showed that compared with the untreated control group, the TNF-α-pretreated group had a significantly increased number of implanted cells within the peri-infarct region of the HI-injured brain following transplantation into HI-injured brain in neonatal mouse, suggesting therapeutically beneficial effects of TNF-α pretreatment on the survival and engraftment of implanted hNPCs in vivo. Thus, preventing apoptosis of stem/progenitor cells without genetic manipulation is a safe and effective approach to enhance cell survival in ischemic conditions. Our chemical pretreatment using TNF-α may provide a safe and simple approach to improve the therapeutic efficacy of hNPC transplantation in ischemic brain injury.
TNF-α is involved in diverse pathophysiological processes regulating the development, survival, and function of cells19,20,] and is induced in response to various neurodegenerative disorders such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, amyotrophic lateral sclerosis, and stroke, in which it is considered to have a proinflammatory function17,34. On the other hand, several studies have shown that TNF-α contributes to neuroprotection and tissue regeneration after stroke, status epilepticus, and inflammation by affecting the survival, differentiation, and proliferation of neural stem/progenitor cells19,20,21,22. TNF-α, which was mainly used at a concentration of 20 ng/ml in our study, significantly affected hNPC viability and cytotoxicity after OGD exposure. Paradoxically, similar concentrations of TNF-α were reported to trigger apoptotic death of neural stem/progenitor cells20,35. Lower doses of TNF-α do not usually induce apoptosis, but higher doses of TNF-α lead to shock-like symptoms23. Most in vitro experiments have used 10–100 ng/ml TNF-α36, and the sensitivity of various cell types to various TNF-α doses has been evaluated under different experimental conditions37,38. These findings show that the biological effects of TNF-α are greatly dose-dependent and differ among the various cell types.
As for the molecular mechanisms underlying the effects of TNF-α treatment on human fetal brain-derived NPCs, the ratios of p-NF-κB p65 to NF-κB p65 and p-p38 MAPK to p38 MAPK were notably higher in TNF-α-treated hNPCs than in untreated control hNPCs, whereas the ratios of p-JNK to JNK and p-ERK1/2 to ERK1/2 were not altered by TNF-α treatment. Signaling via JNKs, also known as stress-activated MAP kinases, is activated by cytokines such as TNF-α or by stress stimuli, including ultraviolet irradiation, redox stress, and osmotic stress39,40; JNK signaling activation plays a crucial role in the control of many cellular events, such as growth and apoptosis41. ERK1/2, which is involved in the regulation of cell proliferation, differentiation, and motility42, can also be activated by TNF-α, and the ERK1/2 pathway is critical for transducing TNF-α signaling to increase matrix metalloproteinase production in trabecular meshwork cells43. The aforementioned reports indicate that TNF-α is able to activate the JNK and ERK1/2 pathways; however, these pathways might not be involved in the effects of TNF-α on hNPCs. The differential activation of these pathways as shown in our study suggests that signaling induction depends on various factors, including the type and status of the cells and the nature of the stimulus 44,45.
The major finding of the current study is that TNF-α protects hNPCs against OGD by activating the NF-κB signaling pathway, and we demonstrated that inhibition of the NF-κB pathway by BAY11-7082 prior to TNF-α treatment significantly decreased cytoprotection induced by TNF-α compared with the degree of cytoprotection in hNPCs treated with TNF-α alone. Previous studies suggest that NF-κB transcription factors play a role in the regulation of apoptosis in diverse cell lines46. The protective role of NF-κB was shown by analysis of p65−/− mice at day 15 of gestation, which showed massive degeneration of the liver by apoptosis47, and by suppression of lymphocyte apoptosis through NF-κB-induced activation of several cell survival genes, including B-cell lymphoma-extra large (Bcl-xl)48. Activation of p38 MAPK, one of the signaling molecules activated by TNF-α in our study, has also been reported to prevent cell death in response to H2O2 treatment via induction of antioxidant genes49. On the other hand, our study showed that although p38 MAPK was activated by TNF-α in hNPCs, inhibition of TNF-α-induced p38 MAPK activation by SB203580 before TNF-α treatment did not significantly alter OGD-induced cell death or cytotoxicity compared with the levels in hNPCs treated with TNF-α alone. These findings indicate that TNF-α supports the role of the NF-κB pathway as an important survival factor in hNPCs.
We observed the influence of TNF-α-induced NF-κB activation on survival-related gene expression in hNPCs by NGS analysis and qRT-PCR. TNF-α pretreatment significantly increased the expression of the survival-related genes cIAP2 and SOD2 in hNPCs, according to NGS analysis. TNF-α-induced mRNA expression of both cIAP2 and SOD2 was lower to a highly significant degree in hNPCs pretreated with the NF-κB inhibitor before TNF-α treatment than in hNPCs treated with TNF-α alone. By contrast, preincubation with a p38 MAPK inhibitor moderately reduced TNF-α-induced SOD2 mRNA expression in hNPCs but did not reduce TNF-α-induced cIAP2 mRNA expression. These data suggest that NF-κB-induced cytoprotection is due to cIAP2 upregulation. NF-κB influences the cytoprotective process by regulating the expression of anti-apoptotic genes (IAPs), which directly inhibit the activity of caspases38. cIAP2, a potent inhibitor of apoptotic death, is a member of the IAP family, which is important for suppressing apoptosis50,51. The IAP family mainly includes cellular IAP1 (cIAP1), cIAP2, X-linked IAP (XIAP), and survivin52,53. TNF-α markedly increased the expression of cIAP2, but not of cIAP1, XIAP, or survivin, in hNPCs. cIAP2-null macrophages are highly sensitive to apoptosis in an LPS-induced proinflammatory environment54, and ischemic preconditioning inhibits caspase-3 activation by preserving cIAP2 in hippocampal neurons55. These findings suggest that cIAP2 was required for TNF-α-mediated cell survival in our study. To demonstrate that cIAP2 is a key factor in this study and protects hNPCs under ischemic conditions, we used a LV-mediated shRNA system to knockdown cIAP2 in hNPCs. We found a pivotal link between TNF-α and cIAP2 in hNPCs: TNF-α treatment increased cIAP2 expression. TNF-α-induced cIAP2 expression was inhibited by an NF-κB inhibitor and cIAP2-specific shRNA, both of which significantly blocked TNF-α-induced cytoprotection under ischemic conditions. In line with our results, TNF-α upregulates cIAP2 expression via activation of the NF-κB pathway in hepatocytes56. Although p38 MAPK, which was one of the signaling molecules activated by TNF-α in our study, was also reported to increase cIAP2 expression via TNF-α in human endothelial cells57, our study showed that inhibition of TNF-α-induced p38 MAPK activation by SB203580 did not affect TNF-α-mediated cIAP2 expression. On the basis of this finding, p38 MAPK is not involved in modulation of cIAP2 expression in hNPCs. Taken together, our results show that TNF-α-induced activation of NF-κB plays a crucial role in cIAP2 expression in hNPCs and that, consequently, TNF-α pretreatment protects hNPCs under ischemic conditions.
In conclusion, we demonstrate that TNF-α pretreatment protects hNPCs against OGD, which was used to mimic ischemic conditions; the pretreatment exerted an important preventive effect against OGD-induced apoptotic processes through cIAP2 upregulation by NF-κB signaling activation. The current study indicates that TNF-α pretreatment is a promising new approach to enhance hNPC survival under ischemic conditions and may lead to further improvements in the therapeutic efficacy of hNPCs by protecting hNPCs transplanted to ischemic injury sites. Although future studies are needed to verify its effect in vivo, the beneficial effect of TNF-α pretreatment on cell survival is a safe and simple approach to improve the viability of transplanted hNPCs in cerebral ischemia.
References
Breunig, J. J., Haydar, T. F. & Rakic, P. Neural stem cells: historical perspective and future prospects. Neuron 70, 614–625 (2011).
Temple, S. The development of neural stem cells. Nature 414, 112–117 (2001).
Kriegstein, A. & Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Annu. Rev. Neurosci. 32, 149–184 (2009).
Lee, I. S., Jung, K., Kim, M. & Park, K. I. Neural stem cells: properties and therapeutic potentials for hypoxic-ischemic brain injury in newborn infants. Pediatr. Int. 52, 855–865 (2010).
Bliss, T. M., Andres, R. H. & Steinberg, G. K. Optimizing the success of cell transplantation therapy for stroke. Neurobiol. Dis. 37, 275–283 (2010).
Andres, R. H. et al. Human neural stem cells enhance structural plasticity and axonal transport in the ischaemic brain. Brain 134, 1777–1789 (2011).
Darsalia, V. et al. Cell number and timing of transplantation determine survival of human neural stem cell grafts in stroke-damaged rat brain. J. Cereb. Blood Flow. Metab. 31, 235–242 (2011).
Hicks, A. U. et al. Transplantation of human embryonic stem cell-derived neural precursor cells and enriched environment after cortical stroke in rats: cell survival and functional recovery. Eur. J. Neurosci. 29, 562–574 (2009).
Nakagomi, N. et al. Endothelial cells support survival, proliferation, and neuronal differentiation of transplanted adult ischemia-induced neural stem/progenitor cells after cerebral infarction. Stem Cells 27, 2185–2195 (2009).
Liu, H. et al. Neuroprotection by PlGF gene-modified human mesenchymal stem cells after cerebral ischaemia. Brain 129, 2734–2745 (2006).
Lee, H. J., Kim, M. K., Kim, H. J. & Kim, S. U. Human neural stem cells genetically modified to overexpress Akt1 provide neuroprotection and functional improvement in mouse stroke m. PLoS ONE 4, e5586 (2009).
Lee, I. S. et al. Neurogenin-2-transduced human neural progenitor cells attenuate neonatal hypoxic-ischemic brain injury. Transl. Res. 183, 121–136.e19 (2017).
Sakata, H. et al. Neural stem cells genetically modified to overexpress cu/zn-superoxide dismutase enhance amelioration of ischemic stroke in mice. Stroke 43, 2423–2429 (2012).
Wang, F. et al. GDNF-pretreatment enhances the survival of neural stem cells following transplantation in a rat model of Parkinson’s disease. Neurosci. Res. 71, 92–98 (2011).
Sakata, H. et al. Interleukin 6-preconditioned neural stem cells reduce ischaemic injury in stroke mice. Brain 135, 3298–3310 (2012).
Rosenblum, S. et al. BDNF pretreatment of human embryonic-derived neural stem cells improves cell survival and functional recovery after transplantation in hypoxic-ischemic stroke. Cell. Transplant. 24, 2449–2461 (2015).
Hallenbeck, J. M. The many faces of tumor necrosis factor in stroke. Nat. Med. 8, 1363–1368 (2002).
Schwabe, R. F. & Brenner, D. A. Mechanisms of liver injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G583–G589 (2006).
Turrin, N. P. & Rivest, S. Tumor necrosis factor alpha but not interleukin 1 beta mediates neuroprotection in response to acute nitric oxide excitotoxicity. J. Neurosci. 26, 143–151 (2006).
Bernardino, L. et al. Tumor necrosis factor-alpha modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells 26, 2361–2371 (2008).
Peng, H. et al. HIV-1-infected and/or immune-activated macrophage-secreted TNF-alpha affects human fetal cortical neural progenitor cell proliferation and differentiation. Glia 56, 903–916 (2008).
Lan, X. et al. TNF-alpha affects human cortical neural progenitor cell differentiation through the autocrine secretion of leukemia inhibitory factor. PLoS ONE 7, e50783 (2012).
Wajant, H., Pfizenmaier, K. & Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 10, 45–65 (2003).
Cuschieri, J. & Maier, R. V. Mitogen-activated protein kinase (MAPK). Crit. Care Med. 33, S417–S419 (2005).
Svendsen, C. N. et al. A new method for the rapid and long term growth of human neural precursor cells. J. Neurosci. Methods 85, 141–152 (1998).
Uchida, N. et al. Direct isolation of human central nervous system stem cells. Proc. Natl Acad. Sci. USA 97, 14720–14725 (2000).
Piper, D. R. et al. Identification and characterization of neuronal precursors and their progeny from human fetal tissue. J. Neurosci. Res. 66, 356–368 (2001).
Kim, H. T. et al. Human neurospheres derived from the fetal central nervous system are regionally and temporally specified but are not committed. Exp. Neurol. 199, 222–235 (2006).
Carpenter, M. K. et al. In vitro expansion of a multipotent population of human neural progenitor cells. Exp. Neurol. 158, 265–278 (1999).
Goldberg, M. P. & Choi, D. W. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J. Neurosci. 13, 3510–3524 (1993).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Greig, N. H. et al. New therapeutic strategies and drug candidates for neurodegenerative diseases: p53 and TNF-alpha inhibitors, and GLP-1 receptor agonists. Ann. N. Y. Acad. Sci. 1035, 290–315 (2004).
Cacci, E., Claasen, J. H. & Kokaia, Z. Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J. Neurosci. Res. 80, 789–797 (2005).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Polunovsky, V. A., Wendt, C. H., Ingbar, D. H., Peterson, M. S. & Bitterman, P. B. Induction of endothelial cell apoptosis by TNF alpha: modulation by inhibitors of protein synthesis. Exp. Cell Res. 214, 584–594 (1994).
Wang, C. Y., Mayo, M. W. & Baldwin, A. S. Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274, 784–787 (1996).
Davis, R. J. Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 (2000).
Ip, Y. T. & Davis, R. J. Signal transduction by the c-Jun N-terminal kinase (JNK)--from inflammation to development. Curr. Opin. Cell Biol. 10, 205–219 (1998).
Tournier, C. et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 (2000).
Kim, E. K. & Choi, E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 (2010).
Alexander, J. P. & Acott, T. S. Involvement of the Erk-MAP kinase pathway in TNFalpha regulation of trabecular matrix metalloproteinases and TIMPs. Invest. Ophthalmol. Vis. Sci. 44, 164–169 (2003).
Deng, Y., Ren, X., Yang, L., Lin, Y. & Wu, X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 115, 61–70 (2003).
Lin, A. Activation of the JNK signaling pathway: breaking the brake on apoptosis. Bioessays 25, 17–24 (2003).
Barkett, M. & Gilmore, T. D. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18, 6910–6924 (1999).
Beg, A. A., Sha, W. C., Bronson, R. T., Ghosh, S. & Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 376, 167–170 (1995).
Khoshnan, A. et al. The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+lymphocytes. J. Immunol. 165, 1743–1754 (2000).
Gutierrez-Uzquiza, A., Arechederra, M., Bragado, P., Aguirre-Ghiso, J. A. & Porras, A. p38alpha mediates cell survival in response to oxidative stress via induction of antioxidant genes: effect on the p70S6K pathway. J. Biol. Chem. 287, 2632–2642 (2012).
Liston, P. et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 379, 349–353 (1996).
Liston, P., Young, S. S., Mackenzie, A. E. & Korneluk, R. G. Life and death decisions: the role of the IAPs in modulating programmed cell death. Apoptosis 2, 423–441 (1997).
Schimmer, A. D. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 64, 7183–7190 (2004).
Deveraux, Q. L. & Reed, J. C. IAP family proteins—suppressors of apoptosis. Genes Dev. 13, 239–252 (1999).
Conte, D. et al. Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol. Cell. Biol. 26, 699–708 (2006).
Tanaka, H. et al. Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J. Neurosci. 24, 2750–2759 (2004).
Schoemaker, M. H. et al. Cytokine regulation of pro- and anti-apoptotic genes in rat hepatocytes: NF-kappaB-regulated inhibitor of apoptosis protein 2 (cIAP2) prevents apoptosis. J. Hepatol. 36, 742–750 (2002).
Furusu, A., Nakayama, K., Xu, Q., Konta, T. & Kitamura, M. MAP kinase-dependent, NF-kappaB-independent regulation of inhibitor of apoptosis protein genes by TNF-alpha. J. Cell Physiol. 210, 703–710 (2007).
Acknowledgements
This study was supported by grants from the National Research Foundation (NRF; 2013M3A9B4076545) and the Korean Health Technology R&D Project (HI14C1564 and HI16C1089). The authors thank Yonsei-Carl Zeiss Advanced Imaging Center at Yonsei University College of Medicine for providing technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, M., Jung, K., Kim, IS. et al. TNF-α induces human neural progenitor cell survival after oxygen–glucose deprivation by activating the NF-κB pathway. Exp Mol Med 50, 1–14 (2018). https://doi.org/10.1038/s12276-018-0033-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-018-0033-1
This article is cited by
-
Astrocytic Extracellular Vesicles Regulated by Microglial Inflammatory Responses Improve Stroke Recovery
Molecular Neurobiology (2024)
-
Effects of cytokines on nuclear factor-kappa B, cell viability, and synaptic connectivity in a human neuronal cell line
Molecular Psychiatry (2021)
-
A Genome-Wide Analysis of the Gene Expression and Alternative Splicing Events in a Whole-Body Hypoxic Preconditioning Mouse Model
Neurochemical Research (2021)
-
Neurovascular Unit as a Source of Ischemic Stroke Biomarkers—Limitations of Experimental Studies and Perspectives for Clinical Application
Translational Stroke Research (2020)
-
Recent Expansions on Cellular Models to Uncover the Scientific Barriers Towards Drug Development for Alzheimer’s Disease
Cellular and Molecular Neurobiology (2019)

{kind=link}
{kind=link}
{kind=link}
{kind=link}