
Abstract
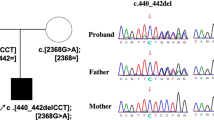
Congenital disorders of glycosylation (CDGs) are inherited metabolic diseases affecting protein and lipid glycosylation. DDOST-CDG is a rare, newly identified type of CDGs, with only one case reported so far. In this study, we report a Chinese patient with a homozygous pathogenic variant in DDOST (c.1187G>A) and who presented with feeding difficulty, lactose intolerance, facial dysmorphism, failure to thrive, strabismus, high myopia, astigmatism, hypotonia, developmental delay and situs inversus totalis. Serum transferrin isoelectrofocusing demonstrated defective glycosylation in our patient. This finding further identifies DDOST as a genetic cause of CDGs and expands the clinical phenotype of DDOST-CDG.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol. 2020;21:729–49.
Starosta RT, Boyer S, Tahata S, Raymond K, Lee HE, Wolfe LA, et al. Liver manifestations in a cohort of 39 patients with congenital disorders of glycosylation: pin-pointing the characteristics of liver injury and proposing recommendations for follow-up. Orphanet J Rare Dis. 2021;16:20.
Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J Inherit Metab Dis. 2011;34:853–8.
Hipgrave Ederveen AL, de Haan N, Baerenfaenger M, Lefeber DJ, Wuhrer M. Dissecting Total Plasma and Protein-Specific Glycosylation Profiles in Congenital Disorders of Glycosylation. Int J Mol Sci. 2020;21:7635.
Schachter H, Freeze HH. Glycosylation diseases: quo vadis? Biochim Biophys Acta. 2009;1792:925–30.
Sparks SE, Krasnewich DM. Congenital disorders of N-linked glycosylation and multiple pathway overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., editors. GeneReviews(®). Seattle (WA): University of Washington, Seattle. Copyright © 1993–2021, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved; 1993.
Mohanty S, Chaudhary BP, Zoetewey D. Structural Insight into the Mechanism of N-Linked Glycosylation by Oligosaccharyltransferase. Biomolecules. 2020;10:624.
Bryant EM, Millichap JJ, Spinelli E, Calhoun JD, Miller C, Giannelli J, et al. Oligosaccharyltransferase complex-congenital disorders of glycosylation: aa novel congenital disorder of glycosylation. Am J Med Genet A. 2020;182:1460–5.
Jones MA, Ng BG, Bhide S, Chin E, Rhodenizer D, He P, et al. DDOST mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am J Hum Genet. 2012;90:363–8.
Molinari F, Foulquier F, Tarpey PS, Morelle W, Boissel S, Teague J, et al. Oligosaccharyltransferase-subunit mutations in nonsyndromic mental retardation. Am J Hum Genet. 2008;82:1150–7.
Shrimal S, Ng BG, Losfeld ME, Gilmore R, Freeze HH. Mutations in STT3A and STT3B cause two congenital disorders of glycosylation. Hum Mol Genet. 2013;22:4638–45.
Blommaert E, Péanne R, Cherepanova NA, Rymen D, Staels F, Jaeken J, et al. Mutations in MAGT1 lead to a glycosylation disorder with a variable phenotype. Proc Natl Acad Sci USA. 2019;116:9865–70.
Conte F, Morava E, Bakar NA, Wortmann SB, Poerink AJ, Grunewald S, et al. Phosphoglucomutase-1 deficiency: Early presentation, metabolic management and detection in neonatal blood spots. Mol Genet Metab. 2020;131:135–46.
Deng H, Xia H, Deng S. Genetic basis of human left-right asymmetry disorders. Expert Rev Mol Med. 2015;16:e19.
Fakhro KA, Choi M, Ware SM, Belmont JW, Towbin JA, Lifton RP, et al. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci USA. 2011;108:2915–20.
Metzler M, Gertz A, Sarkar M, Schachter H, Schrader JW, Marth JD. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J. 1994;13:2056–65.
Boskovski MT, Yuan S, Pedersen NB, Goth CK, Makova S, Clausen H, et al. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature. 2013;504:456–9.
Alam SMD, Tsukamoto Y, Ogawa M, Senoo Y, Ikeda K, Tashima Y, et al. N-glycans on EGF domain-specific O-GlcNAc transferase (EOGT) facilitate EOGT maturation and peripheral endoplasmic reticulum localization. J Biol Chem. 2020;295:8560–74.
Acknowledgements
We thank the patient and his family for their collaboration.
Author information
Authors and Affiliations
Contributions
SP contributed to clinical data collection and writing of the manuscript. XM and HL guided the completion of this manuscript. JG and WX participated in the serum transferrin isoelectrofocusing. BX performed the DNA analysis. All authors approved the submitted version for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pi, S., Gong, J., Xiao, W. et al. The second DDOST-CDG patient with lactose intolerance, developmental delay, and situs inversus totalis. J Hum Genet 67, 103–106 (2022). https://doi.org/10.1038/s10038-021-00974-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-021-00974-2
This article is cited by
-
Functional classification of DDOST variants of uncertain clinical significance in congenital disorders of glycosylation
Scientific Reports (2023)
-
Epidemiology of congenital disorders of glycosylation (CDG)—overview and perspectives
Journal of Rare Diseases (2022)
