
Abstract
Renal cell carcinoma is one of the most common urinary system tumors in adults, it is usually asymptomatic in its early stage and the patients are often diagnosed late. MicroRNA has a higher diagnostic accuracy than traditional markers and may become a new type of early diagnostic biomarker for kidney cancer. Three computational methods and several bioinformatic methods including PPI network, overall survival analysis and enrichment analysis were used to identify the significant differentially expressed miRNAs. Thirteen miRNAs that were significantly differentially expressed in RCC patients were identified, 10 of them have been proved to be associated with kidney cancer in other studies, miR-576, miR-616 and miR-133a-2 are three newly discovered biomarkers of RCC in this study. We found that the target genes of miR-576 (CUL3 and RAC1) are involved in the regulation of multiple cancer-related biological pathways, and the target gene of miR-616 (ASB13 and FBXW2) has been reported to be associated with the development of other cancers. Our findings may have guiding significance for the early diagnosis of renal cell carcinoma.
Similar content being viewed by others

Introduction
Renal cell carcinoma (RCC) is one of the most common urinary system tumors in adults, accounting for ~90% of all malignant renal tumors [1]. In 2017, there were about 63,990 new cases and 14,400 deaths in the United States [2]. The incidence of RCC has been rising at a rate of 2–4% a year [3]. Due to its asymptomatic nature at early stage, many patients are diagnosed in advanced or terminal stage, resulting in distant metastases of tumor cells to bone, lung, brain, and other important organs [4, 5]. The 5-year survival rate of metastatic RCC patients is <10%, while which of early patients is more than 70% [6, 7]. Traditional diagnostic methods have some disadvantages such as low sensitivity, poor specificity and invasiveness [8, 9]. Therefore, a new kind of early diagnostic method is urgently needed.
MicroRNA (miRNA) is a kind of small non-coding RNA, several miRNAs have been shown to play important roles in influencing gene expression at the post-transcriptional levels [10], and can extensively regulate the stability and translation of mRNAs in multiple biological processes such as cell cycle, proliferation, and apoptosis [11, 12].
Faragalla et al. [13] found that miR-21 is upregulated in clear cell RCC (ccRCC) and papillary RCC, and can distinguish between ccRCC and chromophobe RCC (chRCC) with 90% specificity and 83% sensitivity. In the case of unclear morphological features and overlapping histological features among tumor subtypes, miR-21 can be the key factor to improve the accuracy of cancer diagnosis. In addition to early diagnosis, miRNA can also be used as a therapeutic target for cancer. Kelly Gaudelot et al. [14] used anti-miR-21-based silencing strategy to improve the chemotherapy efficacy of ccRCC. PTEN is a potential inhibitor of the PI3K/Akt pathway, by silencing miR-21, PTEN can be upregulated and thus inhibit the PI3K/Akt pathway, and further inhibit the development of RCC.
More and more studies have shown that miRNAs participate in the development of many diseases, and can be a useful biology molecule in the early diagnosis, therapeutic intervention and prognosis evaluation of tumors [15, 16].
In order to find possible biomarkers for the early diagnosis of RCC, we performed a computational identification and evaluation analysis to identify the significant differences in expression data of miRNAs from stage-I renal cancer tissues and normal adjacent tissues. Then several bioinformatic methods were used to construct a biological network of miRNA-mediated target genes, and to found some key target genes and their corresponding miRNAs. The biological function of these target genes were then enriched and analyzed to find the roles they played in the signal transduction pathways that may participate in the development process of RCC.
Materials and methods
Collection and pre-processing of miRNA expression data
Level 3 miRNA-seq isoform quantification data that associated with RCC (including ccRCC and papillary RCC) were downloaded from The Cancer Genome Atlas (TCGA). Then miRNA expression data of clinical stage I (including phase IA and phase IB) were selected, the data from cancer tissues and normal adjacent tissues were also paired up according to their patient IDs for further processing. A total of 50 patients’ clinical data were used in this study, each patient’s clinical data includes 1881 miRNA expression data.
For data preprocessing, firstly, the miRNAs with missing values of more than 20% in all samples were removed. In additional, local least square imputation (LLS impute) method was used to estimate the rest of miRNAs’ missing values [17]. Lastly, z-score method was used to replace abnormal values and standardize the data [18].
Computational identification of significant differentially expressed (SDE) miRNAs
In order to improve the recognition accuracy of SDE miRNAs, Kolmogorov-Smirnov test (K-S test) [19, 20], volcano plot [21] and significance analysis of microarrays (SAM) software [22] were used to identify the SDE miRNAs between the tumor tissue samples and normal adjacent tissue samples. After that, VENN diagram was used to take the intersection of the three results, and heat map was used to visually show the expression pattern of the miRNAs. K-S test is a non-parametric test [19], which can test whether a single population distribution is related to a certain theoretical distribution, and whether there is a significant difference between the two population distributions [20]. It can be divided into single-sample K-S test and paired K-S test. The null hypothesis is that there is no significant difference in the distribution of the two populations from the two independent samples. The null hypothesis was rejected when the significance level ɑ = 0.05. When D ≥ Dcrit (the critical value of D for the significance level ɑ), it indicates that there are differences in the expression of miRNAs between paired samples.
where F1(x) and F2(x) represent the cumulative distribution functions of the miRNA in the tumor tissue samples and normal adjacent tissue samples.
Volcano plot is a type of scatter-plot that is used to quickly identify changes in large data sets composed of replicate data. Our criteria for the data selection are |log2 fold change (FC) | ≥ 1 and p-value < 0.05.
SAM is a method that can identify SDE miRNAs and classify the up (or down)-regulated miRNAs directly in the results [22]. The Excel add-in SAM package (Stanford University, CA) was used, with FDR (false discovery rate) < 0.05 and |log2 FC| ≥ 1 as the cut-off criteria to select SDE miRNAs.
Afterwards, the R package “pheatmap” was used to build a heat map in order to visually display the SDE miRNAs [21].
Obtaining candidate biomarkers
The SDE miRNAs were obtained from the intersection of the above three methods, and the MEDCALC software (MedCalc Software, Mariakerke, Belgium) was used to build the receiver operating characteristic (ROC) curve and calculate the AUC score [23]. In this study, a miRNA with AUC > 0.85 was considered to be a candidate biomarker.
Bioinformatics analysis of candidate biomarkers
miRNA can regulate gene expression at the post-transcriptional level by pairing with the 3′-untranslated region (UTR) of complementary mRNA, inhibiting the translation of mRNA, thus influence the expression level and function of genes involved in the occurrence and development of cancer [24, 25]. To analyze possible target genes of candidate miRNA biomarkers, we used the target prediction algorithms of miRWalk2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/) and miRTarbase 6.0 (http://mirtarbase.mbc.nctu.edu.tw/php/index.php). The target genes predicted by six or more of the 12 online prediction tools of miRWalk, or experimentally validated with strong evidence from miRTarbase were selected as significant target genes. In order to determine whether above candidate genes are associated with RCC, gene-kidney-related enrichment analysis was performed using the enrichment function module of DAVID (https://david.ncifcrf.gov/). Lastly, the GO enrichment analysis and KEGG pathway enrichment analysis were performed using DAVID with the threshold of p < 0.05.
Construction and analysis of protein–protein interaction network
To evaluate the interactive relationships among these target genes associated with RCC, a protein–protein interaction (PPI) network were constructed using STRING (https://string-db.org/cgi/input.pl) online tool (combined score > 0.8), and was visualized using Cytoscape v3.5.1.
Function and pathway enrichment analysis of key sub-networks
The Molecular Complex Detection (MCODE) module of Cytoscape was used to screen the key sub-networks of PPI network with important biological significance in RCC, the cut-off values are as follows: MCODE scores > 30 and number of nodes > 30. After that, DAVID was used to perform the GO enrichment and KEGG pathway analyse of target genes in each key sub-network.
Survival analysis of the hub genes
The CentiScaPe 2.2 module of Cytoscape was used to analyze the topological characteristics of PPI network and search for key nodes and their target genes—hub genes. Then RCC samples from the TCGA database and mRNA z-score data from the cBioPortal database were downloaded, for each hub gene, all samples were divided into the high-expression group and the low-expression group depending on whether the expression is above or below the median in patients with RCC. Then an overall survival (OS) analysis was performed with Graph Pad Prism 6 software, the Kaplan-Meier survival curve visually showed the differences of OS rate between hub genes high-expression group and low-expression group. The log-rank test was used to find the hub genes that significantly affected the OS rate of two groups of patients (p-value < 0.01).
Biological pathway analysis of hub genes
In order to verify the relationship between hub genes and RCC, we used the online enrichment tool of KEGG (http://www.genome.jp/kegg/tool/map_pathway2.html) to perform the pathway enrichment analysis of hub genes. The results can also provide us a better understanding of the mechanism of RCC.
Results
Identification of the candidate biomarkers
K-S test, volcano plot and SAM were used to identify the SDE miRNAs between tumor samples and normal adjacent tissues. 271 SDE miRNAs were identified by K-S test (p-value < 0.05). Hundred and thirteen miRNAs with 58 upregulated and 55 downregulated were identified by volcano plot (|log2 fold-change| ≥ 1, p-value < 0.05). 85 miRNAs with 80 upregulated and 5 downregulated were identified by SAM (|score (d)| ≥ 1.5, |log2 fold-change| ≥ 1, q-value < 0.01). In order to improve the accuracy, the intersecting results from these three methods were selected for later analysis (Fig. 1). Finally, ROC curves and AUC score were used to screen potential biomarkers, 13 miRNAs with AUC > 0.85 were selected as potential biomarkers (Fig. 2).

The VENN diagram of three methods. Fifty-six SDE miRNAs (including 51 upregulated and 5 downregulated miRNAs) were identified by all three methods

ROC curve and AUC score of 13 potential biomarkers. The AUC score of miR-21, miR-616, miR-362, and miR-155 are larger than 0.9, which indicates that they may have a higher diagnostic value
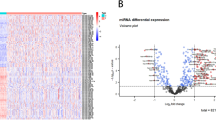
A heat map analysis was applied for these 13 miRNAs to show the different expression pattern between the tumor tissue samples and normal adjacent tissue samples (Fig. 3).

Heat map of 13 potential biomarker miRNAs. The miRNA IDs are displayed on the right of the heat map, and the tissue number are displayed on the bottom of the heat map. Upregulation in metastasis is depicted as red squares and downregulation as green squares
Prediction and enrichment analysis of miRNA-mediated genes
In order to understand how these 13 miRNAs participate in the pathogenesis of RCC, we predicted the potential target genes of these miRNAs. The total number of predicted target genes was 5357, of which 1967 genes were enriched in RCC. Then, the online tools of DAVID were used to conduct the GO and KEGG pathway enrichment analysis on these 1967 genes. Five most enriched GO terms in biological processes are modification-dependent macromolecule catabolic process, modification-dependent protein catabolic process, proteolysis involved in cellular protein catabolic process, cellular protein catabolic process and protein catabolic process. The most enriched GO terms in molecular function and cellular component are acid-amino acid ligase activity and ubiquitin ligase complex. Five most enriched KEGG pathway terms are ubiquitin mediated proteolysis, oocyte meiosis, progesterone-mediated oocyte maturation, Wnt signaling pathway, and RCC.
PPI network analysis of miRNA-mediated genes
In order to understand the interaction between these genes, the online analysis tool of STRING were used to construct a PPI network of the proteins encoded by these genes (combine score > 0.8), this network has 824 nodes and 3618 edges. To find the key nodes and their genes, we further selected 20 nodes using CentiScaPe (degree ≥ 35, betweenness centrality > average) as hub nodes (corresponding coding genes as hub genes). Further, the distinct modules of the whole network and their interacting proteins were identified by the MCODE add-on of Cytoscape software. Among these modules, a key sub-network with 38 nodes were selected (MCODE score > 30, nodes > 30) (Fig. 4), and an enrichment analysis was conducted using DAVID. The genes involved in the sub-networks are mainly associated with proteasome, ECM–receptor interaction, protein digestion and absorption, and focal adhesion.

The key sub-network of the PPI network. The previously selected hub nodes are colored in red, there are totally 10 hub nodes in this sub-network
Survival analysis of the hub gene
In order to find the relationship between the expression of these 20 hub genes and the development of RCC, the hub genes were divided into downregulated group and upregulated group based on whether the expression level of the gene is below or above the median number, after that, the survival data of 499 RCC patients were downloaded and the OS analysis was conducted. The results showed that among the 20 hub genes, 14 were significantly associated with the survival rate of RCC patients (Fig. 5).

Overall survival curve of 14 survival-related hub genes. Black curves represent the downregulated genes and red curves represent the upregulated genes.Among these 14 hub genes, three genes are upregulated, including ANAPC5, RAC1 and NCBP2, while the other 11 genes are downregulated
Integrated miRNA-mRNA-pathway analysis of RCC
Using the online tools of KEGG, a pathway enrichment analysis of the 14 survival-related hub genes was conducted, the results showed that only RNF111 did not participate in any pathway, the remaining 13 hub genes involved in 147 biological pathways, including 40 signal transduction pathways and 107 metabolic pathways. To further demonstrate the regulatory relationship among the potential biomarker miRNAs, target genes and these biological pathways, an interaction network was constructed (Fig. 6).

The interaction network of potential biomarker miRNAs, target mRNAs and biological pathways. The red nodes are miRNAs, the green nods are target genes, the blue nodes are pathways and the key pathways (with five or more genes enriched) are colored in yellow. Nodes with credible interactions were linked with black lines, the interaction among key pathways, genes and miRNAs were colored in red. The 8 key pathways are pathways in cancer, Rap1 signaling pathway, prostate cancer, focal adhesion, proteoglycans in cancer, ubiquitin mediated proteolysis, colorectal cancer and PI3K-Akt signaling pathway
Discussion
Among the 13 potential biomarker miRNAs, 10 miRNAs (miR-142, miR-15, miR-155, miR-185, miR-21, miR-340, miR-362, miR-210, miR-93, and miR-942) were reported to be associated with RCC [26,27,28,29,30,31,32], nine of them (except miR-210) were involved in the regulation of the 147 pathways mentioned in the results section. In these 147 pathways, four of the top 15 pathways have been reported to be involved with the regulatory of RCC and other cancer, they are PI3K/Akt pathway [33], Wnt signaling pathway [34], MAPK/ERK pathway [35], and ubiquitin mediated proteolysis [36]. Although the target genes of miR-210 are not involved in these 147 pathways, there are many reports that miR-210 is associated with the development of renal cancer [29].
Eight miRNAs (miR-21, miR-155, miR-185, miR-362, miR-15a, miR-93, miR-142, and miR-576) and the genes they regulate (EGFR, HSP90AA1, PIK3R3 and RAC1) are involved in the regulation of PI3K/Akt pathway. It is known that the PI3K/Akt signaling pathway plays an important role in the development of tumors by inhibiting the downstream apoptotic genes, activating anti-apoptotic genes, promoting cell survival, and mediating hematopoiesis and angiogenesis [35]. An initial stimulation of a growth factor causes activation of cell surface receptors and phosphorylation of PI3K. Activated PI3K further phosphorylates lipids on the cell membrane to form a second messenger PIP3 (phosphatidylinositol-3,4,5-triphosphate). Akt, a serine/threonine kinase, is recruited to the cell membrane by interaction with these phosphatidylinositol docking sites. Activated Akt mediates downstream responses by phosphorylating a range of intracellular proteins such as Bad, Caspase-9, mTOR, NF-κB, GSK-3, FKHR, thus regulate multiple biological progress including cell survival, cell growth, proliferation, cell migration and angiogenesis, and eventually lead to cancer [37].
Seven miRNAs (miR-15a, miR-362, miR-142, miR-155, miR-340, miR-576, and miR-942) and the genes they regulate (BTRC, CTNNB1, RAC1, and SKP1) are involved in the regulation of Wnt/β-catenin signaling pathway. Previous studies have found that abnormal activation of the Wnt signaling pathway is associated with the development of multiple cancers [38]. When the Wnt signal pathway is activated, Wnt binds with its specific receptors, Frizzled and LRP, and the signal is transmitted from the extracellular to the intracellular dishevelled (Dsh or DV1). Under the synergistic effect of Axin, Dsh inhibits the degradation of hygro-catenin, subsequently, β-catenin enters the nucleus and interacts with TCF (T cell transcription factor)/LEF to activate the expression of downstream related genes, such as c2myc and cyclin D1, and eventually leads to malignant transformation [39].
Seven miRNAs (miR-21, miR-155, miR-15a, miR-93, miR-142, miR-185, and miR-576) and the genes they regulate (EGFR, MAPK1, and RAC1) are involved in the regulation of MAPK signaling pathway. Extracellular growth factors (EGF) activate tyrosine kinase receptors (RTK), providing binding sites for the adapter protein Grb2, and recruiting SOS proteins to the cell membrane, SOS proteins then activate Ras by consuming GTP. Ras-GTP binds directly to a silk/threonine kinase Raf, forming an instantaneous signal, activated Raf kinases further phosphorylate MEK to activate it [40]. Activated MEK phosphorylates ERK1/ERK2, and the phosphorylated ERK enters the nucleus, phosphorylates transcription factors such as elk-1 and myc [41], then regulates gene expression related to cell growth and differentiation, resulting in the promotion of proliferation and evolution of cancer cells [42].
Eight miRNAs (miR-93, miR-185, miR-15a, miR-362, miR-142, miR-576, miR-21, and miR-942) and the genes they regulate (ANAPC5, BTRC, CUL3, NEDD4, and SKP1) are involved in the regulation of ubiquitin mediated proteolysis. The ubiquitin (Ub)-proteasome degradation system can specifically recognize and target the substrate protein, participate in important processes such as cell growth, proliferation and apoptosis, and plays an important role in maintaining and regulating the dynamic balance of cells [43, 44]. The system consists of ubiquitin, ubiquitin-activating enzyme E1, ubiquitin-conjugating enzyme E2, ubiquitin ligase E3, 26S proteasome and ubiquitin dissociation enzyme (DUBs). With ATP, through the catalysis of E1, E2 and E3, ubiquitin is finally connected with e-nh2, a lysine side chain of the target protein, through the isopeptide bond, and then enters the 26S proteasome as the recognition signal to degrade the target protein.
However, there are three miRNAs were not reported to be associated with RCC before, they are miR-576, miR-616 and miR-133a-2.
Although miR-576 has not been reported to be associated with renal cancer, this study found that its corresponding target genes, CUL3 and RAC1, are involved in the regulation of several cancer-related biological pathways such as PI3K/Akt signaling pathway and ubiquitin proteasome degradation. To be more specific, CUL3 involved in the formation of E3 ubiquitin ligase in the ubiquitin mediated proteolysis pathway, and RAC1 encodes G protein linkage receptor Rac1 which participates in the PI3K/Akt signaling pathway, thus it may be a new early diagnostic biomarker.
MiR-616 was neither reported to be associated with RCC, nor involved in the regulation of any one of 147 biological pathways. However, this study found that two proteins encoded by the target genes of miR-616, ASB13 and FBXW2, are the key nodes in the key sub-network of the PPI network, suggesting that they may have interactions with other proteins in the network. Also, ASB13 has been reported to be associated with diffuse large B cell lymphoma [45] and breast cancer [46], and FBXW2 has been reported to have relationship with lung cancer [47, 48]. The evidence indicates that miR-616 may affect the development of renal cancer in an indirect way. Thus it may also be defined as a new early diagnostic biomarker.
The target genes of miR-133a-2 have not appeared in the key sub-network, also, it has not been reported to be associated with RCC, so the relationship between miR-133a-2 and RCC needs to be further verified.
In conclusion, by using multiple computational methods and bioinformatic methods, we managed to find several potential early diagnostic biomarker and the relationship between these potential biomarker miRNAs and RCC. There are totally 13 miRNAs (miR-142, miR-15, miR-155, miR-185, miR-21, miR-340, miR-362, miR-576, miR-93, miR-942, miR-616, miR-133a-2, and miR-210), including two new early diagnostic biomarker miRNA, miR-576 and miR-616, were found by this study, and they will have guiding significance for the early diagnosis of RCC. Also, the mechanism of how these miRNAs participate in the regulation of multiple cancer-related pathways will provide new ideas and theoretical support for the study of mechanisms contained in kidney cancer and the prediction of therapeutic targets.
References
Cairns P. Renal cell carcinoma. Cancer Biomark. 2011;9:461.
Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2018. CA Cancer J Clin. 2018;68:7–30.
Butz H, Szabo PM, Nofech-Mozes R, Rotondo F, Kovacs K, Mirham L, et al. Integrative bioinformatics analysis reveals new prognostic biomarkers of clear cell renal cell carcinoma. Clin Chem. 2014;60:1314.
Sacco E, Pinto F, Sasso F, Racioppi M, Gulino G, Volpe A, et al. Paraneoplastic Syndromes in Patients with Urological Malignancies. Urol Int. 2009;83:1.
Flanigan RC, Campbell SC, Clark JI, Picken MM. Metastatic renal cell carcinoma. Curr Treat Option On. 2003;4:385–90.
Cohen HT, McGovern FJ. Renal-cell carcinoma. New Engl J Med. 2005;353:2477.
De Mulder PHM, van Herpen CML, Mulders PAF. Current treatment of renal cell carcinoma. Ann Oncol. 2004;15:v319.
Wasim AS, Mumtaz F. Limitations of CT scanning in Bosniak staging of renal cystic carcinoma. J Surg Case Rep. 2018;4:1–4.
Björklund M, Heikkilä P, Koivunen E. Peptide inhibition of catalytic and noncatalytic activities of matrix metalloproteinase-9 blocks tumor cell migration and invasion. J Biol Chem. 2004;279:29589.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390.
Braicu C, Calin GA, Berindan-Neagoe I. MicroRNAs and cancer therapy - from bystanders to major players. Curr Med Chem. 2013;20:3561.
Faragalla H, Youssef YM, Scorilas A, Khalil B, White NMA, Mejia-Guerrero S, et al. The clinical utility of miR-21 as a diagnostic and prognostic marker for renal cell carcinoma. J Mol Diagn. 2012;14:385.
Gaudelot K, Gibier J, Pottier N, Hémon B, Van Seuningen I, Glowacki F, et al. Targeting miR-21 decreases expression of multi-drug resistant genes and promotes chemosensitivity of renal carcinoma. Tumor Biol. 2017;39:568834959.
Zhan C, Yan L, Wang L, Jiang W, Zhang Y, Xi J, et al. Identification of reference miRNAs in human tumors by TCGA miRNA-seq data. Biochem Bioph Res Co. 2014;453:375.
Wang H, Peng R, Wang J, Qin Z, Xue L. Circulating microRNAs as potential cancer biomarkers: the advantage and disadvantage. Clin Epigenetics. 2018;10:19.
Yoon D, Lee E, Park T. Robust imputation method for missing values in microarray data. BMC Bioinf. 2007;8(Suppl 2):S6.
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, et al. Normalization for cDNA microarray data: a robust composite method addressing single andmultiple slide systematic variation. Nucleic Acids Res. 2002;30:e15.
Yuan L, Chen L, Qian K, Qian G, Wu C, Wang X. et al. Co-expression network analysis identified six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC). Genomics Data. 2017;14:132.
Su Q, Wang Y, Jiang X, Chen F, Lu W. A cancer gene selection algorithm based on the K-S test and CFS. Biomed Res Int. 2017;2017:1.
Li S, Wang S, Guo Z, Wu H, Jin X, Wang Y, et al. miRNA profiling reveals dysregulation of RET and RET-regulating pathways in Hirschsprung’s Disease. PLoS ONE. 2016;11:e150222.
Yasmin F, Tun HM, Konya TB, Guttman DS, Chari RS, Field CJ, et al. Cesarean section, formula feeding, and infant antibiotic exposure: separate and combined impacts on gut microbial changes in later infancy. Front Pediatr. 2017;5:200.
Zhou Z, Cheng Y, Jiang Y, Liu S, Zhang M, Liu J, et al. Ten hub genes associated with progression and prognosis of pancreatic carcinoma identified by co-expression analysis. Int J Biol Sci. 2018;14:124.
Monteys AM, Spengler RM, Wan J, Tecedor L, Lennox KA, Xing Y, et al. Structure and activity of putative intronic miRNA promoters. RNA (New Y, N Y). 2010;16:495.
Juan D, Alexe G, Antes T, Liu H, Madabhushi A, Delisi C, et al. Identification of a microRNA panel for clear-cell kidney cancer. Urology. 2010;75:835.
Rasti A, Mehrazma M, Madjd Z, Keshtkar AA, Roudi R, Babashah S. Diagnostic and prognostic accuracy of miR-21 in renal cell carcinoma: a systematic review protocol. BMJ Open. 2016;6:e9667.
Ji H, Tian D, Zhang B, Zhang Y, Yan D, Wu S. Overexpression of miR-155 in clear-cell renal cell carcinoma and its oncogenic effect through targeting FOXO3a. Exp Ther Med. 2017;13:2286.
von Brandenstein M, Pandarakalam JJ, Kroon L, Loeser H, Herden J, Braun G, et al. MicroRNA 15a, inversely correlated to PKCα, is a potential marker to differentiate between benign and malignant renal tumors in biopsy and urine samples. Am J Pathol. 2012;180:1787.
Liu TY, Zhang H, Du SM, Li J, Wen XH. Expression of microRNA-210 in tissue and serum of renal carcinoma patients and its effect on renal carcinoma cell proliferation, apoptosis, and invasion. Genet Mol Res. 2016;15:15017746.
Hao JF, Ren KM, Bai JX, Wang SN, Shao B, Cao N, et al. Identification of potential biomarkers for clear cell renal cell carcinoma based on microRNA-mRNA pathway relationships. Special Issue 2. J Cancer Res Ther. 2014;10:C167–77.
Shi J, Zhuang Y, Liu XK, Zhang YX, Zhang Y. TGF-beta induced RBL2 expression in renal cancer cells by down-regulating miR-93. Clin Transl Oncol. 2014;16:986.
Prior C, Perez-Gracia JL, Garcia-Donas J, Rodriguez-Antona C, Guruceaga E, Esteban E, et al. Identification of tissue microRNAs predictive of sunitinib activity in patients with metastatic renal cell carcinoma. PLoS ONE. 2014;9:e86263.
Li W, Dou Z, We S, Zhu Z, Pan D, Jia Z, et al. Long noncoding RNA BDNF-AS is associated with clinical outcomes and has functional role in human prostate cancer. Biomed Pharm. 2018;102:1105.
Liu Z, Liu XW, Liu SA, Lv JJ, Fu Q. Clinical significance of changes of expression of the Wnt/beta-catenin signaling pathway in renal clear cell carcinoma. Eur Rev Med Pharm Sci. 2016;20:4840.
Gao Y, Li J, Qiao N, Meng Q, Zhang M, Wang X, et al. Adrenomedullin blockade suppresses sunitinib-resistant renal cell carcinoma growth by targeting the ERK/MAPK pathway. Oncotarget. 2016;7:63374.
Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2012;44:17.
Guo H, German P, Bai S, Barnes S, Guo W, Qi X, et al. The PI3K/AKT pathway and renal cell carcinoma. J Genet Genomics. 2015;42:343.
Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387.
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11.
Lim W, Jeong W, Song G. Delphinidin suppresses proliferation and migration of human ovarian clear cell carcinoma cells through blocking AKT and ERK1/2 MAPK signaling pathways. Mol Cell Endocrinol. 2016;422:172.
Turney KD, Parrish AR, Orozco J, Gandolfi AJ. Selective activation in the MAPK pathway by Hg(II) in precision-cut rabbit renal cortical slices. Toxicol Appl Pharm. 1999;160:262.
Huang D, Ding Y, Luo WM, Bender S, Qian CN, Kort E, et al. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res. 2008;68:81.
Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays. 2000;22:442.
Vodermaier HC. APC/C and SCF: controlling each other and the cell cycle. Curr Biol. 2004;14:R787.
Blenk S, Engelmann J, Weniger M, Schultz J, Dittrich M, Rosenwald A, et al. Germinal center B cell-like (GCB) and activated B cell-like (ABC) type of diffuse large B cell lymphoma (DLBCL): analysis of molecular predictors, signatures, cell cycle state and patient survival. Cancer Inf. 2007;3:399.
Chi C, Murphy LC, Hu P. Recurrent copy number alterations in young women with breast cancer. Oncotarget. 2018;9:11541.
Yang F, Xu J, Li H, Tan M, Xiong X, Sun Y. FBXW2 suppresses migration and invasion of lung cancer cells via promoting β-catenin ubiquitylation and degradation. Nat Commun. 2019;10:1382.
Xu J, Zhou W, Yang F, Chen G, Li H, Zhao Y, et al. The β-TrCP-FBXW2-SKP2 axis regulates lung cancer cell growth with FBXW2 acting as a tumour suppressor. Nat Commun. 2017;8:14002.
Acknowledgements
This work was supported by the Beijing Municipal Education Commission (Grant No. KM201410005030), the Importation and Development of High-Caliber Talents Project of Beijing Municipal Institutions (Xiaoyi Zhang), the practical training plan of Beijing Municipal Education Commission (Xiaoyi Zhang), the National Natural Science Foundation of China (Grant No. 31100523).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tang, T., Du, X., Zhang, X. et al. Computational identification and analysis of early diagnostic biomarkers for kidney cancer. J Hum Genet 64, 1015–1022 (2019). https://doi.org/10.1038/s10038-019-0640-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0640-2
