
Abstract
Individuals affected with autosomal recessive cutis laxa type 2B and 3 usually show translucent skin with visible veins and abnormal elastic fibers, intrauterine and/or postnatal growth restriction and a typical triangular facial gestalt. Here we describe three unrelated individuals in whom such a cutis laxa syndrome was suspected, especially after electron microscopy revealed immature and less dense dermal elastic fibers in one of them. However, one of these children also displayed optic atrophy and two hypogammaglobulinemia. All had elevated liver enzymes and acute liver failure during febrile episodes leading to early demise in two of them. The only surviving patient had been treated with immunoglobulins. Through exome sequencing we identified mutations in NBAS, coding for a protein involved in Golgi-to-ER transport. NBAS deficiency causes several rare conditions ranging from isolated recurrent acute liver failure to a multisystem disorder mainly characterized by short stature, optic nerve atrophy and Pelger-Huët anomaly (SOPH). Since we subsequently verified Pelger-Huët anomaly in two of the patients the diagnosis SOPH syndrome was unequivocally proven. Our data show that SOPH syndrome can be regarded as a differential diagnosis for the progeroid forms of cutis laxa in early infancy and that possibly treatment of the hypogammaglobulinemia can be of high relevance for the prognosis.
Similar content being viewed by others

Introduction
Genetic disorders with progeroid features are clinically heterogeneous and the causative genetic defects can affect different cellular processes. They are usually characterized by thin and translucent skin, atrophy of the subcutaneous fat tissue, a typical facial gestalt and short stature leading to the progeroid appearance. The disease course and severity is variable ranging from the severe Hutchinson Gilford progeria syndrome to the milder progeroid conditions associated with cutis laxa due to mutations either in ALDH18A1 encoding pyrroline-5-carboxylate synthase (MIM; #138250) or PYCR1 encoding pyrroline-5-carboxylate reductase 1 (MIM; #179035) [1,2,3,4,5]. Besides the above mentioned features the latter disorders are also characterized by intrauterine growth restriction, cataracts or corneal clouding, and variable developmental problems as well as movement disorders. Both enzymes are localized within the mitochondria where they are involved in the conversion from glutamic acid to proline [2, 5].
Another condition with intrauterine growth restriction is SOPH (short stature, optic nerve atrophy and Pelger-Huët anomaly) syndrome (MIM; #614800) due to NBAS (Neuroblastoma amplified sequence) mutations [6,7,8]. The NBAS protein localizes to the Golgi apparatus where it is involved in retrograde trafficking processes [9]. Furthermore, there is evidence that NBAS is involved in the nonsense mediated decay (NMD) pathway in different species [10]. SOPH syndrome is a multisystem condition mainly characterized by a severe growth restriction leading to short stature, atrophy of the optic nerve, Pelger-Huët anomaly and a variable degree of liver disease [7]. Additionally, affected individuals show elevated liver enzymes and hypogammaglobulinemia [6]. Multiple cases have been described presenting with a highly variable phenotype. The first description of this condition by Maksimova et al. demonstrated that the homozygous missense mutation p.(Arg1914His) in the Yakut population is the cause of the observed phenotype. This mutation was also found frequently in other populations, usually in a compound heterozygous state with other pathogenic alleles [6,7,8, 11, 12]. Interestingly, mutations in NBAS can cause another condition leading to isolated recurrent acute liver failure (ALF) (MIM; #616483) [13,14,15]. This condition is characterized by recurrent episodes of acute liver failure in early infancy. In most cases liver failure was precipitated by febrile episodes [14].
In the current study we report on three unrelated affected individuals showing features compatible with these conditions. However, in early infancy also a significant clinical overlap with connective tissue disorders was observed.
Materials and methods
Patients
In this study we report on two affected individuals referred to our centre due to a connective tissue condition with progeroid features. In the third patient a condition associated with NBAS mutations was suspected, however, also in this case, a clinical overlap with a connective tissue condition was observed. The parents gave their written consent for molecular testing and for publication of photographs in Fig. 1. From patient 3 a skin biopsy was taken and blood smears were analysed from patients 1 and 3. In all families, DNA was isolated from peripheral blood samples. The ethical board of the Charité-Universitaetsmedizin Berlin approved the study (EA2/145/07).

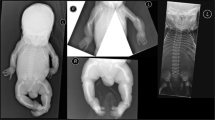
Clinical appearance of the affected individuals. Individual 1 at the age of 7 months, individual 2 at the age of 10 months, and individual 3 at age of 19 years: a–c Affected individuals show a triangular shape of the face, a prominent forehead due to a high frontal hairline, and prominent ears. a, b Individuals 1 and 2 additionally show sparse hair. Furthermore, they show thin skin on their forehead with visible veins leading to a progeroid appearance. Note also hypertelorism in individual 2. d–f The affected individuals also show translucent skin leading to visible veins. g-i Affected individual 3. He shows severe short stature and a short trunk and a general paucity of subcutaneous fat tissue. j Granulocyte staining from the affected individual 3. The control cells show a regular segmentation of the granulocyte nucleus. However, the cells from the affected individual show a hyposegmentation of their nuclei in line with a Pelger-Huët anomaly
Mutation screening
Sequencing of PYCR1 and ALDH18A1 was performed as previously described in the DNA from the affected individuals 1 and 3 [2]. DNA samples from index patients 1 and 3 were enriched for exome Sequencing using the SureSelect XT, Clinical research exome from Agilent. Next Generation Sequencing was performed on a HiSeq 1500 system from Illumina. Data were processed as described previously [16] and variants were prioritized using the GeneTalk platform [17]. The disease causing potential was evaluated using MutationTaster, SIFT and Polyphen 2 [18]. In patient 2, coding genomic regions were enriched with a SureSelect Human All Exon Kit V6 (Agilent technologies, Santa Clara, California) for subsequent sequencing as 2 × 125 bp paired-end reads on an HiSeq2500 system (Illumina, San Diego, California). Generated sequences were analyzed using the megSAP pipeline (https://github.com/imgag/megSAP). Clinical variant prioritization included different filtering steps including a search for rare (MAF < 0.1% in ExAC, gnomAD, 3,000 in-house exome datasets), recessive-type variants according to an in-house standard operating procedure. Potential disease-causing variants were validated in all available DNA samples using Sanger Sequencing of the affected exons and the flanking intron regions of NBAS (NM_015909.3), using BigDye Terminator cycle sequencing kit (Applied Biosystems) and run on ABI 3730 DNA Analyzer (Applied Biosystems). Additionally, co-segregation analysis was performed using all available parental DNA samples to confirm compound heterozygosity. All primer sequences are available on request.
Electron microscopy
A skin biopsy of patient 3 from the upper arm/near elbow was fixed overnight at room temperature in 3% glutaraldehyde solution in 0.1 M cacodylate buffer pH 7.4. It was cut into pieces of approximately 1 mm3, washed in buffer, postfixed for 1 h at 4 °C in 1% aqueous osmium tetroxide, rinsed in water, dehydrated through graded ethanol solutions, transferred into propylene oxide, and embedded in epoxy resin (glycidether 100). Semithin and ultrathin sections were cut with an ultramicrotome (Reichert Ultracut E). Semithin sections of 1 µm were stained with methylene blue. 60–80 nm ultrathin sections were treated with uranyl acetate and lead citrate, and examined with an electron microscope.
Web Resources
Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.gov/Omim/ (for ARCL3A/B, DBS, SOPH, NBAS).
Results
Clinical presentation
Patient 1
The male affected individual was born to a non-consanguineous couple from Turkey as their first child. Birth parameters at 36 weeks of gestation were: weight: 1730 g (−2.5 SD), height: 44 cm (−2 SD), head circumference: 31 cm (−1.7 SD). At the age of seven months, the patient showed a progeroid appearance due to a triangular face with a high frontal hairline and proptotic eyes with blue sclerae. Cranial X-ray showed a large anterior fontanel (8 × 7 cm) and Wormian bones (Supplementary Figure 1A, B). Additionally, the patient presented with loose, redundant and thin skin leading to visibility of the subcutaneous veins (Fig. 1a, d and Table 1). Furthermore, a reduction of the subcutaneous fat tissue was documented. Additional evaluation revealed a hepatomegaly and echocardiography showed patent ductus arteriosus. His psychomotor development was delayed: He was able to sit without support at 32 months. He showed elevated values for ALT (alanine aminotransferase), AST (aspartate aminotransferase) and alkaline phosphatase (AP) already at two months of age and these enzymes remained increased till 32 months while gamma-glytamyl transferase was only repeatedly raised (Supplementary Table 1). Humoral and cellular immunity showed no alterations. The karyotype was normal (46, XY). A condition associated with cutis laxa was suspected. Diagnostic investigation of serum transferrin and alpha-1-antitrypsin revealed no alteration of glycosylation. Pelger-Huët anomaly was confirmed after identification of the causative mutation (data not shown). At 2 years and 8 months the patient was referred to intensive care unit. He showed severe febrile convulsions and strikingly increased liver transaminases indicating hepatic insufficiency (Supplementary Table 1). The patient died due to this acute liver failure.
Patient 2
The male patient was born at 37 weeks of gestation to healthy non-consanguineous Caucasian parents. Fetal growth retardation was diagnosed. Birth weight was 2130 g (−2.2 SD), length was 46 cm (−1.8 SD), head circumference was 32.5 cm (−1.3 SD). The Apgar scores were 9, 9, 10 at 1, 5, and 10 min, respectively. Fontanels were found enlarged. Echocardiography showed VSD and ASD II. Examination at 10 months revealed a weight of 7120 g (−2 SD), OFC of 46 cm (mean), a broad forehead with a high frontal hairline, large fontanels, hypertelorism, thin lips and bilateral clinodactyly V (Fig. 1b, e). Subcutaneous fat was reduced and skin appeared thin and translucent (Fig. 1e). His psychomotor development was delayed: he started to sit at 8 months and to walk at 19 months. He spoke his first words at the age of 16 months and further speech development was delayed. At age of 20 months, the patient had an acute infection followed by a liver crisis. After this infection the liver enzyme levels remained elevated. Results of liver biopsy testing revealed unremarkable histology and no signs of any storage material. Hypogammaglobulinemia was also documented. Short stature and dystrophy were also observed at the age of 24 months: weight was 10 kg (−2 SD), length 79 cm (−2.3 SD). OFC was 49.5 cm (mean). When he had fever he developed repeated episodes of acute liver failure and a liver transplantation was considered but he died at 26 months with signs of hepatic encephalopathy. Liver crisis was accompanied by massively elevated ALT 13330 U/l (normal, <41 U/l), and AST 25310 U/l (normal, 30–71 U/l). Cranial computer tomography revealed no brain abnormalities. A radiograph of his thorax showed a hypoplastic left rib and no further congenital abnormalities. Due to the recurrent acute liver failure in association with the other clinical manifestations we suspected a NBAS related condition. Exome sequencing based diagnostics was initiated.
Patient 3
The patient is the first child of healthy unrelated Caucasian parents. Growth retardation was seen prenatally. He was born at 37th week of gestation with a birth length of 43 cm (−3 SD), birth weight of 1910 g (−2.7 SD) and an OFC of 28.5 cm (−4 SD). Wide fontanels and a hypospadia were noted after birth. Chronic hepatopathy and muscular hypotonia have been documented since his neonatal period. Blood measurements revealed elevated liver enzymes. By abdominal ultrasound we found hepatomegaly. Results of liver biopsy showed fatty infiltration of the parenchyma. During the next years, hepatomegaly was not detectable anymore. However, transaminases remained elevated in repeated tests, e.g., AST 59 U/l (normal range 2–47 U/l) and ALT 98 (normal range 2–40 U/l) at age of 4 years. His motor and mental development was retarded. He was able to sit at 8 months and to walk at 22 months. At two years of age, he was severely growth retarded (75 cm, −3.4 SD), dystrophic (8 kg, −3.5 SD) and microcephalic (46 cm, −2.5 SD). Wrinkled skin of the dorsum of the hands and translucent skin were present. He started to speak at 2.5 years. A skin biopsy showed immature and less dense elastic fibers suggesting a connective tissue disorder (Fig. 2). Skull radiograph at age two years showed multiple occipital Wormian bones. Bilateral optic atrophy and myopia were found at 10 years of age. At the age of 14 years, hypogammaglobulinemia was detected necessitating substitution of immunoglobulins. At age 15 years, insulin-dependent diabetes mellitus occurred. At age of 19 years his facial appearance included a triangular shape with deep set eyes and prominent ears. Furthermore, he had thin and translucent skin with visible veins leading to an overall progeroid appearance (Fig. 1c, f).

Electron microscopy of skin biopsy from Patient 3 in comparison to an age-matched control (a, c) In the control individual, the dermal region looks normal with regular collagen bundles (Col) and elastic fibers (Eln). b, d The dermal region in the patient’s biopsy shows normal collagen bundles. However, the elastic fibers have an irregular shape and reduced electron density in comparison to the control. Scale bar: A, B = 500 nm and C, D = 1000 nm
At this age he was still severely growth retarded leading to a body length of 125 cm (−10.7 SD) and severely dystrophic (weight 27 kg, BMI 17.3 mg/m2) (Fig. 1g–i). Brain MRI at different ages (5 months, 5 years) showed hypoplasia of the corpus callosum and a hypoplastic cerebellar vermis (Supplementary Figure 1C). Diagnostic investigation of serum transferrin and alpha-1-antitrypsin revealed no abnormalities. Pelger-Huët anomaly was confirmed after mutation identification (Fig. 1j). Over the years, different clinical diagnoses were considered, among them Costello syndrome, osteogenesis imperfecta, gerodermia osteodysplastica and autosomal recessive cutis laxa type 2B.
Identification of the causative genetic defect
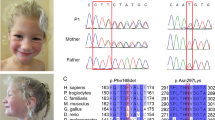
Due to the clinical overlap with PYCR1- or ALDH18A1-related cutis laxa we first sequenced the coding region of these genes in the affected individuals 1 and 3 without detecting any mutation. Subsequent exome sequencing revealed in Patient 1 two heterozygous variants affecting NBAS (NM_015909.3). The first variant, chr2:g.15679455 C > T; c.405 G > A predicts a premature termination codon p.(Trp135*). The second alteration, chr2:g.15378794 C > T; c.5741 G > A leads to the substitution p.(Arg1914His). The first mutation has not been described so far in literature and is not annotated in gnomAD. The second has been described previously and was found in gnomAD, mainly in the South Asian population, with a frequency of 0.0028% only in a heterozygous state [7]. To investigate whether these changes are in trans, we sequenced the parental DNA samples and found the alteration p.(Trp135*) inherited from the father, whereas the mutation p.(Arg1914His) was transmitted from the mother (Fig. 3a).

Identified NBAS mutations in Patients 1, 2 and 3. a Pedigree from family 1 including Sanger sequencing results. The affected individual shows compound heterozygosity for the mutations p.(Trp135*) and p.(Arg1914His). b Pedigree from family 2 including Sanger sequencing results. The mutations p.(Ile984Leufs*8) and p.(His1053Arg) were found compound heterozygous in the affected individual. c Pedigree from family 3 including Sanger sequencing results. The mutations p.(Arg1914His) and p.(Glu2189*) were found to be compound heterozygous in the affected individual. d Interspecies alignment of NBAS. Histidine1053 resides in a, during evolution, highly conserved stretch of NBAS indicating its importance. The already described NBAS mutation p.(Leu1055Pro) is in close proximity to the alteration found in patient 2, indicating a similar importance and causality for the observed phenotype
In Patient 2, we identified the heterozygous alteration chr2:g.15542413delT; c.2950delA in NBAS predicting a frameshift and thereby a premature termination of translation after eight additional codons p.(Ile984Leufs*8). A second heterozygous variant chr2:g.15534450 T > C; c.3158 A > G causes the substitution p.(His1053Arg). The frameshift and the missense allele were found in gnomAD with a total frequency of 0.012% and 0.0007% respectively, both in a heterozygous state only. The frameshift variant was found in the mother whereas the missense variant was absent. Unfortunately, no DNA was available from the father (Fig. 3b).
In Patient 3 we identified two heterozygous mutations in NBAS. The first mutation chr2:g. 15378794 C > T, c.5741 G > A causes the substitution p.(Arg1914His) also identified in Patient 1. The second alteration chr2:g.15330396dupA; c.6564dupT predicts a premature termination of translation p.(Glu2189*). This mutation has not been described previously and is absent from gnomAD. The missense mutation p.(Arg1914His) was also detected in the mother (Fig. 3c).
In all the three affected individuals, the mutations identified were in a compound heterozygous state shown by analysis of at least one parental DNA sample and can thereby be regarded as the cause of the observed SOPH phenotype. The missense mutation p.(His1053Arg) has not been described so far to be causative for SOPH syndrome. The affected position is highly conserved during evolution shown by an interspecies alignment and it resides close to an already described mutation for ALF (Fig. 3d) [13].
Discussion
In this study, we present the molecular and clinical data of three affected individuals with biallelic NBAS mutations. All displayed variable pre and postnatal growth retardation and dystrophy. Furthermore, they presented with developmental delay, triangular face with a broad forehead and a high frontal hairline, thin lips as well as translucent skin and atrophy of the subcutaneous fat tissue. Hepatomegaly and elevated liver enzymes were present in all and repeated liver crises cumulating in liver failure led to early death in two of three affected individuals. The signs of Pelger-Huët anomaly as well as optic atrophy were found in Patients 1 and 3.
All patients showed a liver crisis in the first years of life, however, it was recurrent only in Patients 1 and 2, who died between two and three years of life. In both of them severe febrile convulsions and strongly increased liver transaminases indicated hepatic insufficiency, which can be regarded as the cause of their early demise. In all cases, the severe liver failure seems to be triggered by infections, which is in line with literature. In patient 1 the liver enzymes were always high indicating constant damaging processes to the liver also in a phase without infection. In a clinically overlapping case described by Kortüm et al. this phenomenon was also observed [6].
In Patient 3 after birth elevated liver enzymes and hepatomegaly were detectable. Later in life hepatomegaly vanished, but transaminases remained high. One obvious difference to the two other patients with lethal course was the treatment of the hypogammaglobulinemia by IgG substitution. Patient 1 described by Segarra et al. was treated similarly and showed no further liver crisis [8]. Thus, we may speculate that this therapy strengthened the immune system and prevented infections accompanied by fever that might trigger an acute liver crisis.
Many mutations within NBAS have been described in the literature leading to a wide range of clinical symptoms. Unfortunately, no correlation with the severity of the phenotype is possible so far. Only one consanguineous Lebanese family with three affected individuals carrying a homozygous NBAS nonsense mutation was described previously. This mutation very likely leads to complete absence of NBAS and might be the cause for the severe progression of the phenotype and the early demise of the affected individuals [19].
The cases described by Maksimova harboring the missense mutation p.(Arg1914His) in a homozygous state seem relatively mildly affected since no liver involvement was reported [7]. In fibroblasts from two patients carrying different nonsense alleles but the same p.(Arg1914His) missense mutation, a comparable amount of remaining NBAS protein was detectable indicating that the altered protein remains stable [11]. One might speculate that the presence of the p.(Arg1914His) mutation in a homozygous state leads to a relatively high level of remaining NBAS function probably explaining the milder phenotype. In almost all other described cases, a biallelic combination of a nonsense mutation and a missense alteration was shown. The mutations usually reside within the middle part of the protein or the N terminal region [8]. Only some were found very close to the C-terminal region. To our knowledge, the most downstream mutation is the p.(Val2145_Glu2237del) in patient 12 (FXI) described by Staufner et al. [13]. Taking into account that the second allele in this individual is a nonsense mutation at position 943, the inframe deletion seems to provide the necessary NBAS activity to be compatible with life. Our patient 3 is severely affected, however, in comparison to the other cases the phenotype is somewhat milder in terms of liver failure and survival. We speculate that the combination of the already known p.(Arg1914His) allele together with a very late truncating mutation, which might have some residual functionality, and the fact that the patient was supplemented with IgG might be an explanation for his long term survival.
The mutation p.(His1053Arg) identified in patient 2 has not been described previously. Interestingly, a mutation at position 1055 was shown in combination with an inframe deletion of one amino acid in patient 3 (FIII) affected by ALF [13]. In light of the discussion above, one can speculate that the deletion of the one amino acid in combination with the observed missense alteration in the ALF patient leads to an even higher remaining NBAS activity compared to our patient and thereby to a different phenotype without any skeletal involvement [13].
NBAS related disorders range from ALF via SOPH to a combined condition of ALF and SOPH [6, 7, 13]. However, the spectrum of possible clinical diagnoses is even broader. Affected individuals are sometimes classified to have atypical osteogenesis imperfecta [11], acrofrontofacionasal dysostosis type 1 [20] or, like in our patients, a condition from the spectrum of progeroid cutis laxa disorders. Therefore, we compared the cases available in literature carrying mutations in either PYCR1, ALDH18A1 or NBAS (Table 1). This comparison revealed that especially in early infancy, affected individuals show overlapping features such as intrauterine growth restriction, a delayed fontanel closure, large ears, a triangular face with a broad forehead, reduced subcutaneous fat tissues, redundant and thin skin and sparse hair. The term senile skin has been used frequently in literature to describe the skin of patients with NBAS mutations [7, 8]. This reflects the translucent skin with reduced subcutaneous fat tissue also observed in the group of progeroid cutis laxa conditions [1, 2, 5, 21]. Also the abnormal elastic fibers found in Patient 3 suggest a connective tissue condition; however, the alterations observed are not as severe as in typical cutis laxa patients. Furthermore, hepatomegaly, hepatic dysfunction, acute liver failure, optic atrophy, hypogammaglobulinemia were never described in patients with progeroid cutis laxa and can help to differentiate these conditions from each other.
The partial overlap of the NBAS and proline cycle pathologies suggest a similar mechanism. Interestingly, the enzymes PYCR1 and P5CS are localized within the mitochondria whereas NBAS is involved in Golgi-to-ER trafficking. Impaired Golgi function was also observed in ATP6V1A-, ATP6V1E1-, and ATP6V0A2-related autosomal recessive cutis laxa (ARCL) and in gerodermia osteodysplastica [22,23,24,25,26]. However, in contrast to our patients, these disorders show glycosylation anomalies, but no liver problems. On the other hand, defects in the functionally related genes ATP6AP1 and CCDC115 only lead to liver disease and glycosylation defects, but in most cases no connective tissue changes [27, 28]. However, also one patient with a defect in ATP6AP1 has been described showing cutis laxa in early infancy [29]. Further research is needed to understand how perturbations of Golgi, ER and mitochondrial compartments might converge in a similar pathophysiological output explaining the overlapping features of the abovementioned disorders. Additionally, the impact of NBAS deficiency on extracellular matrix secretion and assembly should be investigated to gain insights into the connective tissue phenotype in SOPH syndrome patients.
In conclusion, we identified three affected individuals with compound heterozygous NBAS mutations underlining that SOPH syndrome can be regarded as a differential diagnosis for the progeroid forms of cutis laxa in early infancy. Our data further support a positive effect of IgG supplementation on the disease progression in SOPH patients.
References
Baumgartner MR, Rabier D, Nassogne MC, Dufier JL, Padovani JP, Kamoun P, et al. Delta1-pyrroline-5-carboxylate synthase deficiency: neurodegeneration, cataracts and connective tissue manifestations combined with hyperammonaemia and reduced ornithine, citrulline, arginine and proline. Eur J Pedia. 2005;164:31–6.
Fischer-Zirnsak B, Escande-Beillard N, Ganesh J, Tan YX, Al Bughaili M, Lin AE, et al. Recurrent de novo mutations affecting residue Arg138 of pyrroline-5-carboxylate synthase cause a progeroid form of autosomal-dominant cutis laxa. Am J Hum Genet. 2015;97:483–92.
Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–24.
Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, Perry MB, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358:592–604.
Reversade B, Escande-Beillard N, Dimopoulou A, Fischer B, Chng SC, Li Y, et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet. 2009;41:1016–21.
Kortum F, Marquardt I, Alawi M, Korenke GC, Spranger S, Meinecke P, et al. Acute liver failure meets SOPH syndrome: a case report on an intermediate phenotype. Pediatrics. 2017;139:e1–8.
Maksimova N, Hara K, Nikolaeva I, Chun-Feng T, Usui T, Takagi M, et al. Neuroblastoma amplified sequence gene is associated with a novel short stature syndrome characterised by optic nerve atrophy and Pelger-Huet anomaly. J Med Genet. 2010;47:538–48.
Segarra NG, Ballhausen D, Crawford H, Perreau M, Campos-Xavier B, van Spaendonck-Zwarts K, et al. NBAS mutations cause a multisystem disorder involving bone, connective tissue, liver, immune system, and retina. Am J Med Genet A. 2015;167A:2902–12.
Aoki T, Ichimura S, Itoh A, Kuramoto M, Shinkawa T, Isobe T, et al. Identification of the neuroblastoma-amplified gene product as a component of the syntaxin 18 complex implicated in Golgi-to-endoplasmic reticulum retrograde transport. Mol Biol Cell. 2009;20:2639–49.
Longman D, Hug N, Keith M, Anastasaki C, Patton EE, Grimes G, et al. DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res. 2013;41:8319–31.
Balasubramanian M, Hurst J, Brown S, Bishop NJ, Arundel P, DeVile C, et al. Compound heterozygous variants in NBAS as a cause of atypical osteogenesis imperfecta. Bone . 2017;94:65–74.
Park JW, Lee SJ. Foveal hypoplasia in short stature with optic atrophy and Pelger-Huet anomaly syndrome with neuroblastoma-amplified sequence (NBAS) gene mutation. J AAPOS. 2017.
Staufner C, Haack TB, Kopke MG, Straub BK, Kolker S, Thiel C, et al. Recurrent acute liver failure due to NBAS deficiency: phenotypic spectrum, disease mechanisms, and therapeutic concepts. J Inherit Metab Dis. 2016;39:3–16.
Haack TB, Staufner C, Kopke MG, Straub BK, Kolker S, Thiel C, et al. Biallelic mutations in NBAS cause recurrent acute liver failure with onset in infancy. Am J Hum Genet. 2015;97:163–9.
Regateiro FS, Belkaya S, Neves N, Ferreira S, Silvestre P, Lemos S, et al. Recurrent elevated liver transaminases and acute liver failure in two siblings with novel bi-allelic mutations of NBAS. Eur J Med Genet. 2017;60:426–32.
Ehmke N, Graul-Neumann L, Smorag L, Koenig R, Segebrecht L, Magoulas P, et al. De novo mutations in SLC25A24 cause a craniosynostosis syndrome with hypertrichosis, progeroid appearance, and mitochondrial dysfunction. Am J Hum Genet. 2017;101:833–43.
Kamphans T, Krawitz PM. GeneTalk: an expert exchange platform for assessing rare sequence variants in personal genomes. Bioinformatics. 2012;28:2515–6.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Capo-Chichi JM, Mehawej C, Delague V, Caillaud C, Khneisser I, Hamdan FF, et al. Neuroblastoma Amplified Sequence (NBAS) mutation in recurrent acute liver failure: confirmatory report in a sibship with very early onset, osteoporosis and developmental delay. Eur J Med Genet. 2015;58:637–41.
Palagano E, Zuccarini G, Prontera P, Borgatti R, Stangoni G, Elisei S, et al. Mutations in the neuroblastoma amplified sequence gene in a family affected by acrofrontofacionasal dysostosis type 1. Bone. 2018;114:125–36.
Fischer B, Callewaert B, Schroter P, Coucke PJ, Schlack C, Ott CE, et al. Severe congenital cutis laxa with cardiovascular manifestations due to homozygous deletions in ALDH18A1. Mol Genet Metab. 2014;112:310–6.
Chan WL, Steiner M, Witkos T, Egerer J, Busse B, Mizumoto S, et al. Impaired proteoglycan glycosylation, elevated TGF-beta signaling, and abnormal osteoblast differentiation as the basis for bone fragility in a mouse model for gerodermia osteodysplastica. PLoS Genet. 2018;14:e1007242.
Fischer B, Dimopoulou A, Egerer J, Gardeitchik T, Kidd A, Jost D, et al. Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum Genet. 2012;131:1761–73.
Hennies HC, Kornak U, Zhang H, Egerer J, Zhang X, Seifert W, et al. Gerodermia osteodysplastica is caused by mutations in SCYL1BP1, a Rab-6 interacting golgin. Nat Genet. 2008;40:1410–2.
Kornak U, Reynders E, Dimopoulou A, van Reeuwijk J, Fischer B, Rajab A, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H + -ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32–34.
Van Damme T, Gardeitchik T, Mohamed M, Guerrero-Castillo S, Freisinger P, Guillemyn B, et al. Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa. Am J Hum Genet. 2017;100:216–27.
Jansen EJ, Timal S, Ryan M, Ashikov A, van Scherpenzeel M, Graham LA, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun. 2016;7:11600.
Jansen JC, Cirak S, van Scherpenzeel M, Timal S, Reunert J, Rust S, et al. CCDC115 deficiency causes a disorder of golgi homeostasis with abnormal protein glycosylation. Am J Hum Genet. 2016;98:310–21.
Dimitrov B, Himmelreich N, Hipgrave Ederveen AL, Luchtenborg C, Okun JG, Breuer M, et al. Cutis laxa, exocrine pancreatic insufficiency and altered cellular metabolomics as additional symptoms in a new patient with ATP6AP1-CDG. Mol Genet Metab. 2018;123:364–74.
Acknowledgements
We are grateful to the patients and their families whose cooperation made this study possible. The study was funded by the Deutsche Forschungsgemeinschaft (FI 2240/1-1) to B.F.-Z. This project has received funding from EURO-CDG-2 and the European Community’s Seventh Framework Programme under grant agreement no. 602300 (SYBIL) to U.K. This study was supported by the German Bundesministerium für Bildung und Forschung (BMBF) through the Juniorverbund in der Systemmedizin “mitOmics” (FKZ 01ZX1405C) to TBH.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Fischer-Zirnsak, B., Koenig, R., Alisch, F. et al. SOPH syndrome in three affected individuals showing similarities with progeroid cutis laxa conditions in early infancy. J Hum Genet 64, 609–616 (2019). https://doi.org/10.1038/s10038-019-0602-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0602-8

{kind=link}