
Abstract
Background:
Mutations in the X-linked gene MED12 cause at least three different, but closely related, entities of syndromic intellectual disability. Recently, a new syndrome caused by MED13L deleterious variants has been described, which shows similar clinical manifestations including intellectual disability, hypotonia, and other congenital anomalies.
Methods:
Genotyping of 1,256 genes related with neurodevelopment was performed by next-generation sequencing in three unrelated patients and their healthy parents. Clinically relevant findings were confirmed by conventional sequencing.
Results:
Each patient showed one de novo variant not previously reported in the literature or databases. Two different missense variants were found in the MED12 or MED13L genes and one nonsense mutation was found in the MED13L gene.
Conclusion:
The phenotypic consequences of these mutations are closely related and/or have been previously reported in one or other gene. Additionally, MED12 and MED13L code for two closely related partners of the mediator kinase module. Consequently, we propose the concept of a common MED12/MED13L clinical spectrum, encompassing Opitz-Kaveggia syndrome, Lujan-Fryns syndrome, Ohdo syndrome, MED13L haploinsufficiency syndrome, and others.
Similar content being viewed by others

Main
Regulation of gene expression in eukaryotes is an extremely complex process that is controlled by several mechanisms at multiple stages (1). Alterations of transcriptional regulation are involved in the pathogenesis of many human diseases and most of the affected signaling pathways finally target the core transcriptional machinery (2).
MED12 (Mediator Complex Subunit 12) and MED13L (Mediator Complex Subunit 13 like) encode two subunits of the macromolecular complex known as “Mediator transcription co-activator complex”. The mediator interacts with general transcription factors and RNA polymerase II to regulate transcription. It is the essential coactivator acting as a bridge molecule between transcription factors bound at upstream DNA regulatory elements and the transcriptional machinery. This complex consists of four distinct modules (3,4): the head, middle, and tail modules and all together form the core Mediator Complex that interacts directly with RNA polymerase II and both general and gene-specific transcription factors (5,6), and a fourth “kinase” module that reversibly associates with the core complex which is composed by MED12, MED13, CDK8, and CCNC or their paralogs MED12L, MED13L, and CDK19 (7,8). This module was primarily thought to be involved in transcriptional repression by blocking the binding of the core Mediator Complex to RNA polymerase II because Mediators containing this module are less active than in its absence (4,9). However, there are growing evidences that the kinase module is also involved in transcriptional activation (10).
MED12 gene variants cause at least three different syndromes: Opitz-Kaveggia (FG) (MIM #305450), Lujan-Fryns (MIM #309520), and Ohdo (Maat-Kievit-Brunner type, or OSMKB) (MIM #300895). They all have similar clinical manifestations with multiple congenital anomalies, macrocephaly, hypotonia, and moderate to severe intellectual disability (11). MED13L mutations were initially described in patients with congenital heart defect (transposition of the great arteries). However, according to recent studies, the cardiac phenotype is an unclear feature associated with variants of the MED13L gene (12,13,14,15,16). Recently, Adegbola and colleagues have described the clinical features caused by MED13L deleterious variants. All individuals had intellectual disability and share characteristic facial features, delayed speech and language development, hypotonia, poor coordination, and behavioral problems. None of the patients had a complex congenital heart defect (17).
Here, we report on one patient with a de novo MED12 mutation and two patients with de novo MED13L mutations, not previously described. The clinical features in these patients and in the ones previously reported indicate that mutations in both genes result in similar phenotypes.
Results
All three patients had a normal karyotype and were negative for fragile-X mutation. For the three patients, whole genome dosage analysis was performed by 44K oligonucleotide array CGH (G4426B Agilent Technologies, Palo Alto, CA). No pathogenic variants were detected.
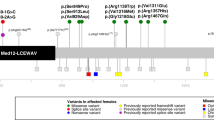
By applying next-generation sequencing, we identified two novel heterozygous potentially relevant missense variants and one nonsense mutation in the MED12 or MED13L genes ( Figure 1 ). Patient 1 showed the de novo mutation c.887G>A in the MED12 gene (NM_005120.2), which causes the aminoacid change arginine to glutamine at position 296 of the protein. Patient 2 carries a de novo variant c.5695G>A in the MED13L gene (NM_015335.4), which is predicted to substitute the glycine at position 1899 by arginine. Both substitutions affect highly conserved aminoacids. In silico predictions of functional relevance suggested a pathogenic effect for both variants (see Table 1 ) which, together with the fact of occurring de novo, are highly suggestive of their pathogenic effect. Patient 3 showed the nonsense mutation c.2524C>T (NM_015335.4), which generates a premature stop codon at exon 14 of the MED13L gene. These mutations were not detected in available databases, including the 1000 Genomes database (EMBL-EBI, Hinxton, Cambridge, UK), dbSNP142, the Exome Variant Server (EVS) (Washington, MO), and the Exome Aggregation Consortium (ExAC) (Cambridge, MA). All variants were submitted to the DECIPHER database (Hinxton, Cambridge, UK).

Distribution of point mutations along MED12 and MED13L genes. Schematic presentation of the MED12 (a) and MED13L (b) genes (intronic regions are not drawn to scale). The mutations identified in the patients reported here are represented by a cross, and previously published mutations are indicated by a rhomb (if mutations are single nucleotide variant), dotted line (if mutations are duplications) or dashed line (if mutations are deletions).
Discussion
In this study, we report one patient with MED12 and two patients with MED13L de novo mutations. All of them display common characteristic facial features, have intellectual disability, speech impairment, motor developmental delay with muscular hypo- or hypertonia and behavioral difficulties. Furthermore, most of the additional clinical features of these patients were previously reported in association to other mutations in the MEDs genes (see Table 2 ). As it can be observed, there is a wide clinical spectrum among the patients carrying a mutation in MED12 or among those with mutation in MED13L. However, the range of clinical manifestations that can be present when MED12 or MED13L are mutated is strikingly similar. For instance, it is worthy to note that all the features shared by patients 2 and 3, both of them with mutation in the MED13L gene (hypotonia, low-set ears, strabismus, hypertelorism, epicantal folds, down-slanting palpebral fissures and depressed nasal bridge, in addition to intellectual disability) are equally reported not only among patients with MED13L mutations, but also in the Opitz-Kaveggia or Lujan-Fryns syndromes (see Table 2 ).
Our results support the findings reported by Adegbola et al. (17) regarding the absence of congenital heart disease associated with mutations in the MED13L gene and, most importantly, emphasizes the clinical similarities between mutations in the MED12 and MED13L.
Based exclusively on the clinical features, patients with mutations in the MED12 gene were previously classified in three different clinical syndromes, although all of them share in common, to a greater or lesser degree, the same clinical findings: somatic growth disorders (affecting weight and/or height), altered size of the head, micrognathia, philtrum anomalies, some dysmorphic facial features, genitourinary anomalies, abnormal hands and feet, in addition to problems in the central nervous system (agenesis of corpus callosum, seizures, autistic and/or hyperactive behavior, intellectual disability and developmental delay) (reviewed in refs. (11,18,19)). A very similar scenery has been found among the patients with mutations in the MED13L gene, who present a wide phenotypic spectrum which basically includes the same clinical features found in association to mutations in the MED12 (13,17,20,21).
According to Daniels and colleagues (22), the mediator kinase module is formed by MED12, MED13, and CDK8, but also can be formed by their respective paralogs MED12L, MED13L, and CDK19, or by a combination of both. These interactions and the possible formation of different kinase modules can explain the phenotypic spectrum presented by our patients and other patients with MED12 and MED13L mutations. Our hypothesis is that mutations in the different genes of the mediator kinase module may result in similar clinical features, whenever these mutations affect the same protein interactions within the mediator complex, and conversely, mutations in the same gene, affecting other domains implicated in distinct protein interactions, may have different clinical consequences. In other words, mutations affecting MED12 and MED13L are equally responsible for the same phenotypic spectrum, which includes different syndromic entities. All these findings indicate that this transcription mechanism is a complex process involving many protein complexes and that deregulation of one of the components can affect has clinical consequences.
Deleterious variants in other mediator-complex genes have been described: a pericentric inversion that cause haploinsufficiency of CDK19, associated with bilateral congenital retinal folds, microcephaly, and mild intellectual disability (23); MED17 missense variants have been linked to postnatal progressive microcephaly with seizures and brain atrophy (24); MED23 variants cause autosomal recessive non-syndromic ID (25), and a homozygous MED25 variant causes autosomal recessive adult onset axonal Charcot-Marie-Tooth neuropathy (CMT2B2, MIM #605589) (26).
In addition to these genes with mutations causing disease, other components of the mediator complex are good candidate genes for pathology. MED1 gene was disrupted in a mouse model in which null mutants died at an early gestational stage due to heart failure and exhibited impaired neuronal development with extensive brain apoptosis (27). Haploinsufficient mice showed growth retardation, pituitary hypothyroidism and widely impaired transcription in certain organs. On the other hand, a homozygous transversion in the MED20 gene was found in two affected sisters from a consanguineous Austrian family, both having infantile-onset basal ganglia degeneration and brain atrophy (28). The patients had a progressive neurodegenerative disorder characterized by severely delayed psychomotor development, spasticity, and dystonia.
Chromatin immunoprecipitation assays showed that depletion of MED26 led to a reduced phosphorylation of the RNA pol II C-terminal domain and a reduced recruitment of RNA pol II and the SEC components AFF4 and CDK9 to both the promoter regions and bodies of the MYC and HSP70 genes (29). The authors hypothesized that MED26 may function as a molecular switch that in a first instance interacts with the RNA pol II initiation complex through direct interactions with TFIID, and, in a second phase, exchanges TFIID with RNA pol II elongation factors to facilitate productive elongation. TFIID multi-protein complex consists of the TATA binding protein (TBP) and 12 additional TBP-associated factors (TAFs) (30). Variants observed in proteins of this complex (TAF1, TAF2, and TBP) have been implicated in several syndromic forms of intellectual disability and developmental delay (31,32,33,34), as well as in X-linked dystonia-parkinsonism (35,36).
Kagey et al. (37) disclosed that Mediator and cohesin (SMC1A, MIM#300040) connect, physical and functionally, the enhancers and promoters of active genes in murine embryonic stem cells. Mediator complex interacts with cohesin and form a ring complex that connect two DNA segments because a DNA looping is formed between the enhancers and promoters occupied by Mediator and cohesin. The cohesin-loading factor NIPBL (MIM #608667) is associated with mediator-cohesin complexes, providing a mechanism to upload cohesin at promoters. Mutations in the NIPBL and SMC1A genes cause Cornelia de Lange syndrome, a genetically heterogeneous syndrome characterized by distinct facial features, hirsutism, developmental delay, intellectual disability, and limb abnormalities (38). It is worthy to note that the phenotypic spectrum of Cornelia de Lange syndrome includes clinical features that are widely shared by MEDs syndromes, further suggesting a functionally-related molecular etiology. Some phenotypic features that overlap between MEDs syndromes and Cornelia de Lange syndrome include anteverted nares, long philtrum, high palate, low-set ears, hearing impairment, strabismus, and intellectual disability.
Other protein complex that may interact with mediator complex is the KMT2D complex. This complex has an important role in histone methylation and transcription activation by recruitment of RNA polymerase II complex. Mutations in KMT2D cause Kabuki syndrome (39). It is important to note that Patient 1 presented some resemblances with Kabuki syndrome, including microcephaly, absence of palmar digital triradius, highly-arched eyebrows and other facial features (strabismus, long and flat philtrum and cleft palate), genitourinaty anomalies (cryptorchidism), hirsutism, gastrointestinal problems, hypotonia, and intellectual disability. It is thus conceivable that other patients with a phenotype suggestive of Kabuki syndrome could be due to MED12 or MED13L mutations.
In conclusion, here we present the clinical and laboratory findings of three patients with novel mutations in two distinct genes of the mediator kinase module, MED12 and MED13L. The phenotypic consequences of these mutations are clearly similar and allow us to propose the concept of MED12/MED13L clinical spectrum, encompassing Opitz-Kaveggia syndrome, Lujan-Fryns syndrome, Ohdo syndrome, MED13L haploinsufficiency syndrome, and others.
The clinical similarities of MEDs syndromes with other genetic conditions caused by alterations in genes that are components of the initiation machinery of transcription, including the cohesin complex, the TFIID complex or even the KMT2D complex, are also remarkable. Altogether, these clinical similarities caused by different genes, coding for functionally related proteins, fully agree with the recently proposed concept of “transcriptomopathies”, a class of disorders that may reflect a global disturbance in transcriptional regulation (40).
Methods
Subjects
Written informed consent for the study was obtained from all the participants. This study was reviewed and approved by the Institutional Ethics Committee of our hospital and research was carried out in compliance with the Helsinki Declaration.
Genomic DNA from the patients and their parents was isolated from peripheral blood using QIAamp DNA Mini Kit and the QIAcube automated extractor (QIAGEN, Hilden, Germany). DNA quality and concentration were measured using the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Rockland, DE) and was stored at −20 °C.
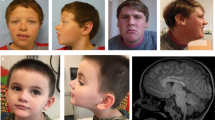
Patient 1. Patient 1 is a 7-y-old male with multiple congenital anomalies, dysmorphic features and psychomotor delay (DECIPHER no. 323184). He was the second child of non-consanguineous healthy parents. He was born at 37th weeks of gestation by spontaneous vaginal delivery, with a weight of 1,850 g (<10 percentile), length of 45 cm (10–25 percentile) and Occipito-Frontal Circumference of 32.5 cm (25–50 percentile). During pregnancy, oligoamnios and intrauterine growth retardation were observed. At birth, he showed the Pierre-Robin sequence (which required tracheostomy) characteristic facial features, and hypertonia. On clinical examination at 21 mo of age, he had sparse hair, arched eyebrows, ocular hypertelorism, blepharophimosis, bulbous nose with split tip and depressed root, long and flat philtrum, cleft palate, micro-retrognathia, and abnormal ears. He had also brachicephaly and microcephaly, absent digital triradius, bilateral cryptorchidism, overlapping fourth toe, and sandal gap with a subcutaneous fold. At neurological examination he showed psychomotor delay: he was not able to hold his head until 6 mo of age, unable to sit until 14 mo and at 21 mo his speech was absent. magnetic resonance imaging and electroencephalogram were reported as normal. Several dismorphic features were considered reminiscent of Kabuki syndrome. In the last examination at age 7 y, he had a weight of 19 kg (3–10 percentile), height of 114 cm (3–10 percentile), and Occipito-Frontal Circumference of 49 cm (<3 percentile). He had strabismus, astigmatism, high grade of hypermetropia (6 diopters), a small open mouth, chronic constipation, and hirsutism ( Figure 2 ). Neurologically, a moderate-severe intellectual disability was noticed, with a very limited use of expressive language and a hyperkinetic behavior.

Pictures of the patients illustrating the typical facial features and other relevant clinical features of patients 1 (a and b) with MED12 mutation, and patient 2 (c and d) and 3(e, f and g) with MED13L mutations.
Patient 2. Patient 2 is an 8-y-old male with multiple congenital anomalies, dysmorphic features and psychomotor delay (DECIPHER no. 323183). He was the second child of non-consanguineous healthy parents. He was born at 35th weeks of gestation, with a weight of 1,880 g (10–25 percentile), length of 43 cm (10–25 percentile), Occipito-Frontal Circumference of 31 cm (25–50 percentile), and hypotonia. During pregnancy, increased nuchal translucency and intrauterine growth retardation were noticed with normal fetal karyotype. He was delivered by cesarean section due to alteration in Doppler-echography. Twenty-four hours after birth, he developed respiratory difficulties that required intubation for 6 d, stayed at intensive care unit for 15 d and hospitalization in the neonatal unit for 1 mo. On physical examination at 17 mo of age, he had asymmetric face, strabismus, left eye ptosis, ocular hypertelorism, downslanting palpebral fissures, bilateral epicanthus, wide and depressed nasal root, tented upper lip with frequent drooling, and low set ears ( Figure 2 ). He also had unilateral hearing loss, patent ductus arteriosus, corrected at 12 mo of age, and atopic dermatitis. Neurological examination showed psychomotor delay, as he did not have head control until 7 mo of age, unable to sit until 17 mo, and absence of speech at 17 mo. His behavioral disturbance fitted into the autism spectrum disorder with autoaggression. Electroencephalogram did not show relevant anomalies. Magnetic resonance imaging showed a prominence of subarachnoid space, predominantly frontal, ventriculomegaly and mega cisterna magna. Signs of periatrial hyperintense areas coexisting with dilated perivascular spaces of Virchow-Robin were considered a normal variant of terminal areas of myelination.
Patient 3. Patient 3 is a 24-y-old male affected with multiple congenital anomalies, dysmorphic features and psychomotor delay (DECIPHER no. 323185). He was the first child of non-consanguineous healthy parents. He was born at 40th weeks of gestation by cesarean section, with a weight of 2,690 g (10–25 percentile), length of 48 cm (25–50 percentile), and Occipito-Frontal Circumference of 35 cm (75–90 percentile). During pregnancy, no problems were detected. Since birth, he had hypotonia with feeding problems. Neurological examination showed psychomotor delay, he did not had head control until the age of 12 mo old and started to walk at 26 mo. On physical examination at 14 y old, he had hypertrichosis, frontal bossing, low-set hair, synophrys, strabismus, downslanting palpebral fissures, epicanthus, bulbous nose, wide and depressed nasal root, narrow palate, wide gingival fold, abnormal implantation of teeth, low-set ears, and retrognathia ( Figure 2 ). Parents referred that he always had chewing difficulties, but normal swallowing. He also had growth retardation, long and thin fingers, kyphosis, pes cavus deformity, joint hyperlaxity, one café-au-lait spot and unilateral renal agenesis. Mild intellectual disability was reported. In the last physical examination at 24 y of age, he had long face with prominent maxilla and retrognathia, big nose, small ears, middle grade of myopia (5 diopters) and hirsutism. He is able to read and write simple sentences.
Capture Array Design, Next-Generation Sequencing, and Analysis Pipeline
A custom SureSelect oligonucleotide probe library was designed to capture 19,878 coding exons of 614 pathogenic and 642 candidate genes associated with intellectual disability (manuscript in preparation). The design includes all the transcripts reported for each target gene in different databases (RefSeq, Ensembl, CCDS, Gencode, VEGA). The SureSelect DNA Standard Design Wizard (Agilent Technologies) was used for probe design with a 2X tiling density and a moderately stringent masking. A total of 71,994 probes, covering 5.073 Mbp (99.48% coverage of targets), were synthesized by Agilent Technologies (Santa Clara, CA). Sequence capture, enrichment, and elution were performed according to the manufacturer’s instructions. The libraries were sequenced on an Illumina HiSeq 2000 platform with a paired-end run of 2 × 90 bp, following the manufacturer’s protocol to generate at least a 100× effective mean depth.
Variation calling was performed with the DNAnexus platform (DNAnexus, Mountain View, CA) through the following pipeline: Fasq paired reads were aligned to the reference human genome UCSC hg19 using the BWA-MEM algorithm from the BWA software package. Mappings were deduplicated using Picard, realigned around sites of known indels, and their quality was recalibrated by looking at covariance in quality metrics with frequently observed variation in the genome. After recalibration, variants were called with the GATK Unified Genotyper module. This pipeline follows the Broad Institute’s recommendations for best practices in variant calling. Variants on regions with low mappability or variants in which there was not at least one sample with read depth ≥10 were filtered out. Annotation of nucleotide variants was performed by the Ion Reporter Software (Life Technologies, Carlsbad, CA).
Prioritization of Variants
To evaluate the putative clinical impact of the variants, the following criteria were applied: (i) an allele frequency <0.01 in the 1000 genomes or EVS databases; (ii) stop gain, frameshift and splicing variants were a priori considered as most likely to be pathogenic; (iii) for missense mutations, amino acid conservation and prediction of pathogenicity (SIFT, Polyphen-2 and Grantham); (iv) a de novo occurrence (dominant inheritance), the presence of two mutant alleles in the same gene, each from a different parent (recessive inheritance), or maternal inheritance of X-linked variants; (v) the absence of the variant in other samples (in-house database); (vi) phenotypic consistency with clinical signs associated to mutations in the same gene when available. To evaluate the possible effect of synonymous or intronic variants in gene splicing, we used the Human Splicer Finding web tool.
Validation by Sanger Sequencing
Relevant variants were resequenced by Sanger sequencing. After PCR amplification from DNA of the patient and his/her parents using specific primers, bidirectional sequencing was performed using the BigDye Terminator v1.1 kit following manufacturer’s protocol on an ABI PRISM 3500 automated sequencer (Life Technologies). All primers for amplification and sequencing were selected with exon-primer (primers and PCR conditions are available on request).
Statement of Financial Support
This work was supported by grant PI14/00350 (ISCIII - Acción Estratégica en Salud 2013–2016; FEDER -Fondo Europeo de Desarrollo Regional). A.C-L. is supported by grant X Convocatoria Anual de Ayudas a proyectos de investigación en la áreas de psiquiatría de la infancia y adolescencia y neurociencias en el niño (Fundación Alicia Koplowitz- 2014).
Disclosure
The authors declare no conflict of interest.
References
Liu X, Bushnell DA, Kornberg RD. RNA polymerase II transcription: structure and mechanism. Biochim Biophys Acta 2013;1829:2–8.
Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell 2013;152:1237–51.
Dotson MR, Yuan CX, Roeder RG, et al. Structural organization of yeast and mammalian mediator complexes. Proc Natl Acad Sci USA 2000;97:14307–10.
Taatjes DJ, Näär AM, Andel F 3rd, Nogales E, Tjian R. Structure, function, and activator-induced conformations of the CRSP coactivator. Science 2002;295:1058–62.
Taatjes DJ. The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci 2010;35:315–22.
Conaway RC, Conaway JW. Origins and activity of the Mediator complex. Semin Cell Dev Biol 2011;22:729–34.
Borggrefe T, Davis R, Erdjument-Bromage H, Tempst P, Kornberg RD. A complex of the Srb8, -9, -10, and -11 transcriptional regulatory proteins from yeast. J Biol Chem 2002;277:44202–7.
Malumbres M, Harlow E, Hunt T, et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol 2009;11:1275–6.
Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev 2009;23:439–51.
Conaway RC, Conaway JW. Function and regulation of the Mediator complex. Curr Opin Genet Dev 2011;21:225–30.
Graham JM Jr, Schwartz CE. MED12 related disorders. Am J Med Genet A 2013;161A:2734–40.
Lei L, Lin H, Zhong S, et al. Analysis of mutations in 7 candidate genes for dextro-Transposition of the great arteries in Chinese population. J Thorac Dis 2014;6:491–6.
van Haelst MM, Monroe GR, Duran K, et al. Further confirmation of the MED13L haploinsufficiency syndrome. Eur J Hum Genet 2015;23:135–8.
Redin C, Gérard B, Lauer J, et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet 2014;51:724–36.
Gilissen C, Hehir-Kwa JY, Thung DT, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014;511:344–7.
Cafiero C, Marangi G, Orteschi D, et al. Novel de novo heterozygous loss-of-function variants in MED13L and further delineation of the MED13L haploinsufficiency syndrome. Eur J Hum Genet 2015;23:1499–504.
Adegbola A, Musante L, Callewaert B, et al. Redefining the MED13L syndrome. European Journal of Hum Genet 2015; 00: 1–9.
Schwartz CE, Tarpey PS, Lubs HA, et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet 2007;44:472–7.
Vulto-van Silfhout AT, de Vries BB, van Bon BW, et al. Mutations in MED12 cause X-linked Ohdo syndrome. Am J Hum Genet 2013;92:401–6.
Yamamoto T, Shimojima K. A novel MED12 mutation associated with non-specific X-linked intellectual disability. Hum Genome Var 2015;2:15018.
Utami KH, Winata CL, Hillmer AM, et al. Impaired development of neural-crest cell-derived organs and intellectual disability caused by MED13L haploinsufficiency. Hum Mutat 2014;35:1311–20.
Daniels DL, Ford M, Schwinn MK, et al. Mutual exclusivity of MED12/MED12L, MED13/13L, and CDK8/19 paralogs revealed within the CDK-mediator kinase module. J Proteomics Bioinform S2: 004. doi:10.4172/jpb.S2-004
Mukhopadhyay A, Kramer JM, Merkx G, et al. CDK19 is disrupted in a female patient with bilateral congenital retinal folds, microcephaly and mild mental retardation. Hum Genet 2010;128:281–91.
Kaufmann R, Straussberg R, Mandel H, et al. Infantile cerebral and cerebellar atrophy is associated with a mutation in the MED17 subunit of the transcription preinitiation mediator complex. Am J Hum Genet 2010;87:667–70.
Hashimoto S, Boissel S, Zarhrate M, et al. MED23 mutation links intellectual disability to dysregulation of immediate early gene expression. Science 2011;333:1161–3.
Leal A, Huehne K, Bauer F, et al. Identification of the variant Ala335Val of MED25 as responsible for CMT2B2: molecular data, functional studies of the SH3 recognition motif and correlation between wild-type MED25 and PMP22 RNA levels in CMT1A animal models. Neurogenetics 2009;10:275–87.
Ito M, Yuan CX, Okano HJ, Darnell RB, Roeder RG. Involvement of the TRAP220 component of the TRAP/SMCC coactivator complex in embryonic development and thyroid hormone action. Mol Cell 2000;5:683–93.
Vodopiutz J, Schmook MT, Konstantopoulou V, et al. MED20 mutation associated with infantile basal ganglia degeneration and brain atrophy. Eur J Pediatr 2015;174:113–8.
Takahashi H, Parmely TJ, Sato S, et al. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 2011;146:92–104.
Papai G, Weil PA, Schultz P. New insights into the function of transcription factor TFIID from recent structural studies. Curr Opin Genet Dev 2011;21:219–24.
Rooms L, Reyniers E, Scheers S, et al. TBP as a candidate gene for mental retardation in patients with subtelomeric 6q deletions. Eur J Hum Genet 2006;14:1090–6.
Hellman-Aharony S, Smirin-Yosef P, Halevy A, et al. Microcephaly thin corpus callosum intellectual disability syndrome caused by mutated TAF2. Pediatr Neurol 2013;49:411–416.e1.
Abu-Amero KK, Hellani A, Salih MA, et al. Ophthalmologic abnormalities in a de novo terminal 6q deletion. Ophthalmic Genet 2010;31:1–11.
O’Rawe JA, Wu Y, Dörfel MJ, et al. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am J Hum Genet 2015;97:922–32.
Makino S, Kaji R, Ando S, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet 2007;80:393–406.
Herzfeld T, Nolte D, Grznarova M, Hofmann A, Schultze JL, Müller U. X-linked dystonia parkinsonism syndrome (XDP, lubag): disease-specific sequence change DSC3 in TAF1/DYT3 affects genes in vesicular transport and dopamine metabolism. Hum Mol Genet 2013;22:941–51.
Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010;467:430–5.
Deardorff MA, Kaur M, Yaeger D, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet 2007;80:485–94.
Ho HH, Eaves LC. Kabuki make-up (Niikawa-Kuroki) syndrome: cognitive abilities and autistic features. Dev Med Child Neurol 1997;39:487–90.
Yuan B, Pehlivan D, Karaca E, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest 2015;125:636–51.
Acknowledgements
The authors sincerely thank all the participants in the present study.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Caro-Llopis, A., Rosello, M., Orellana, C. et al. De novo mutations in genes of mediator complex causing syndromic intellectual disability: mediatorpathy or transcriptomopathy?. Pediatr Res 80, 809–815 (2016). https://doi.org/10.1038/pr.2016.162
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.162
This article is cited by
-
Cyclin-dependent kinases and rare developmental disorders
Orphanet Journal of Rare Diseases (2020)
-
Report of a de novo c.2605C > T (p.Pro869Ser) change in the MED13L gene and review of the literature for MED13L-related intellectual disability
Italian Journal of Pediatrics (2020)
-
MED13L-related intellectual disability: involvement of missense variants and delineation of the phenotype
neurogenetics (2018)
