
Abstract
Introduction:
The NaKCl cotransporter NKCC1 facilitates intraneuronal chloride accumulation in the developing brain. Bumetanide (BUM), a clinically available diuretic, inhibits this chloride transporter and augments the antiepileptic effects of phenobarbital (PB) in neonatal rodents. In a neonatal cerebral hypoxia–ischemia (HI) model, elicited by right carotid ligation, followed by 90 min 8% O2 exposure in 7-d-old (P7) rats, PB increases the neuroprotective efficacy of hypothermia (HT). We evaluated whether BUM influenced the neuroprotective efficacy of combination treatment with PB and HT.
Methods:
P7 rats underwent HI lesioning; 15 min later, all received PB (30 mg/kg), and 10 min later, half received BUM (10 mg/kg, PB-HT+BUM) and half received saline (PB-HT+SAL). One hour after HI, all were cooled (30 °C, 3 h). Contralateral forepaw sensorimotor function and brain damage were evaluated 1–4 wk later.
Results:
Forepaw functional measures were close to normal in the PB-HT+BUM group, whereas deficits persisted in PB-HT+SAL controls; there were corresponding reductions in right cerebral hemisphere damage (at P35, % damage: PB-HT+BUM, 21 ± 16 vs. 38 ± 20 in controls).
Discussion:
These results provide evidence that NKCC1 inhibition amplifies PB bioactivity in the immature brain and suggest that coadministration of PB and BUM may represent a clinically feasible therapy to augment the neuroprotective efficacy of therapeutic HT in asphyxiated neonates.
Similar content being viewed by others

Main
In neonates with hypoxic–ischemic encephalopathy, therapeutic hypothermia (HT) (initiated within the first 6 h of life) is associated with reductions in death and neurological impairment at 18 mo (1). However, more than 40% of treated infants have poor neurodevelopmental outcomes, and there is an urgent need to identify interventions that can effectively supplement HT.
In experimental models of neonatal hypoxic–ischemic (HI) brain injury, a broad range of therapeutic agents including phenobarbital (PB) augment hypothermic neuroprotection (2,3). In a well-characterized model of neonatal HI brain injury, elicited by unilateral carotid artery ligation, followed by 90 min 8% oxygen exposure, in 7-d-old (P7) rats, early post-HI treatment with PB improved the neuroprotective efficacy of delayed onset brief moderate HT. This combination therapy resulted in sustained improvements in sensorimotor function and a greater than 50% reduction in brain damage, in comparison with saline (SAL)-injected HT-treated controls (3).
PB, a γ-amino-butyric acid agonist, is the antiepileptic drug used most frequently to treat neonatal seizures, although its efficacy is limited (4). Recent studies have provided important insights into the mechanisms underlying its limited anticonvulsant efficacy in neonates and have suggested pharmacological strategies to overcome them (5,6,7). In mature neurons, γ-amino-butyric acid triggers membrane hyperpolarization and neuronal inhibition because of the passive influx of chloride down its electrochemical gradient. In neonatal cortex (rodent and human), the developmentally regulated chloride transporter NKCC1 is expressed, and it facilitates intraneuronal chloride accumulation. Because immature neurons have high chloride concentrations, γ-amino-butyric acid triggers chloride efflux and membrane depolarization.
Moreover, NKCC1 expression may be upregulated both by neonatal HI (8) and by seizures (7). Bumetanide (BUM), a clinically available loop diuretic, inhibits NKCC1 and augments the antiepileptic effects of PB in a neonatal rat seizure model (6). A pilot clinical study of BUM as add-on treatment after PB administration for newborn seizures is underway (Clinical trials.gov, NCT00830531).
This study evaluated the impact of BUM on the neuroprotective efficacy of combination therapy with PB and HT in the neonatal HI model. We found that BUM improved the neuroprotective efficacy of treatment with PB and HT, and that HT was essential to achieve optimal benefit from combination drug therapy.
Results
Physiological Measures
Among animals allocated to Protocols 1–4, 152/154 survived until P14; in Protocol 5, 35/36 animals survived until P35. In the first day after lesioning, BUM-treated animals gained less weight than controls (0.01 ± 1 vs. 0.85 ± 0.9 g), but weights did not differ between BUM- and SAL-treated animals at P14 or P35.
Table 1 summarizes sequential temperature measurements for all protocols. In the first two sets of experiments that evaluated combination treatment with BUM and HT (Protocols 1 and 2), during HT mean body temperatures were slightly lower in BUM-treated than in SAL control groups (range: −0.3 to −0.9 °C, P < 0.05, repeated-measures ANOVA). In the third set of experiments, which evaluated combination treatment with two doses of BUM (Protocol 3), temperatures did not differ. In the fourth set of experiments (Protocol 4), the only variable was post-HI temperature management, and mean temperatures during this intervention differed substantially (range: −4.4 to −6 °C, P < 0.001, ANOVA) between groups. Of note, when Protocol 2 was replicated to assess late outcomes (Protocol 5), there were no significant treatment-related temperature differences. In two Protocol 5 experiments, measurements were also obtained 60 min after the end of cooling, when pups were recovering with their dams, and their temperatures did not differ.
Brain Damage on P14
Figure 1 summarizes pathology outcomes from Protocols 1–4 on P14. Right cerebral hemisphere damage did not differ between animals that received injections of BUM (10 mg/kg) or SAL and then underwent the brief HT treatment (Protocol 1, Figure 1a ); in both groups, the mean percentage of right cerebral hemisphere damage was 34%, which was within the range expected after 90-min HI. Combination treatment with PB (30 mg/kg), BUM (10 mg/kg), and HT resulted in attenuation of right cerebral hemisphere damage (18 ± 16%, as compared with 27 ± 17% in animals that received SAL instead of BUM, P < 0.04, Mann–Whitney test) (Protocol 2, Figure 1b ). In the next group of experiments (Protocol 3, Figure 1c ), damage was again attenuated by combination treatment with PB, BUM (10 mg/kg), and HT; in contrast, treatment effects were lost with a lower dose of BUM (2.5 mg/kg) (P < 0.01, Mann–Whitney test). To determine if HT contributed to neuroprotection conferred by the combination of PB and BUM, in Protocol 4 all animals were treated with PB and BUM and half underwent HT (n = 24/group; Figure 1d ). Right cerebral hemisphere damage was lower in the HT-treated group (15 ± 17% vs. 27 ± 20%, P <0.04, Mann–Whitney). Note that results of the three groups of experiments that included a treatment arm that combined PB, BUM (10 mg/kg), and HT, right cerebral hemisphere damage in this treatment arm was similar (18 ± 16%, 14 ± 9%, and 15 ± 17%).

Comparison of right cerebral hemisphere brain damage on P14. All animals were lesioned on P7 (see Methods and Figure 5 ), and brain damage was measured on P14. Percentage damage was calculated from bilateral cerebral hemisphere measurements with the formula: 100 × (left − right/left); group data were compared with nonparametric Mann–Whitney tests. Results are presented as box and whisker plots (boxes extend from the 25th to 75th percentiles, and whiskers from 10th to 90th percentiles); horizontal bars within each box represent group medians. Panel (a) illustrates similar outcomes in bumetanide (BUM)- and saline (SAL)-treated animals that also underwent hypothermia (HT). Panel (b) illustrates that in animals that received phenobarbital (PB), addition of BUM, as compared with SAL, prior to hypothermia (HT), reduced the severity of right cerebral hemisphere damage (*P < 0.04). Panel (c) compares the effects of two doses of BUM (2.5 mg/kg and 10 mg/kg), in groups that both were treated with PB+HT and illustrates that only the higher dose of BUM conferred neuroprotection (†P < 0.01). Panel (d) compares outcomes in animals that received the same drug treatments (PB 30 mg/kg and BUM 10 mg/kg), followed either by HT or normothermia (NT) (see Methods). Brain damage was lower in the HT group (*P < 0.04).
Sensorimotor Function and Pathology on P35
To determine whether neuroprotection conferred by combination treatment was sustained, Protocol 2 was replicated. Animals were lesioned on P7 and all received PB (30 mg/kg); half received BUM and half received SAL injections, and all underwent HT. Sensorimotor function was reevaluated weekly up to P35, and then brain damage was assessed (n = 17–18/group).
Figure 2a summarizes results of left vibrissae–stimulated forepaw placement scores, assessed weekly. Right forepaw placement scores were all consistently normal (10/10) (data not shown). In animals treated with PB, BUM, and HT, mean left forepaw scores ranged from 9.5 to 10/10; in controls, mean scores remained at 5.3–5.5/10 from P14 to P28 and improved modestly at P35 to 7.2/10. Performance differed markedly between groups at each age (P < 0.001, ANOVA; P < 0.01, Bonferroni post hoc t-tests comparing scores at each age).

Measures of contralateral forepaw sensorimotor function. Results of two measures of sensorimotor function in animals that were lesioned on P7, treated with phenobarbital (PB, 30 mg/kg) and bumetanide (BUM) or saline (SAL), and underwent the same hypothermia (HT) treatment. Sensorimotor function was reevaluated weekly up to P35 (n = 17–18/group). Panel (a) summarizes results of left (“contralateral”) forepaw placement scores (10 trials; normal = 10/10). In animals treated with PB+BUM+HT, mean scores ranged from 9.5–10/10. In SAL-treated controls there were persistent deficits (*P < 0.001, ANOVA). All right forepaw scores were normal (data not shown). Panel (b) summarizes bilateral grip strength measurements. Grip strength increased in both groups; right forepaw grip strength did not differ between groups. In the controls, left forepaw strength was reduced compared to BUM-treated group at each age (P < 0.001, ANOVA; †P < 0.01; ‡P < 0.001, Bonferroni post hoc tests comparing left forepaw values at each age). L, left; R, right.
Figure 2b summarizes results of sequential bilateral grip strength measurements. Grip strength increased in both groups from P21 to P35. Right forepaw grip strength was the same in both groups. In the SAL-treated controls, left forepaw strength was reduced as compared with the BUM-treated group at each age (ANOVA, P < 0.001). Bilateral (left/right) forepaw grip strength ratios remained close to 1 in the BUM-treated group (0.94 ± 0.13, 0.97 ± 0.18, and 0.97 ± 0.18 at P21, P28, and P35 respectively); in the controls, with persistent left forepaw deficits, corresponding ratios were 0.6 ± 0.12, 0.6 ± 0.14, and 0.53 ± 0.1.
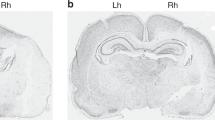
Figure 3 presents illustrative P35 histopathology; Figure 3a,c,e demonstrates a severe lesion with marked right hemisphere atrophy and cortical infarction (from an animal in the PB-HT+SAL group), and Figure 3b,d,f demonstrates a milder lesion with right striatal and hippocampal atrophy (from an animal in the PB-HT+BUM group).

Histopathology. These cresyl-violet stained coronal brain sections illustrate features of histopathology on P35 at corresponding anatomic levels at (a,b) mid-striatum and (c-f) hippocampus. In a,c,e (from an animal treated with PB-HT+SAL), there is marked right-hemisphere atrophy and cortical infarction (arrowheads). In b,d,f (from an animal treated with PB-HT+BUM), there is right-hemisphere atrophy but no infarction. Bar = 1 mm. BUM, bumetanide; HT, hypothermia; PB, phenobarbital; SAL, saline.
Table 2 compares regional volume measurements and percentage damage severity on P35. Left cerebral hemisphere volumes did not differ; right cerebral hemisphere volumes were larger in the BUM-treated group and mean percentage damage was lower (21 ± 16 vs. 39 ± 20%, P < 0.01, Mann–Whitney test). Trends were similar across brain regions (ANOVA; P < 0.0001). Figure 4a compares the pathology scores in the two groups. Although there is a broad range of scores in both groups, median scores are markedly lower in the PB-HT+BUM than in PB-HT+SAL groups (P < 0.001, Mann–Whitney test). Figure 4b evaluates the relationship between pathology scores and right cerebral hemisphere percentage damage in all brain samples and demonstrates that there is a close correlation between the two measures of brain injury (r2 = 0.88; P < 0.0001).

Brain injury measures on P35. (a) Comparison of histopathology scores on P35 in animals that were lesioned on P7, treated with phenobarbital (PB, 30 mg/kg) and bumetanide (BUM) or saline (SAL), and underwent the same hypothermia (HT) treatment (n = 17–18/group). Horizontal bars are median scores; boxes represent 25th and 75th percentiles, and whiskers are minimum and maximum values (*P = 0.001, Mann–Whitney test). (b) Illustrates the strong relationship between pathology scores and percentage right cerebral hemisphere damage values and includes data from both groups (open squares are values from the PB-HT+BUM group and filled triangles are from the PB-HT+SAL group; r2 = 0.88, P < 0.0001).
In summary, in BUM-PB-HT–treated animals, sensorimotor function was preserved, and mean right cerebral hemisphere damage was less than 20%. There were no gender differences in treatment effects.
Discussion
Combined treatment with PB, BUM, and brief moderate HT resulted in sustained improvements in sensorimotor function and reduced brain damage in this neonatal hypoxic–ischemic brain injury model. Although we did not perform a detailed dose–response study, the results demonstrated that the beneficial effects of BUM were dose-dependent. Of note, in the PB-SAL-HT control group, the PB dose administered to both groups was a 25% lower dose than the dose that was administered in the prior study in which PB, in combination with HT, substantially reduced brain damage (3).
BUM enhances the anticonvulsant efficacy of PB in neonatal rodent brain (6), and the timing of drug administration in our protocols suggests that treatment may have limited postischemic seizures. We could not directly evaluate seizure activity because of the technical challenges inherent in performing and analyzing electroencephalograms in P7 rats. The issue of whether or not neonatal seizures amplify ischemic brain injury has been controversial and difficult to address both experimentally and clinically (9,10,11). In the sheep model of fetal asphyxia that provided a foundation for clinical trials of neonatal HT, HT lost its protective efficacy if initiation of cooling was delayed until after seizure onset (12).
In the first group of experiments that evaluated combination treatment with PB, BUM, and HT (Protocol 2), during HT mean body temperatures were slightly lower (range: −0.3 to 0.9 °C) in the BUM than in the SAL control group; temperatures were the same 90 min after the end of cooling. When the protocol was replicated to assess late outcomes (Protocol 5), there were no temperature differences between groups, and degree of HT could not account for sustained neuroprotection.
NKCC1 activation may contribute to ischemic brain injury and BUM can attenuate ischemic injury by diverse mechanisms, including effects on cerebral vessels, astrocytes, and neurons (8,13,14). Although BUM may have modest intrinsic neuroprotective properties, we found no attenuation of injury in animals treated with BUM in combination with HT.
One of the important considerations in evaluating the translational potential of these results is whether these treatments are safe. Although both drugs have been used for many years to treat neonates, there is experimental evidence that both PB and BUM may have adverse effects on brain development. In rodents, several antiepileptic drugs, including PB, and anesthetics (i.e., agents that suppress synaptic activity) cause dose-dependent apoptotic neurodegeneration in the developing brain (15). The relevance of these findings to human neonates remains uncertain. Of interest in the context of this study, HT may suppress this mechanism of drug-induced neuronal damage (16). There is also recent evidence that chronic BUM administration during the pre- and early postnatal period can have adverse effects on brain development and disrupt cortical excitatory synapse formation in mice (17). The authors of this study raised concerns regarding risks of treatment of neonates with BUM. Yet it must be emphasized that deleterious effects were noted only with chronic fetal and early postnatal NKCC1 inhibition. Whether these findings are relevant to acute BUM treatment of human infants or in the setting of heightened NKCC1 expression, as occurs with hypoxia–ischemia, is uncertain. Moreover, the potential risks of chronic PB and/or BUM therapy are likely to be substantially outweighed, if brief treatment confers neuroprotection in the setting of neonatal hypoxic–ischemic brain injury.
Our findings raise many questions. We could not assess whether treatment effects were mediated by prevention of seizures. Nor did we evaluate the comparative safety and efficacy of the combination of PB and BUM with either higher doses of PB alone, or with other anticonvulsants such as topiramate. Nonetheless, our results provide support for the hypothesis that drug therapy could augment hypothermic neuroprotection in neonates with hypoxic–ischemic encephalopathy. It could be feasible to administer drugs either intrapartum in a high risk situation, or in the early postnatal period, after resuscitation, and prior to, or concurrently with initiation of HT.
Implementation of combination therapy neonatal neuroprotection clinical trials will be complex and expensive. Yet both infants who have poor outcomes with HT treatments and infants who are ultimately classified as having “good” outcomes, but who may have the potential for better long-term function, could benefit. Our data provide evidence that rational combination therapies, such as PB plus BUM plus HT, have potential to improve outcomes after neonatal hypoxic–ischemic brain injury.
Methods
Surgery
P7 Sprague-Dawley rats (11–12/experiment, gender-balanced; Charles River, Portage, MI), anesthetized with isofluorane, underwent right common carotid artery ligation; 90 min later, they were exposed to 8% oxygen (balance nitrogen) for 90 min (3,18). This lesioning procedure typically results in 30–40% tissue loss of the right cerebral hemisphere, as compared with the weight or volume of the left cerebral hemisphere, 1–5 wk later. Strengths of the model include relative simplicity and reproducibility, low mortality, and ability to integrate functional and pathological outcome measures. Weaknesses include intra- and interexperiment variation in severity of damage, the limited behavioral repertoire of rodents, and inability to replicate important elements of neonatal intensive care unit practice. All procedures were approved by the University of Michigan Committee on Use and Care of Animals, and efforts were made to minimize the numbers of animals used.
Temperature Management
In P7 rats, ambient temperature determines body and brain temperature (19). During hypoxia, animals remained in acrylic containers that were partially submerged in a water-bath (36.5 °C, 90 min). Then they were placed in recovery incubators (36.5 °C, 60 min). One hour later, they were moved to circulating air incubators set at 30 °C (“HT”; 3 h); the incubator was partitioned to prevent huddling. This delayed cooling intervention, alone, confers no benefit on sensorimotor or histology outcomes (18).
In experiments in which the impact of HT was evaluated, controls remained in the recovery incubators (36.5 °C, 3 h; described in the protocol as “normothermia”). All animals were then returned to the dams. Rectal temperatures were measured intermittently (YSI thermometer 43T with probe 554; YSI, Yellow Springs, OH) before surgery, at the end of hypoxia, 15 min later, 60 min later (just before HT), 15 and 30 min after cooling began, and at the end of HT; in two experiments, temperatures were also measured 60 min after pups were returned to the dams.
Drugs
PB, 40 mg/kg, together with delayed onset HT, attenuates brain damage in this model (3). The dose was reduced to 30 mg/kg in this study; this dose is commonly used to treat neonatal seizures (4). Animals received intraperitoneal injections of PB (30 mg/kg) at 15 min after the end of hypoxia, and a second injection (of BUM or SAL) 10 min later. As BUM has a very short half-life in rats, a high dose (10 mg/kg) was selected (20), and preliminary experiments (data not shown) demonstrated that this dose of BUM, itself, in combination with HT (3 h) had no effects on survival or severity of brain damage. In this study, we reevaluated BUM plus HT and also evaluated a lower dose (2.5 mg/kg) in combination with PB and HT. Drugs, purchased from Sigma Chemicals, St. Louis, MO, were dissolved in SAL.
Outcome Measures
All experiments evaluated survival and weight gain. In initial experiments, brain damage was evaluated 1 wk after lesioning. Subsequently, one combination treatment protocol was replicated, and sensorimotor function and brain damage were evaluated up to 4 wk later.
Brain damage. To quantify damage severity, coronal 20-µm brain sections were stained with cresyl violet; bilateral cross-sectional areas of striatum, neocortex, hippocampus, and cerebral hemisphere were measured on regularly spaced sections from the level of the anterior genu to the posterior genu of the corpus callosum; these were captured and analyzed in ImageJ (http://rsbweb.nih.gov/ij/) using the dot-grid method. Bilateral volumes were estimated by multiplying the sum of areas by the distance between sections.
In experiments that evaluated P35 outcomes, neuropathology was also scored (range: 0–4) in seven brain regions by an observer unaware of treatment group assignment (F.S.S.), as previously described (3).
Sensorimotor testing. Bilateral vibrissae-stimulated forepaw placement was tested (3); the number of successful placements in 10 trials/side was recorded. In lesioned animals, contralateral deficits in forepaw placement are detectable at P14. One point was assigned for full forepaw extension; quality and speed of movement were not scored. Animals were tested at weekly intervals from P14 to P35.
Forepaw grip strength (maximal force applied) was measured using a Grip Strength Meter (Columbus Instruments, Columbus, OH) on P21, P28, and P35. Three measurements were taken for each forepaw; mean values/side and ratios of left/right forepaw strength were calculated.
Study Design
All animals underwent the same lesioning procedure; each experiment included equal numbers of animals of each gender in treatment and control groups. Initial studies (Protocols 1–4) ( Figure 5 ) evaluated brain damage on P14; then the protocol to evaluate the impact of addition of BUM to combination treatment with PB and HT was replicated, and sensorimotor and pathology outcomes were evaluated up to P35 (Protocol 5).

Summary of four treatment protocols. This graphic outlines all components of the four treatment protocols studied. In all experiments, all animals underwent the same hypoxic–ischemic (HI) lesioning procedure on P7 (see Methods), and all treatments were administered after the end of hypoxia exposure (injections at 15 or 25 min later, and hypothermia (HT), beginning at 60 min later). Only one phenobarbital (PB) dosage was included (30 mg/kg; PB 30). Two bumetanide (BUM) doses were studied, 10 mg/kg (BUM 10) and 2.5 mg/kg (BUM 2.5). NT, normothermia; SAL, physiological saline.
Protocol 1 evaluated the impact of BUM in combination with delayed-onset brief moderate HT. Half of the animals received i.p. injections of BUM (10 mg/kg) and half received SAL injections, and all animals underwent the same HT treatment (n = 17–18/group).
Protocol 2 evaluated the impact of BUM on the neuroprotective efficacy of combined treatment with PB and HT. All animals received PB; half received BUM (10 mg/kg) and half received SAL injections; all underwent HT (n = 24/group).
Protocol 3 compared the two doses of BUM, 2.5 mg/kg and 10 mg/kg, on the neuroprotective efficacy of combined treatment with PB and HT. All animals received PB; half then received BUM (10 mg/kg) and half received BUM (2.5 mg/kg) injections, and all underwent HT (n = 11/group).
Protocol 4 evaluated whether HT contributed to the neuroprotection conferred by combination treatment with PB and BUM. All animals received PB and BUM; half underwent HT and half remained in a “normothermia” (36.5 °C) incubator for 3 h (n = 24/group).
Protocol 5 replicated Protocol 2 (all animals received PB; half received BUM and half received SAL injections; all underwent HT) and included sensorimotor testing up to P35 and pathology assessment (n = 18/group).
Data Analysis
Serial temperature measurements for each protocol were compared with repeated-measures ANOVA. Percentage right cerebral hemisphere brain damage values on P14 and P35 were calculated from bilateral volume measurements with the formula (100 × (left − right)/left), and group values were compared with nonparametric Mann–Whitney tests. Serial vibrissae scores, and bilateral grip strength measurements were compared with ANOVA; post hoc Bonferroni multiple comparison tests were applied to compare values at each age. Regional volumes and regional percentage damage values on P35 were compared by two-way ANOVA and post hoc t-tests. Pathology scores on P35 were compared with Mann–Whitney tests. Linear regression modeling was applied to evaluate the relationship between pathology scores and right cerebral hemisphere percentage damage.
Statement of Financial Support
This study was supported by grant HD 060348 from the National Institutes of Health (to F.S.S.) and a grant from the GorgEffen Gift Fund (to J.D.E.B.).
References
Edwards AD, Brocklehurst P, Gunn AJ, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 2010;340:c363.
Cilio MR, Ferriero DM . Synergistic neuroprotective therapies with hypothermia. Semin Fetal Neonatal Med 2010;15:293–8.
Barks JD, Liu YQ, Shangguan Y, Silverstein FS . Phenobarbital augments hypothermic neuroprotection. Pediatr Res 2010;67:532–7.
Silverstein FS, Jensen FE . Neonatal seizures. Ann Neurol 2007;62:112–20.
Dzhala VI, Talos DM, Sdrulla DA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med 2005;11:1205–13.
Dzhala VI, Brumback AC, Staley KJ . Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol 2008;63:222–35.
Dzhala VI, Kuchibhotla KV, Glykys JC, et al. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci 2010;30:11745–61.
Dai Y, Tang J, Zhang JH . Role of Cl- in cerebral vascular tone and expression of Na+-K+-2Cl- co-transporter after neonatal hypoxia-ischemia. Can J Physiol Pharmacol 2005;83:767–73.
Silverstein FS . Do seizures contribute to neonatal hypoxic-ischemic brain injury? J Pediatr 2009;155:305–6.
Wirrell EC, Armstrong EA, Osman LD, Yager JY . Prolonged seizures exacerbate perinatal hypoxic-ischemic brain damage. Pediatr Res 2001;50:445–54.
Yager JY, Armstrong EA, Jaharus C, Saucier DM, Wirrell EC . Preventing hyperthermia decreases brain damage following neonatal hypoxic-ischemic seizures. Brain Res 2004;1011:48–57.
Gunn AJ, Bennet L, Gunning MI, Gluckman PD, Gunn TR . Cerebral hypothermia is not neuroprotective when started after postischemic seizures in fetal sheep. Pediatr Res 1999;46:274–80.
Yan Y, Dempsey RJ, Flemmer A, Forbush B, Sun D . Inhibition of NaKCl cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res 2003;961:22–31.
Pond BB, Berglund K, Kuner T, Feng G, Augustine GJ, Schwartz-Bloom RD . The chloride transporter Na-K-Cl cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci 2006;26:1396–406.
Bittigau P, Sifringer M, Ikonomidou C . Antiepileptic drugs and apoptosis in the developing brain. Ann N Y Acad Sci 2003;993:103–14; discussion 123–4.
Creeley CE, Olney JW . The young: neuroapoptosis induced by anesthetics and what to do about it. Anesth Analg 2010;110:442–8.
Wang DD, Kriegstein AR . Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb Cortex 2011;21:574–87.
Liu Y, Barks JD, Xu G, Silverstein FS . Topiramate extends the therapeutic window for hypothermia-mediated neuroprotection after stroke in neonatal rats. Stroke 2004;35:1460–5.
Yager J, Towfighi J, Vannucci RC . Influence of mild hypothermia on hypoxic-ischemic brain damage in the immature rat. Pediatr Res 1993;34:525–9.
Brandt C, Nozadze M, Heuchert N, Rattka M, Löscher W . Disease-modifying effects of phenobarbital and the NKCC1 inhibitor bumetanide in the pilocarpine model of temporal lobe epilepsy. J Neurosci 2010;30:8602–12.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, Y., Shangguan, Y., Barks, J. et al. Bumetanide augments the neuroprotective efficacy of phenobarbital plus hypothermia in a neonatal hypoxia–ischemia model. Pediatr Res 71, 559–565 (2012). https://doi.org/10.1038/pr.2012.7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2012.7
This article is cited by
-
Neuroprotective Effects of Lacosamide and Memantine on Hyperoxic Brain Injury in Rats
Neurochemical Research (2020)
-
Evaluating anti-epileptic drugs in the era of therapeutic hypothermia
Pediatric Research (2019)
-
Effects of therapeutic hypothermia on white matter injury from murine neonatal hypoxia–ischemia
Pediatric Research (2017)
-
Treatment temperature and insult severity influence the neuroprotective effects of therapeutic hypothermia
Scientific Reports (2016)
-
Treatment during a vulnerable developmental period rescues a genetic epilepsy
Nature Medicine (2015)
