
Abstract
In mice, secondary alveolar septal formation primarily occurs during a brief postnatal period and is accompanied by transient expansion of the interstitial lung fibroblast (LF) population. PDGF-A, which solely signals through PDGF-receptor-alpha (PDGF-Rα), is required for expansion, but the receptor's relevant downstream targets remain incompletely defined. We have evaluated the proliferation, apoptosis, and differential response to the selective protein tyrosine kinase inhibitor, imatinib, by pdgfrα-expressing LF (pdgfrα-LF) and compared them with their nonexpressing LF counterparts. Our objective was to determine whether diminished signaling through PDGF-Rα-mediated pathways regulates the decline in myofibroblasts, which accompanies septal thinning and ensures more efficient alveolar gas exchange. Using quantitative stereology and flow cytometry at postnatal d 12 and 14, we observed that imatinib caused a selective suppression of proliferation and an increase in apoptosis. The number of the alpha smooth muscle actin (αSMA) producing pdgfrα-LF was also reduced. Using cultures of neonatal mouse LF, we showed that imatinib did not suppress PDGF-Rα gene expression but reduced PDGF-A-mediated Akt phosphorylation, potentially explaining the increase in apoptosis. Our findings are relevant to bronchopulmonary dysplasia in which positive pressure ventilation interferes with myofibroblast depletion, septal thinning, and capillary maturation.
Similar content being viewed by others


Main
Mammalian secondary pulmonary alveolar septa form from existing saccules through eruption (lifting off from the primary septum), elongation, and thinning, thereby increasing the gas exchange surface (1). This requires coordinated expansion of the epithelial and endothelial surfaces as well as a supporting structural core. The structural core includes interstitial fibroblasts, which contain stabilizing cytoskeletal elements and secrete extracellular matrix structural proteins. Distinct populations of interstitial lung fibroblasts (LF) have been characterized based on Thy-1 (CD90) (2), the abundance of intracellular neutral lipid droplets, (3) or the presence of alpha-smooth muscle actin (αSMA, ACTA2) (4). All these phenotypic variants involve differences in PDGF-Rα signaling, which is critical for secondary alveolar septation (2,5).
PDGF-A solely signals through PDGF-Rα, suggesting that PDGF-Rα regulates one or more required interstitial fibroblast processes, for example proliferation, migration, extracellular matrix production, or cell loss. Because lung pdgf-rα mRNA declines during P1 through P14 (5), whereas PDGF-AA mRNA remains constant (6), the decline in pdgf-rα expressing LF (pdgfrα-LF) does not result from diminished PDGF-AA ligand. However, the abundance of pdgfrα-LF is influenced by receptor activity. When PDGF-Rα is constitutively activated, the pdgfrα-LF subpopulation and septal thickness both increase (7). This suggests that there is a link between PDGF-Rα receptor signaling and the abundance of LF bearing the receptor. To explore the mechanism(s) behind this link, we reasoned that if PDGF-Rα kinase activity regulates the abundance of PDGF-Rα LF, then selectively manipulating protein tyrosine kinase activity should preferentially alter the abundance of LF expressing the receptor. We have used the protein tyrosine kinase inhibitor, imatinib, to study the function of pdgfrα-LF at the beginning of alveolar thinning, P12–P14.
We have examined the diminution in PDGF-Rα-LF, which accompanies septal thinning. Others showed that unbridled signaling by PDGF-A through PDGF-Rα results in excessive thickening of the septal core, which leads to late gestational or early postnatal death from respiratory failure (7,8). We hypothesized that PDGF-Rα signaling must be dampened to achieve the optimal septal thickness for gas exchange. We further hypothesized that pdgf-rα expressing myofibroblasts diminish through decreased proliferation and increased apoptosis. Although we took an indirect approach, it enabled septal formation to be studied in its entirety, which is not possible when PDGF-A signaling is either completely negated (5) or constitutively activated (7).
Although septal thinning is essential, it is unclear how it is coordinated with formation of a mature capillary bed (9). Some cellular features of the thinning process have been described. There is preferential apoptosis of the lipid interstitial fibroblast subpopulation (3). Septal thinning is accompanied by remodeling of the capillary network with a reduction in septal interstitial extracellular matrix proteins and closer apposition between capillaries and the type-1 epithelium (9). Septal thickening that accompanied overexpression of connective tissue growth factor during P1–P14 seemed to inhibit alveolar capillary formation (10). Understanding the thinning process is important because thickened walls are observed in mechanically ventilated lungs of children (11) and baboons (12) with bronchopulmonary dysplasia (BPD).
MATERIALS AND METHODS
Reagents.
Imatinib and oridonin were obtained from LC Laboratories (Woburn, MA) and Calbiochem (La Jolla, CA), respectively; other reagents were described (13). The concentration of imatinib that produces a 50% reduction in tyrosine phosphorylation (IC50) is 0.25 μM for Ableson kinase (Abl) or its close family member, Abelson-related gene (Arg), and 0.3 μM for the PDGF receptors (14). In contrast, no inhibition is observed for other tyrosine kinases (for example, the insulin receptor) at more than 100 μM. Imatinib inhibits cKit, but this is not expressed in resident LF (15). The optimal inhibitory doses of imatinib in lung and LF are known (16).
Animals.
Mice bearing the PDGF-Rα-GFP construct were described (13). Production of green-fluorescent protein (GFP) is controlled by the endogenous pdgf-rα promoter. GFP expression in PDGF-Rα-GFP mice spatially and temporally recapitulated endogenous pdgf-rα expression (17). The mice used in this study carried one pdgf-rα-GFP and one functional pdgf-rα allele and are phenotypically identical to WT mice, except for nuclear GFP marker (17). Protocols have been approved by the Iowa City Veterans Affairs Medical Center animal use committee (13). Aqueous imatinib, 5 mg/kg, was administered by oral gavage twice daily for up to 5 successive days, ending at either P12 or P14. This dose of imatinib is optimal for specifically inhibiting PDGF-Rα, Abl, or cKit in mice (15,18).
Lung fibroblasts were isolated on d P12 or P14 using a modified method (13). The lung tissue was digested for 1 h, interrupted by trituration, and the medium was replenished every 15 min. Adherent cells were dislodged using TrypLE Express (Invitrogen, Carlsbad, CA).
Fluorescence-activated cell sorting analysis of αSMA, Ki67, and annexin-V.
Freshly isolated LF were immumnostained for Ki67 and αSMA and subjected to fluorescence-activated cell sorting (FACS) and the 2.4G2 MAb was added to block mouse Fc receptors (13). At least 20,000 gated events were analyzed. The background fluorescence from the nonimmune IgG controls was subtracted. For analysis of annexin V, LF were fixed for 5 min at 4°C with 0.25% paraformaldehyde and not permeabilized. The washed LF were supplemented with AlexaFluor-647-conjugated annexin V (1:100 dilution, Invitrogen #A23204), made 30 nM in 4′,6-diamidino-2-phenylindole (DAPI).
Tissue preparation and LSCM.
Lungs were uniformly inflated, fixed, sectioned, and stained for αSMA (19). After blocking mouse Fc-receptors with 2.4G2, apoptotic cells were identified using 0.5 μg/mL anticleaved poly-ADP ribose polymerase (PARP, clone F21-852; BD Biosciences, Franklin Lakes, NJ) and 7.5 μg/mL DyLight 549 anti-mouse IgG1 (Jackson ImmunoResearch, West Grove, PA). Nuclei were stained with To-Pro3. Image z-stacks (3 μm and 2 μm intervals for αSMA and cleaved PARP, respectively) were acquired from randomly selected fields using a Zeiss LSM710. The laser intensities and detector gains were optimized and remained constant throughout the imaging session.
Cell culture.
Mouse LF primary were isolated at P12 cultures and subcultured once (13). The medium was changed to OptiMEM-1 supplemented with 2.5% fetal bovine serum (FBS) and CaCl2 to a final calcium concentration of 3 mM 24 h before adding 10 μM imatinib, PDGF-AA (20 ng/mL), or oridonin (2.5 μg/mL). Imatinib and PDGF-AA were added 40 and 10 min, respectively, before cell lysis. LF from PDGF-Rα-GFP+ mice was adhered to vitronectin-coated coverslips. After 24 h, the medium was changed to OptiMEM containing 2% FBS. Sixteen hours later, 5 ng of TGFβ1 per milliliter or imatinib was added to some coverslips, whereas others remained untreated (control). Nuclei containing GFP or Ki67 and all nuclei stained with DAPI were enumerated using fluorescence microscopy.
Analysis of ACTA2 and pdgf-rα mRNA using real-time quantitative PCR.
RNA was isolated, reverse-transcribed, and αSMA, pdgf-rα (Mm00440701_m1), and β-2-microglobulin mRNA were quantified using Taqman Gene Expression Assays (13).
Phospho Akt and PDGF-Rα immunoblotting.
Washed cell layers were harvested in 10 mM Tris-HCl, pH 7.4, 5 mM EDTA, 50 mM NaCl, 50 mM sodium fluoride, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 2 mM Na3VO4, and 20 μg/mL aprotinin. Equal quantities of protein were subjected to SDS-PAGE and transferred to nitrocellulose. Rabbit polyclonal antibodies anti-phospho (Ser 473) Akt (#9271), anti-Akt (#9272, Cell Signaling Technology, Beverly, MA) or anti-phospho (Tyr 742) PDGF-Rα (Invitrogen), and anti-PDGF-Rα (Santa Cruz Biotechnology, Santa Cruz, CA) were diluted 1:1000 and detected using enhanced chemiluminescence.
Analyzing LSCM images.
Laser scanning confocal microscopy (LSCM) z-stacks were analyzed using IPLab (BD Biosciences) (13). The pixels containing Cy3 dye were segmented using a red intensity range from 25 to 255 and saturation ranging from 150 to 255. The accuracy of these ranges was confirmed by visual inspection. The composite pixel area, which contained red and met the segmentation criteria, was ascertained. The mean areas from 12 fields per lung were analyzed, using four control and four imatinib-treated mice.
The z-stacks were analyzed using the optical fractionator and StereoInvestigator software (MicroBrightField, Williston, VT). The cells were classified as follows: nuclear GFP+ (pdgf-rα expressing), ToPro3+ (all nuclei), cleaved PARP surrounding nuclei with ToPro3 (but without GFP), and cleaved PARP around nuclei with GFP. The approximate numbers of cells counted in the total dissector volume for each type of cell were 350, 1100, 2, and 15, respectively. Knowing the dissector volume and the lung-displacement volume, we calculated the number of GFP+ cells per lung. Six regions were analyzed, and the data were averaged for each mouse, using four mice for each age and imatinib treatment condition.
Stereology using paraffin embedded tissue.
To assess the surface area of alveoli and alveolar ducts, and the arithmetic mean thickness of alveolar and alveolar duct walls, three lung slices from each mouse were embedded in paraffin, sectioned at 4.5-μm intervals, and stained with hematoxylin and eosin. The surface area of the alveoli and alveolar ducts was calculated using the cycloids for Sv probe (StereoInvestigator, units, μm−1) multiplied by the displacement volume. Six fields from three separate regions of the lung (18 fields) were evaluated for each of the four control and imatinib-treated mice. The thickness of alveoli and alveolar ducts was ascertained using the point sampled intercept probe. The arithmetic mean length (thickness) was calculated as la = π/4(l) from randomly imaged fields (20). Nine regions from four controls and from four imatinib-treated mice were analyzed at P12 and P14 by counting 200 wall transections.
Statistical methods.
Data are expressed as the mean ± SEM of the number of different mice used or the number of different experiments performed using cell cultures. ANOVA (one-, two-, or three-way as indicated in the figure legends) was performed using Systat (Chicago, IL) and t test using Microsoft Excel. Post hoc tests are described in the figure legends. Values of p < 0.05 were considered significant.
RESULTS
Pdgfrα-LF diminish after P12.
Our previous studies showed that pdgf-rα expressing alveolar cells increased from P4 to P12 (19). To determine whether this increase was influenced by PDGF-Rα function, we exposed mice to imatinib. Figure 1A shows that imatinib diminished the abundance of GFP+ alveolar cells at P12 and P14, suggesting that the maintenance of pdgf-rα-expressing cells depends on active protein tyrosine kinase signaling (PDGF-R, Abl, or both). The abundance of pdgf-rα-expressing cells diminished between P12 and P14 and their loss was enhanced by imatinib.

Imatinib decreases abundance of pdgfrα-GFP+ cells. (A) PDGF-Rα (GFP+) parenchymal cells were enumerated in LSCM z-stacks from four unexposed control (□) and four imatinib-treated (▪) lungs. Actual count 107 fold more than shown. *p < 0.01, imatinib-treated vs age-matched control. †p < 0.01, P14 vs P12 control. Two-way ANOVA, Tukey test. LF isolated at P12 (B) or P14 (C) from control or imatinib-treated lungs. GFP and Ki67 containing subpopulations enumerated using FACS. *p < 0.01, imatinib-treated vs control. Two-way ANOVA, Tukey test.
To confirm that LF comprised the GFP+ population, we isolated LF and assessed the abundance of GFP+ cells using FACS. Epithelial, macrophage, and endothelial cells comprised 8.1 ± 6% (n = 2), 18.1 ± 1% (n = 3), and 1.6 ± 0.5% (n = 3, mean ± SD), respectively, of the cells isolated at P12 (13). Figure 1B shows that at P12, imatinib decreased the abundance of pdgf-rα GFP+ cells, which we previously showed were myofibroblasts (13). Exposure for 4 d, but not 3 or fewer days was sufficient to decrease the pdgfrα-LF at P14 (Fig. 1C).
PDGF-Rα alters LF proliferation.
If PDGF-Rα-signaling is necessary to maintain LF proliferation, then the effect of imatinib should differentially alter the proliferation of pdgfrα-LF. We evaluated the abundance of Ki67 in freshly isolated LF using FACS. Representative FACS scatter plots for P12 are shown in Figure 2A, which demonstrates that the proportion of proliferative GFP+ LF is lower than that of GFP− LF. Nearly, all of the αSMA-containing LF were in the PDGF-Rα-GFP+ subpopulation.

Imatinib reduces proliferation of pdgfrα-LF. (A) Representative scatter plots from FACS for Ki67 and αSMA. x axis: intensity of GFP; y axis: intensities of antibody binding to Ki67 or αSMA. Quadrants (Q) demarcated various subpopulations. A portion of the 20,000 collected events is shown. Specifically stained GFP+, Ki67+ cells (Q2-1) ranged from 250 to 450. LF isolated from PDGF-Rα-GFP mice at P12 (B) or P14 (C) and resolved into GFP− (□) and GFP+ (▪) by FACS. LF staining for Ki67 from mice exposed to imatinib expressed relative to concurrently stained LF from controls. *p < 0.01, imatinib vs time-matched control. Two-way ANOVA, Tukey test. (D) Same FACS analysis as for (B and C), but data expressed as percentage of cells staining with Ki67 relative to the total LF in pdgfrα-expressing (GFP+, ▪) or nonexpressing (GFP−, □) subpopulations (subpop). n = 4 experiments. *p < 0.05, three-way ANOVA, Tukey test.
Figure 2B and C shows that exposure to imatinib suppressed proliferation in LF expressing PDGF-Rα (GFP+, solid bars) relative to LF lacking PDGF-Rα (GFP−) at P12 and P14, respectively. Observing that imatinib selectively suppresses proliferation in pdgfrα-LF indicates that the effect of imatinib depends on the potential for cellular signaling through PDGF-Rα. Figure 2D shows that Ki67+ cells comprise a smaller proportion of the pdgfrα-expressing (GFP+, solid bars) than of the GFP− subpopulation. In controls, the abundance of Ki67+ LF declined from P12 to P14 only in the GFP+, but not the GFP− subpopulation. TGFβ1 did not increase and imatinib did not decrease proliferating Ki67+ LF, which comprised 7.5 ± 1.4, 4.2 ± 2.4, 5.4 ± 0.9, and 1.6 ± 0.9% in control, or in cultures treated with imatinib, TGFβ1, or TGFβ1 and imatinib, respectively (mean ± SEM, n = 3, one-way ANOVA).
LF apoptosis is influenced by PDGF-Rα.
Figure 3A (P12) and B (P14) shows that the proportion of apoptotic cells (bind annexin but exclude DAPI) was lower in the pdgf-rα expressing LF (solid bars) compared with control, suggesting that this subpopulation is more resistant to apoptosis. Exposure to imatinib made the PDGFRα-LF (GFP+) as likely to undergo apoptosis as their nonexpressing (GFP−) counterparts.

Functional kinase protects pdgfrα-LF from apoptosis. Unpermeabilized-LF isolated, as in Figure 2, were subjected to FACS immediately after isolation at P12 (A) or P14 (B). Apoptotic GFP− (□) and GFP+ (▪) LF stained with annexin V but not DAPI. Mean ± SEM, n = 4 experiments. *p < 0.01, GFP+ compared with time-matched GFP−. †p < 0.01, untreated vs imatinib-treated GFP+ LF. Two-way ANOVA, Student-Newman-Keuls test. (C) Representative micrographs (scale bar, 25 μm) from control and imatinib-treated mice stained in parallel with anticleaved PARP (red) and ToPro3 (blue to visualize all nuclei). Yellow arrows, apoptotic GFP− cells containing cleaved PARP (nuclei only visualized with ToPro3); white arrow, apoptotic PDGF-Rα GFP+ cell (green nucleus). Percentage cells containing cleaved-PARP in PDGF-Rα expressing (GFP+, ▪) compared with parenchymal cells not expressing pdgf-rα (GFP−, □) at P12 (D) and P14 (E). †Imatinib vs control GFP−; *Imatinib GFP+ vs imatinib GFP− <0.05. One-way ANOVA, Student Newman-Keuls test.
We also assessed the abundance of cleaved PARP in lung tissue, which circumvented apoptosis resulting from the isolation procedure. Because the GFP− population also included cells that were not LF, our results partially differed from those obtained using FACS. Figure 3C shows representative images obtained using LSCM with lungs from control and imatinib-treated mice at P14. Figure 3D and E demonstrates that apoptosis was greater in the lung parenchymal cells lacking GFP (open bars) than in the PDGF-Rα-GFP+ LF at P12 and P14, respectively. As observed using FACS, apoptosis was lower in the PDGF-Rα-GFP+ population compared with other parenchymal cells. However, using cleaved PARP, imatinib also increased apoptosis in alveolar cells that are not fibroblasts (and were depleted in the LF-enriched population analyzed by FACS). Inhibition of Abl promotes apoptosis in alveolar epithelial and endothelial cells (16,21).
Because PDGF-Rα-signaling enhances survival through the Akt pathway, we assessed whether imatinib disrupted Akt and PDGF-Rα-phosphorylation in primary mouse LF isolated at P12 (22). Figure 4 shows that imatinib significantly reduced both Akt and PDGF-Rα phosphorylation after PDGF-A treatment.

Imatinib reduces phospho-Akt and phospho-PDGF-Rα in cultured mouse LF after stimulation with PDGF-A. Representative immunoblots showing phospho-Akt (pAkt) and Akt (A) or phosphor-PDGF-Rα (pPDGFRα) or PDGF-Rα (C). Bars are means of four (B) or three (D) experiments. †p < 0.01, PDGF-A vs control; *p < 0.01, PDGF-A + imatinib vs PDGF-A. Two-way ANOVA, Student-Newman-Keuls test.
Imatinib reduces alveolar surface area and wall thickness.
Imatinib significantly diminished both the alveolar surface area and arithmetic mean wall thickness at P12 (open bars) and P14 (solid bars; Fig. 5). Lung parenchyma comprised a similar percentage of the total lung volume in control (90.4 ± 0.879, mean ± SEM) and imatinib-exposed (89.5 ± 0.81) mice.

Imatinib reduces alveolar surface area and arithmetic mean wall thickness (la,). Portions of lungs used in Figure 3D at P12 (□) and P14 (▪) were used to analyze alveolar (alveolar and alveolar ducts were not distinguished) surface area (A) wall thickness (B). Four mice were used from each treatment group. *p < 0.05; †p < 0.01, two-way ANOVA, Tukey-test.
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) regulates pdgf-rα transcription in LF.
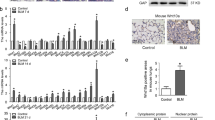
The imatinib-related decrease in PDGF-Rα-GFP+ LF (Fig. 1) could result from diminished PDGF-Rα transcription. Factors that diminish transcriptional activity of the pdgf-rα promoter-regulatory region would also diminish the production GFP, which is directly dependent on pdgf-rα promoter activity. Rather than a decrease, we observed that imatinib increased pdgf-rα mRNA (Fig. 6A). Oridonin disrupts binding of NF-κB to its consensus DNA elements (23). Therefore, we reasoned that oridonin should block an imatinib-induced, NF-κB-dependent, increase in PDGF-Rα mRNA. In the absence of oridonin, imatinib increased pdgf-rα mRNA, whereas in the presence of oridonin, the stimulatory effect of imatinib was overcome (Fig. 6B). Therefore, the imatinib-related decrease in GFP+ alveolar cells is more likely due to a reduction in the PDGF-Rα-GFP+ cell population.

Imatinib increases and disruption of NF-κB-DNA binding abrogates the increase in pdgf-rα mRNA. (A) Mouse LF exposed to 10 μM imatinib for 24 h. RNA normalized to β2-microglobulin mRNA and expressed relative to the control (no imatinib). n = 5 for pdgf-rα (□) and αSMA (▪), *p < 0.01, paired t test compared imatinib and control pdgf-rα mRNA. (B) LF exposure to imatinib and oridonin for 12 h. *p < 0.05, imatinib, or oridonin and imatinib vs control, †p < 0.05, oridonin and imatinib vs imatinib. n = 3. Two-way ANOVA, Tukey test.
Inhibition of PDGF-R and/or Abl reduces alveolar αSMA.
Because pdgf-rα (GFP+)-LF produce αSMA (Fig. 2A), we also expected that imatinib would reduce αSMA containing LF. Figure 7A shows that at P14, there is less αSMA in alveolar ducts from a mouse treated for 5 d with imatinib than an untreated control. Imatinib treatment decreased the pixel area occupied by αSMA (Fig. 7B) (19). Using FACS (Figs. 2A and 7C), we observed that the decrease in αSMA containing cells was only observed in the pdgfrα-LF (GFP+). This indicates that PDGF-R and/or Abl/Arg signaling is required to sustain the production of αSMA. However, Figure 6A shows that imatinib did not directly reduce αSMA-gene expression in cultured LF.

Imatinib reduces alveolar αSMA. (A) Representative images of control and imatinib-treated mice. Lung tissues stained in parallel with Cy3-conjugated anti-αSMA (red). AlxaFluor 633 conjugated DiD (blue) counterstain. White arrows, αSMA; scale bar, 25 μm. (B) Summary αSMA pixel areas using three mice per treatment group. *p < 0.01, imatinib-treated (4 or 5 d) vs control. (C) Comparison of αSMA (using FACS) of untreated control and imatinib-treated within the PDGF-Rα negative (−, GFP−) or within the PDGF-Rα-positive (+, GFP+) subpopulations. Same LF preparations as for P14 in Figure 2C, stained for αSMA. *p < 0.05, two-way ANOVA, Student Newman-Keuls test.
DISCUSSION
PDGF-A, which signals solely through PDGF-Rα, is required for secondary alveolar septal formation and elastic fiber deposition (5). The pdgf-rα expressing LF population increases between P4 and P8 through proliferation (13) and maintains its abundance until at least P12 (19). We have now shown that the pdgfrα-expressing population diminishes after P12 because of apoptosis and diminished proliferation, resulting in fewer and thinner alveolar walls. Protein tyrosine kinase signaling through PDGF-R and/or Abl/Arg is important for sustaining the pdgfrα-LF subpopulation (Fig. 8). Because imatinib increased, rather than decreased pdgfrα gene expression in vitro, it is more likely that the loss of pdgfrα-expressing cells results from a decline in the number of cells rather than a decrease in pdgf-rα transcription. PDGF-A stimulates and imatinib interferes with the ability of PDGF-A to increase PDGF-Rα- and Akt-phosphorylation, suggesting that the antiapoptotic effect of PDGF-A on pdgfrα-LF may involve Akt.

Alveolar myofibroblasts (MF) diminish during secondary septal maturation. From P12 to P14 (proceeding from left to right), signaling through PDGF-Rα diminishes (shaded triangle). Fewer pdgf-rα expressing MF (black nuclei) occupy the central core because attenuation of PDGF-Rα signaling results in decreased proliferation and increased apoptosis (less phospho-Akt, pAkt). Septal thinning enables the capillary network to coalesce and closely appose the epithelial septal covering.
Others demonstrated that without PDGF-A, pdgf-rα expressing LF diminish and secondary septal formation fails (5). However, these studies did not establish whether the effects of PDGF-A were specifically directed toward pdgfrα-LF or toward progenitors, which supplied the pdgfrα-LF population. Our novel findings indicate that protein tyrosine kinase signaling through PDGF-R and/or Abl maintains the pdgfrα myofibroblast subpopulation and that diminished PDGF-Rα signaling may promote septal thinning and maturation of the air-blood interface. This diminution is accelerated by imatinib.
Using imatinib to disrupt protein tyrosine kinase signaling has distinct advantages and some disadvantages. Targeted gene deletions of PDGF-A or PDGF-Rα are gene and pathway specific, but they do not disrupt a particular phase of alveolar development, because the abnormalities begin in utero. Therefore, the observations made postnatally are compounded by abnormalities before P4, when septal eruption begins. Starting imatinib on or after P8 enabled septal eruption and early elongation to proceed normally, so we could specifically study subsequent septal elongation and thinning. A disadvantage is that although imatinib is selective, it likely inhibits PDGF-Rβ and Abl, as well as PDGF-Rα, under our experimental conditions.
Nevertheless, inhibition of PDGF-Rα most likely accounts for our observations. Imatinib inhibited proliferation and enhanced apoptosis in the PDGF-Rα-GFP+ but not the GFP− sub-population, indicating that PDGF-Rα-kinase is necessary for the observed effects. Whether inhibition of PDGF-Rα is sufficient to account for our findings is less certain, because imatinib also inhibits Abl kinase. In fibroblasts, PDGF-receptors primarily activate Abl-kinase by signaling through Src and phospholipaseC-γ, rather than through phosphoinositol-3-kinase, the dominant PDGF-Rα-pathway (24). This suggests that PDGF-Rα may not initiate Abl-mediated signaling in LF. Although Abl is present in cultured mouse LF, its constitutive kinase activity is very low, and we could not demonstrate further reduction by imatinib (data not shown). Although others observed that inhibition of Abl by imatinib reduced TGFβ-mediated renal fibroblast proliferation (25), we have not observed this in mouse LF. We previously showed that proliferation decreased in pdgfrα-expressing LF at P12, whereas Smad-dependent signaling did not (13). Therefore, it is unlikely that imatinib interferes with activation of Abl-kinase by TGFβ, in our mouse LF cultures. Because PDGF-A enhances Akt-phosphorylation, pdgfrα-LF differentially maintains survival as long as signaling through PDGF-Rα is maintained. But as proliferation continues to decline, pdgfrα-LF diminishes so that septal thinning occurs (Fig. 8).
In conclusion, we have shown that pdgf-rα expressing myofibroblasts persist throughout the period of maximal septation (P12) and then diminish. Although pdgfrα-GFP expressing myofibroblasts are less proliferative than their GFP− counterparts at P12, they are more resistant to apoptosis. Timely regulation of cellular proliferation and apoptosis is required to ensure that sufficient myofibroblasts are present not only to establish mechanically competent septa but also to enable septal thinning as the vasculature matures. Aberrant regulation occurs during positive pressure mechanical ventilation of newborn mammals and likely contributes to the development of BPD (12). Targeted and carefully timed strategies to maintain a normal complement of myofibroblasts could improve the outcome for children with BPD. Similar strategies may be relevant to pulmonary emphysema in adults, when the alveolar septa atrophy through apoptosis (26). Alternatively, insufficient myofibroblast apoptosis contributes to pulmonary fibrosis and may be corrected by the administration of imatinib (27).
Abbreviations
- Abl:
-
Abelson kinase
- Arg:
-
Abelson-related gene
- αSMA, ACTA2:
-
alpha-smooth muscle actin
- BPD:
-
bronchopulmonary dysplasia
- DAPI:
-
4′,6-diamidino-2-phenylindole, diacetate
- FACS:
-
fluorescence activated cell sorting
- GFP:
-
green fluorescent protein
- LSCM:
-
laser scanning confocal microscope
- LF:
-
lung fibroblast
- NF-κB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- PDGF-Rα:
-
PDGF-receptor-alpha
- pdgfrα-LF :
-
pdgf-rα expressing LF
- PARP:
-
poly-ADP ribose polymerase
- P12:
-
postnatal d 12
REFERENCES
Mund SI, Stampanoni M, Schittny JC 2008 Developmental alveolarization of the mouse lung. Dev Dyn 237: 2108–2116
Nicola T, Hagood JS, James ML, MacEwen MW, Williams TA, Hewitt MM, Schwiebert L, Bulger A, Oparil S, Chen YF, Ambalavanan N 2009 Loss of Thy-1 inhibits alveolar development in the newborn mouse lung. Am J Physiol Lung Cell Mol Physiol 296: L738–L750
Srinivasan S, Strange J, Awonusonu F, Bruce MC 2002 Insulin-like growth factor I receptor is downregulated after alveolarization in an apoptotic fibroblast subset. Am J Physiol Lung Cell Mol Physiol 282: L457–L467
Mitchell JJ, Reynolds SE, Leslie KO, Low RB, Woodcock-Mitchell J 1990 Smooth muscle cell markers in developing rat lung. Am J Respir Cell Mol Biol 3: 515–523
Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, Hedstrand H, Pekna M, Hellström M, Gebre-Medin S, Schalling M, Nilsson M, Kurland S, Törnell J, Heath JK, Betsholtz C 1996 PGDF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85: 863–873
Buch S, Han RN, Cabacungan J, Wang J, Yuan S, Belcastro R, Deimling J, Jankov R, Luo X, Lye SJ, Post M, Tanswell AK 2000 Changes in expression of platelet-derived growth factor and its receptors in the lungs of newborn rats exposed to air or 60% O(2). Pediatr Res 48: 423–433
Olson LE, Soriano P 2009 Increased PDGFRα activation disrupts connective tissue development and drives systemic fibrosis. Dev Cell 16: 303–313
Li J, Hoyle GW 2001 Overexpression of PDGF-A in the lung epithelium of transgenic mice produces a lethal phenotype associated with hyperplasia of mesenchymal cells. Dev Biol 239: 338–349
Burri PH 2006 Structural aspects of postnatal lung development-alveolar formation and growth. Biol Neonate 89: 313–322
Wu S, Platteau A, Chen S, McNamara G, Whitsett J, Bancalari E 2010 Conditional overexpression of connective tissue growth factor disrupts postnatal lung development. Am J Respir Cell Mol Biol 42: 552–563
Thibeault DW, Mabry SM, Ekekezie II, Zhang X, Truog WE 2003 Collagen scaffolding during development and its deformation with chronic lung disease. Pediatrics 111: 766–776
Coalson JJ 2006 Pathology of bronchopulmonary dysplasia. Semin Perinatol 30: 179–184
Kimani PW, Holmes AJ, Grossmann RE, McGowan SE 2009 PDGF-Rα gene expression predicts proliferation, but PDGF-A suppresses transdifferentiation of neonatal mouse lung myofibroblasts. Respir Res 10: 119–135
Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB 1996 Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2: 561–566
Vuorinen K, Gao F, Oury T, Kinnula VL, Myllärniemi M 2007 Imatinib mesylate inhibits fibrogenesis in asbestos-induced interstitial pneumonia. Exp Lung Res 33: 357–373
Vittal R, Zhang H, Han MK, Moore BB, Horowitz JC, Thannickal VJ 2007 Effects of the protein kinase inhibitor, imatinib mesylate, on epithelial/mesenchymal phenotypes: implications for treatment of fibrotic diseases. J Pharmacol Exp Ther 321: 35–44
Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P 2003 Evolutionary divergence of platelet derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol 23: 4013–4025
Krebs R, Tikkanen JM, Nykänen AI, Wood J, Jeltsch M, Yla-Herttuala S, Koskinen PK, Lemström KB 2005 Dual role of vascular endothelial growth factor in experimental obliterative bronchiolitis. Am J Respir Crit Care Med 171: 1421–1429
McGowan SE, Grossmann RE, Kimani PW, Holmes AJ 2008 Platelet-derived growth factor receptor-alpha-expressing cells localize to the alveolar entry ring and have characteristics of myofibroblasts during pulmonary alveolar septal formation. Anat Rec (Hoboken) 291: 1649–1661
Weibel ER 1979-1980 Stereological Methods. Academic Press, London, pp 9–196
Evensen L, Micklem DR, Blois A, Berge SV, Aarsaether N, Littlewood-Evans A, Wood J, Lorens JB 2009 Mural cell associated VEGF is required for organotypic vessel formation. PLoS One 4: e5798
Horowitz JC, Rogers DS, Sharma V, Vittal R, White ES, Cui Z, Thannickal VJ 2007 Combinatorial activation of FAK and AKT by transforming growth factor-β1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal 19: 761–771
Zhang N, Khachigian LM 2009 Injury-induced platelet-derived growth factor receptor-alpha expression mediated by interleukin-1beta (IL-1beta) release and cooperative transactivation by NF-kappaB and ATF-4: IL-1beta facilitates HDAC-1/2 dissociation from promoter. J Biol Chem 284: 27933–27943
Plattner R, Pendergast AM 2003 Activation and signaling of the Abl tyrosine kinase: bidirectional link with phosphoinositide signaling. Cell Cycle 2: 273–274
Wilkes MC, Leof EB 2006 Transforming growth factor β activation of c-Abl is independent of receptor internalization and regulated by phosphatidylinositol 3-kinase and PAK2 in mesenchymal cultures. J Biol Chem 281: 27846–27854
Aoshiba K, Nagai A 2009 Senescence hypothesis for the pathogenic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc 6: 596–601
Garneau-Tsodikova S, Thannickal VJ 2008 Protein kinase inhibitors in the treatment of pulmonary fibrosis. Curr Med Chem 15: 2632–2640
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by a Merit Review Award from the Department of Veterans Affairs and a Carver Foundation Collaborative Award from the University of Iowa Carver College of Medicine.
Rights and permissions
About this article
Cite this article
McGowan, S., McCoy, D. Fibroblasts Expressing PDGF-Receptor-Alpha Diminish During Alveolar Septal Thinning in Mice. Pediatr Res 70, 44–49 (2011). https://doi.org/10.1203/PDR.0b013e31821cfb5a
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31821cfb5a
This article is cited by
-
Understanding alveolarization to induce lung regeneration
Respiratory Research (2018)
-
Lipidomics reveals dramatic lipid compositional changes in the maturing postnatal lung
Scientific Reports (2017)
-
Understanding the developmental pathways pulmonary fibroblasts may follow during alveolar regeneration
Cell and Tissue Research (2017)
