
Abstract
Hydrogen (H2) has been reported to neutralize toxic reactive oxygen species. Oxidative stress is an important mechanism of neuronal damage after perinatal asphyxia. We examined whether 2.1% H2-supplemented room air (H2-RA) ventilation would preserve cerebrovascular reactivity (CR) and brain morphology after asphyxia/reventilation (A/R) in newborn pigs. Anesthetized, ventilated piglets were assigned to one of the following groups: A/R with RA or H2-RA ventilation (A/R-RA and A/R-H2-RA; n = 8 and 7, respectively) and respective time control groups (n = 9 and 7). Asphyxia was induced by suspending ventilation for 10 min, followed by reventilation with the respective gases for 4 h. After euthanasia, the brains were processed for neuropathological examination. Pial arteriolar diameter changes to graded hypercapnia (5–10% CO2 inhalation), and NMDA (10−4 M) were determined using the closed cranial window/intravital microscopy before and 1 h after asphyxia. Neuropathology revealed that H2-RA ventilation significantly reduced neuronal injury induced by A/R in virtually all examined brain regions including the cerebral cortex, the hippocampus, basal ganglia, cerebellum, and the brainstem. Furthermore, H2-RA ventilation significantly increased CR to hypercapnia after A/R (% vasodilation was 23 ± 4% versus 41 ± 9%, p < 0.05). H2-RA ventilation did not affect reactive oxygen species-dependent CR to NMDA. In summary, H2-RA could be a promising approach to reduce the neurologic deficits after perinatal asphyxia.
Similar content being viewed by others


Main
Perinatal asphyxia induces brain injury that can result in death of the affected infants or severe neurodevelopmental deficits of the survivors. According to World Health Organization (WHO) estimates, perinatal asphyxia is responsible for 8% of the yearly 10.6 million deaths in children younger than 5 years world wide (1). Thus, it is important to seek suitable therapies that can increase survival and attenuate the neurologic deficits. Despite the large number of affected infants, the incentives and involvement of the pharmaceutical industry have been limited so far perhaps due to financial concerns and ethical issues of initiating clinical trials in neonates (2). In fact, there is currently no effective neuroprotective pharmacotherapy to treat perinatal asphyxial encephalopathy. However, recently, the Total Body Hypothermia for Neonatal Encephalopathy Trial study group has shown that moderate whole-body hypothermia improves outcome in full-term infants by decreasing the incidence of severe neurodevelopmental deficits, and thus, body cooling is expected to become standard care very soon in this group of patients (3,4).
Therapeutic hypothermia can be initiated up to 6 h after birth, by the time the patient reaches a neonatal intensive care unit equipped to provide this care. Thus, there would be a window of opportunity for the employment of an immediate neuroprotective therapy until the neuroprotective cooling starts. The management of asphyxiated infants usually starts with resuscitation and involves mechanical ventilation. Reoxygenation of the tissues is a primary goal of resuscitation efforts; however, reoxygenation and reperfusion provide oxygen for the synthesis of reactive oxygen species (ROS) that can result in oxidative stress and subsequent reoxygenation/reperfusion injury (5–9). Resuscitation/ventilation with pure oxygen has been shown to be detrimental in neonatal large animal models (10–12) and in clinical trials as well (13,14). Resuscitation is now recommended with air to be supplemented with oxygen in infants with persistent central cyanosis (15). However, the gas mixture used for resuscitation could perhaps be used to deliver neuroprotective agents to the brain. Molecular hydrogen (H2) has recently been shown to exert antioxidant properties (16). H2 was indeed neuroprotective against hypoxic/ischemic injury in adult and neonatal rats (16–18) and also in a rat model of Parkinson's disease (19). However, the possible protective effect of H2 against perinatal asphyxia has not been tested in a large animal model yet.
Therefore, the purpose of this study was to investigate whether ventilation with H2-supplemented room air (H2-RA) preserves hypoxia-sensitive cerebrovascular reactivity (CR) and attenuates neuronal injury in asphyxiated newborn piglets. We also studied whether H2-RA ventilation in the absence of asphyxia/reventilation (A/R) stress affects CR or brain morphology.
MATERIALS AND METHODS
Animals.
Newborn (aged less than 1 d) piglets of either sex (n = 34) were delivered from a local farm to the laboratory on the morning of the experiments. All procedures were approved by the Animal Care and Use Committee of the University of Szeged. The piglets were anesthetized with sodium thiopental (45 mg/kg i.p., Biochemie, Vienna, Austria) followed by i.v. administration of α-chloralose (30–40 mg/kg i.v., Sigma Chemical Co., St. Louis, MO). Supplemental doses of α-chloralose (3–7 mg/kg/h) were administered to maintain a stable level of anesthesia determined by continuously monitoring the blood pressure and responsiveness to tactile stimuli. The femoral artery and vein were catheterized to monitor arterial pH, blood gases, and blood pressure and to inject drugs and fluids, respectively. The animals were intubated through tracheotomy and artificially ventilated with RA (21% O2 and 79% N2). The ventilation rate (∼28/min) and tidal volume (∼20 mL) were adjusted to maintain physiological blood gas values. Core body temperature was kept in the physiological range (∼38–39°C) with electrical heating pads.
To determine CR, the piglets were equipped with a closed cranial window filled with artificial cerebrospinal fluid (aCSF) to measure pial arteriolar diameter (PAD) by intravital microscopy as described previously (20). Cortical blood flow (CoBF) was monitored using a two-channel laser-Doppler flowmeter (LDF, Periflux 4001, Perimed, Sweden) as described previously (12). In each experiment, a pial arteriole with a baseline diameter of 80–120 μm was selected to study CR. Ten-minute periods of arterial blood pressure and CoBF LDF data were recorded before, during, and after asphyxia and at each hour of the reventilation period. Data were stored on a personal computer and were evaluated off-line using the Perisoft 1.30 software (Perimed).
Experimental protocol.
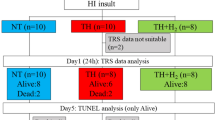
The instrumented piglets were divided into four experimental groups 1) A/R with RA (21% O2, 79% N2) ventilation (A/R-RA; n = 8), 2) A/R with H2-RA (2.1% H2; 21% O2; 76.9% N2) ventilation (A/R-H2-RA; n = 7), 3) time control group ventilated with RA (SHAM (sham-treated)-RA; n = 9); and 4) time control group ventilated with H2-RA (SHAM-H2-RA; n = 7).
First, CR was determined to graded hypercapnia and NMDA. Graded hypercapnia was elicited by ventilating the animals with a gas mixture containing 5% CO2 (21% O2 and balance N2) for 5–8 min, then with 10% CO2 for additional 5–8 min. Ventilation was then switched back to RA, and PAD was allowed to return to baseline diameters. During a 10-minute stabilization period, the cranial window was gently flushed with aCSF, then NMDA dissolved in aCSF (10−4 M, 5 mL) was applied onto the cerebral cortex for 5–6 min, and then the cranial window was repeatedly washed with aCSF. PAD was measured each minute during hypercapnia and NMDA stimulation, and the maximal vasodilation maintained for at least 2 min was used to determine CR to hypercapnia or NMDA, respectively.
After obtaining baseline CR values, asphyxia was induced (groups A/R-RA and A/R-H2-RA) by suspending the artificial ventilation and clamping the endotracheal tube for 10 min. Reventilation started with the gas mixtures according to the protocol of the respective experimental groups. There was no need to apply chest compressions or drugs for resuscitation. In the SHAM groups, ventilation was switched to the respective gas mixtures after a RA ventilation time control period (10 min). After 1 h ventilation, CR to hypercapnia and NMDA were again determined in all experimental groups, then ventilation was continued for an additional 3 h. At the end of the 4-h survival period, the anesthetized piglets were euthanized with KCl (2 M, 10 mL i.v.).
Neuropathology.
An additional group of piglets (NAÏVE, n = 3) were euthanized with sodium thiopental (150 mg/kg i.p.) to provide an absolute control for the neuropathology studies. After euthanasia in all experimental groups, the brains were rapidly and carefully removed and immersion fixed in 4% paraformaldehyde. Tissue blocks from the frontal, parietal, temporal, and occipital cortices, the hippocampus, the cerebellum, the basal ganglia, the pons, and the medulla oblongata were embedded in paraffin, and sections were double stained with hematoxylin and eosin. Two independent observers (O.O. and I.N.), who were blinded to the treatments, quantitatively determined, then averaged the percentage of injured neurons in each region based on the presence of shrunken hyperchromatic (red) neurons with pyknotic nuclei.
Statistical analysis.
Blood chemistry, mean arterial blood pressure (MABP), CoBF, and PAD data are expressed as mean ± SEM and have been analyzed using two-way repeated measures analysis of variance followed by the Student-Newman-Keuls post hoc test (SigmaStat, SPSS, Chicago, IL). Histopathology data were not normally distributed and were analyzed with Kruskal-Wallis Analysis of Variance on Ranks followed by the Dunn's post hoc test for multiple comparisons. p values of <0.05 were considered statistically significant.
RESULTS
Hemodynamics and blood chemistry.
The MABP, pH, and blood gas values were in the physiological range and did not differ significantly between the experimental groups before asphyxia (Tables 1 and 2). Ventilation with gas mixtures containing 5% or 10% CO2 to determine CR to hypercapnia resulted in graded increase in arterial Pco2 with simultaneous drops in pH that were similar between all experimental groups before and after A/R injury (Table 1). Asphyxia caused significant hypoxia, hypercapnia, acidosis, hypotension, and decrease in CoBF that were similar in both groups subjected to A/R (Tables 1 and 2). At the onset of reventilation, MABP rapidly increased then stabilized at near baseline values by the end of first hour (Table 2). MABP values were thus similar when determining CR to hypercapnia and NMDA before and after A/R. However, at 30 min and 1 h after asphyxia, Pco2 was significantly higher in the A/R-RA group compared with the A/R-H2-RA group, albeit pH and pO2 values were not significantly different (Table 1). In contrast, CoBF was ∼20–25% lower in the A/R-H2-RA group compared with the A/R-RA (Table 2). In the SHAM groups, the MABP remained in the normal ranges during the whole course of the experiment (Table 2). CoBF also seemed to be decreased by 15–20% in the SHAM-H2-RA group compared with the SHAM-RA group; however, this difference did not reach statistical significance (Table 2).
H2-RA preserves CR to hypercapnia after A/R injury.
The first stimulation with graded hypercapnia elicited significant, dose-dependent, pial arteriolar vasodilation in all experimental groups (Fig. 1A and B). Baseline PADs were not significantly different between repeated hypercapnia stimulations, and their values were (first versus second stimulation): 88 ± 4 μm versus 94 ± 8 μm for group A/R-RA, 87 ± 4 μm versus 92 ± 7 μm for group A/R-H2-RA, 85 ± 4 μm versus 91 ± 3 μm for group SHAM-RA, and 86 ± 6 μm versus 82 ± 7 μm for group SHAM-H2-RA. A/R significantly attenuated CR to hypercapnia in the A/R-RA (Fig. 1A) but not in the other experimental groups (Fig. 1A and B). Thus, ventilation with H2-RA under normoxic conditions in the SHAM-H2-RA group did not affect CR to hypercapnia. Furthermore, ventilation with H2-RA after asphyxia prevented the attenuation of CR to hypercapnia in the A/R-H2-RA group (Fig. 1A).

H2-RA ventilation preserves cerebrovascular reactivity to hypercapnia 1 hour after A/R. Graded hypercapnia induced by ventilation with 5–10% CO2 and topical application of 10−4 M NMDA elicited similar, significant increases in pial arteriolar diameters in all experimental groups (Panels A and B). In time controls (Panel B), pial arteriolar responses were repeatable and similar between the SHAM-RA (n = 9, white bars) and the SHAM-H2-RA (n = 7, light gray bars) groups. Importantly, H2-RA ventilation did not affect NMDA-induced, NO-dependent vasodilation (Panel B). After A/R, however (Panel A), pial arteriolar dilation to 10% CO2 was significantly reduced in the A/R-RA group (n = 8, black bars). However, in the A/R-H2-RA (n = 7, dark gray bars) group, pial arteriolar dilation to 10% CO2 was significantly higher than in group A/R-RA and not significantly different from the respective baseline response before A/R. p < 0.05, *vs corresponding response before A/R, †corresponding responses after A/R in the A/R-RA vs the A/R-H2-RA groups.
H2-RA ventilation does not affect CR to NMDA.
NMDA induced significant, repeatable pial arteriolar vasodilation in all experimental groups (Fig. 1A and B). Thus, ventilation with H2-RA did not affect CR to NMDA. Baseline PADs were not significantly different between repeated NMDA applications, and their values were (first versus second application): 87 ± 4 μm versus 85 ± 9 μm for group A/R-RA, 85 ± 4 μm versus 91 ± 6 μm for group A/R-H2-RA, 86 ± 4 μm versus 82 ± 4 μm for group SHAM-RA, and 84 ± 5 μm versus 80 ± 6 μm for group SHAM-H2-RA.
H2-RA protects neurons after A/R injury.
The percentage of damaged, red neurons was negligible in any brain region of the untreated control animals (NAÏVE animals): the median value was 0% in seven regions, 2% in three regions, and 3% in two regions (data not shown). In the time control animals (SHAM-RA and SHAM-H2-RA groups), there was also only a minimal (1–4%) neuronal damage depending on the brain region studied, indicating that H2-RA ventilation alone did not affect neuronal viability (Figs. 2A–C and 3). A/R significantly increased neuronal injury in all brain regions studied in the A/R-RA group. However, the increase in injured, red neurons was smaller in the A/R-H2-RA group, showing a clear neuroprotective effect in virtually all brain regions examined (Figs. 2A–C and 3).

H2-RA ventilation attenuates A/R-induced early neuronal injury. The percentage of damaged neurons was quantitatively determined at 4 h after 10 min of asphyxia and in corresponding time control animals. The examined brain regions were Panel A: CA1, CA2, CA3, and stratum granulosum (Str. Gr.) of the hippocampus; Panel B: frontal (Fr. Cx.), parietal (Par Cx.), temporal (Temp Cx.), and occipital (Occ. Cx.) cortices; and Panel C: basal ganglia (Basal ggl.), pons, medulla, and cerebellum. Summated data from the hippocampal (Σ Hippoc.) and cortical subregions (Σ Cortex) are also shown on Panels A and B, respectively. The box plots show the median (horizontal line), the 25th–75th percentile (box), the 10th–90th percentiles (error bars), and any outlying data points (bullets). The experimental groups are represented by gray, gray-hatched, white, and white-hatched boxes for A/R-RA, A/R-H2-RA, SHAM-RA, and SHAM-H2-RA (n = 8, 7, 9, and 7), respectively. A/R resulted in significant increases in neuronal injury in virtually all examined brain regions in the A/R-RA but not in the A/R-H2-RA group compared with the respective time controls. Thus, H2-RA ventilation attenuated the detrimental effect of A/R on neuronal viability. p < 0.05, *vs corresponding time control (A/R-RA vs SHAM-H2-RA and A/R-H2-RA vs SHAM-H2-RA) A/R, †A/R-RA vs the A/R-H2-RA.

Representative photomicrographs from the parietal cortex, the stratum granulosum of the hippocampus, and the cerebellar cortex showing neuroprotection by H2-RA ventilation. Shrunken, hyperchromatic neurons with pyknotic nuclei are more common in samples obtained from animals of the A/R-RA group, when compared with A/R-H2-RA group. Time control groups (SHAM-RA and SHAM-H2-RA) show minimal neuronal damage, the samples are similar to those obtained from untreated (NAÏVE) animals. Scale bar: 100 μM.
DISCUSSION
The major findings of this study are the following: ventilation with H2-RA after asphyxia preserves CR to hypercapnia and protects the brain from A/R-induced neuronal injury in piglets. Furthermore, H2-RA ventilation per se does not affect CR or neuronal viability.
Oxidative stress induced by ROS produced during/after a hypoxic/ischemic episode is a widely accepted pathomechanism of perinatal brain injury (2,21). Thus, administration of antioxidant drugs either to decrease ROS production or to neutralize ROS seems to be a simple, straightforward approach to alleviate brain damage. However, the antioxidant drug needs to be present at the ROS level surge in the early reventilation/reperfusion to exert its effect. For example, the xanthine oxidase inhibitor allopurinol was found ineffective when administered to severely asphyxiated term infants 4 h after birth (22). The authors speculated that drug administration was too late to successfully target the immediate ROS surge after resuscitation. In fact, in a more recent study, the same research group has found that maternal treatment of in utero asphyxiated infants with allopurinol reduced the concentration of the brain injury marker S-100B in infants with therapeutic allopurinol levels (23). The disadvantage of maternal treatment is that placental transport of the drugs can vary greatly, and the drug may not reach therapeutic levels in a significant portion of patients (23). H2-RA ventilation could be an appealing technique for antioxidant neuroprotective therapy because administration could start simultaneously with the resuscitation effort to provide reoxygenation.
ROS also play physiological roles that have been demonstrated in numerous processes including neuronal and vascular signaling (24). Potent antioxidants may interfere with these physiological ROS-mediated mechanisms that can attenuate or even nullify their beneficial effects. Hydrogen gas has been suggested to selectively reduce the most toxic ROS: hydroxyl radicals and peroxinitrate anions (16), thus leaving the less deleterious/physiological ROS intact. This study provides evidence that H2-RA ventilation does not affect a ROS-dependent neuronal-vascular response: NMDA-induced vasodilation. NMDA-induced vasodilation has been first described in the piglet (25), and CR to NMDA can be used to assess the integrity of the neurovascular unit (26). The mechanism of dose-dependent NMDA-induced pial arteriolar dilation and subsequent cortical hyperemia involves the neuronal synthesis and vascular actions of NO (20,27). In this study, the intact CR to NMDA during H2-RA ventilation indicates that H2 does not interfere with the increases in NO levels necessary for the induction of pial arteriolar vasodilation. NMDA-induced vasodilation has been repeatedly shown to be sensitive to global cerebral ischemia/reperfusion, and CR to NMDA (10−4 M) is attenuated by ∼50% 1 h after 10 min ischemia (26,28). Interestingly, we have not observed a decrease in CR to NMDA in this study after 10 min of asphyxia. The only study in the literature investigating the effect of A/R on NMDA-induced vasodilation reported “altered” CR to NMDA based on progressively increasing CR to NMDA in the reventilation period (29). However, in that study, CR to NMDA was not determined before asphyxia. Furthermore, the vasodilation to 10−5 M NMDA used in that study corresponds with our later findings with this dose of NMDA under normoxic conditions (20). Thus, it is conceivable that A/R represents a less severe stress than global ischemia/reperfusion, and CR to 10−4 M NMDA does not change appreciably using this stress.
However, A/R significantly attenuated CR to hypercapnia. In the piglet, hypercapnia-induced pial arteriolar vasodilation is dependent on the function of microvascular endothelial cells (20,30). In this study, H2-RA ventilation preserved CR to hypercapnia after A/R indicating intact endothelial function. The preservation of CR likely results in more adequate microvascular blood flow matching neuronal needs and, thus, likely contributes to the neuroprotective effect of H2-RA ventilation.
Our study is the first in demonstrating the early neuroprotective effect of H2-RA ventilation in a large animal model of perinatal asphyxia. We have previously shown the neurotoxic effect of 100% oxygen ventilation compared with RA ventilation after the same A/R stress used in this study (12). The degree of neuronal injury was similar in the RA ventilated control groups in both studies, albeit in this study we used a quantitative cell counting approach in contrast to the semiquantitative scoring system used in the previous study. Thus, our simple A/R model of perinatal asphyxia is capable to detect both neuroprotective and neurotoxic effects related to the changes in the composition of gas mixture used for ventilation. We emphasize that we observed a beneficial effect of H2-RA ventilation in virtually all brain regions studied. In the previous study, the detrimental effects of 100% oxygen were significant only in the hippocampus and the cerebellum where reactive hyperemia during reventilation was twice higher than in the parietal cortex, suggesting that enhanced oxygen delivery may exacerbate oxidative stress (12). We observed a mild decrease in CoBF during H2-RA ventilation that may also contribute to its neuroprotective effect after A/R by limiting luxury oxygen delivery to the metabolically compromised brain parenchyma. The observed neuroprotective effect of H2-RA ventilation of this study is in accordance with previous data reporting neuroprotection by 2% H2 gas in a neonatal rat hypoxia/ischemia model (18) and also in an adult rat stroke model (16).
The selection of gas used for the resuscitation of infants affected by perinatal asphyxia may represent an ideal way to implement an instant neuroprotective therapy. The importance of careful selection of inhaled oxygen concentration level is already well established, and this study confirms that addition of 2% H2 gas may likely confer neuroprotection by combating ROS-inflicted neuronal damage during the early reperfusion/reoxygenation period. This concentration of H2 (2–2.1%) is safe because H2 has no risk of flammability below 4.7% in RA, and this H2-RA ventilation protocol yielded H2 concentration of ∼20 ng/mL (∼10−5 M) in the arterial blood of adult rats, suggesting an effective antioxidant capacity at this dose (16). Furthermore, other gases may potentially be added to the neuroprotective gas mixture to enhance neuronal survival and function. The anesthethic gas, xenon (50% inhaled concentration) has been found to augment neuroprotection afforded by hypothermia in the neonatal rat hypoxia/ischemia model (31,32), and helium (70% He/30% O2) provided excellent neuroprotection in adult rat stroke models (33,34).
This study has several important limitations. The neuroprotective effect of H2-RA ventilation was demonstrated in α-chloralose-anesthetized piglets using only one A/R insult severity and one H2-RA ventilation protocol. The anesthesia might enhance the neuroprotective potential of H2-RA, and the neuroprotection afforded by H2 can be diminished by increasing insult severity. H2-RA ventilation likely exerts its greatest beneficial effect when administered immediately after reoxygenation, but it is unknown whether delayed administration of H2-RA ventilation would have any protective effect. These clinically relevant topics warrant further studies to elucidate the full neuroprotective potential of H2 perhaps in concert with other inert gases such as helium and therapeutic hypothermia.
In conclusion, H2-RA ventilation after A/R provides early neuroprotection after asphyxia in newborn piglets, at least in part by preservation of neurovascular unit function. Ironically, after fruitless research for a “magic bullet” neuroprotective drug, the optimal combination of the simplest elements: hydrogen, helium, and oxygen with appropriate body temperature control emerges as the most promising neuroprotective treatment in the management of perinatal asphyxia.
Abbreviations
- A/R:
-
asphyxia/reventilation
- aCSF:
-
artificial cerebrospinal fluid
- CoBF:
-
cortical blood flow
- CR:
-
cerebrovascular reactivity
- H2-RA:
-
hydrogen-supplemented room air
- MABP:
-
mean arterial blood pressure
- PAD:
-
pial arteriolar diameter
- RA:
-
room air
- ROS:
-
reactive oxygen species
References
Bryce J, Boschi-Pinto C, Shibuya K, Black RE 2005 WHO estimates of the causes of death in children. Lancet 365: 1147–1152
Degos V, Loron G, Mantz J, Gressens P 2008 Neuroprotective strategies for the neonatal brain. Anesth Analg 106: 1670–1680
Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, Thoresen M, Whitelaw A, Brocklehurst P 2009 Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med 361: 1349–1358
Rutherford M, Ramenghi LA, Edwards AD, Brocklehurst P, Halliday H, Levene M, Strohm B, Thoresen M, Whitelaw A, Azzopardi D 2010 Assessment of brain tissue injury after moderate hypothermia in neonates with hypoxic-ischaemic encephalopathy: a nested substudy of a randomised controlled trial. Lancet Neurol 9: 39–45
Vento M, Asensi M, Sastre J, Garcia-Sala F, Pallardo FV, Vina J 2001 Resuscitation with room air instead of 100% oxygen prevents oxidative stress in moderately asphyxiated term neonates. Pediatrics 107: 642–647
Buonocore G, Perrone S, Longini M, Vezzosi P, Marzocchi B, Paffetti P, Bracci R 2002 Oxidative stress in preterm neonates at birth and on the seventh day of life. Pediatr Res 52: 46–49
Saugstad OD 2005 Room air resuscitation-two decades of neonatal research. Early Hum Dev 81: 111–116
Solberg R, Andresen JH, Escrig R, Vento M, Saugstad OD 2007 Resuscitation of hypoxic newborn piglets with oxygen induces a dose-dependent increase in markers of oxidation. Pediatr Res 62: 559–563
Kumar VH, Patel A, Swartz DD, Wang H, Wynn KA, Nielsen LC, Ryan RM 2010 Exposure to supplemental oxygen and its effects on oxidative stress and antioxidant enzyme activity in term newborn lambs. Pediatr Res 67: 66–71
Munkeby BH, Borke WB, Bjornland K, Sikkeland LI, Borge GI, Halvorsen B, Saugstad OD 2004 Resuscitation with 100% O2 increases cerebral injury in hypoxemic piglets. Pediatr Res 56: 783–790
Temesvári P, Karg E, Bodi I, Nemeth I, Pinter S, Lazics K, Domoki F, Bari F 2001 Impaired early neurologic outcome in newborn piglets reoxygenated with 100% oxygen compared with room air after pneumothorax-induced asphyxia. Pediatr Res 49: 812–819
Domoki F, Zimmermann A, Cserni G, Bori R, Temesvari P, Bari F 2006 Reventilation with room air or 100% oxygen after asphyxia differentially affects cerebral neuropathology in newborn pigs. Acta Paediatr 95: 1109–1115
Davis PG, Tan A, O'Donnell CP, Schulze A 2004 Resuscitation of newborn infants with 100% oxygen or air: a systematic review and meta-analysis. Lancet 364: 1329–1333
Saugstad OD, Ramji S, Vento M 2005 Resuscitation of depressed newborn infants with ambient air or pure oxygen: a meta-analysis. Biol Neonate 87: 27–34
ILCOR 2006 The International Liaison Committee on Resuscitation (ILCOR) consensus on science with treatment recommendations for pediatric and neonatal patients: neonatal resuscitation. Pediatrics 117: e978–e988
Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y, Asoh S, Ohta S 2007 Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med 13: 688–694
Cai J, Kang Z, Liu K, Liu W, Li R, Zhang JH, Luo X, Sun X 2009 Neuroprotective effects of hydrogen saline in neonatal hypoxia-ischemia rat model. Brain Res 1256: 129–137
Cai J, Kang Z, Liu WW, Luo X, Qiang S, Zhang JH, Ohta S, Sun X, Xu W, Tao H, Li R 2008 Hydrogen therapy reduces apoptosis in neonatal hypoxia-ischemia rat model. Neurosci Lett 441: 167–172
Fu Y, Ito M, Fujita Y, Ito M, Ichihara M, Masuda A, Suzuki Y, Maesawa S, Kajita Y, Hirayama M, Ohsawa I, Ohta S, Ohno K 2009 Molecular hydrogen is protective against 6-hydroxydopamine-induced nigrostriatal degeneration in a rat model of Parkinson's disease. Neurosci Lett 453: 81–85
Domoki F, Perciaccante JV, Shimizu K, Puskar M, Busija DW, Bari F 2002 N-methyl-D-aspartate-induced vasodilation is mediated by endothelium-independent nitric oxide release in piglets. Am J Physiol Heart Circ Physiol 282: H1404–H1409
Ferriero DM 2004 Neonatal brain injury. N Engl J Med 351: 1985–1995
Benders MJ, Bos AF, Rademaker CM, Rijken M, Torrance HL, Groenendaal F, van Bel F 2006 Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch Dis Child Fetal Neonatal Ed 91: F163–F165
Torrance HL, Benders MJ, Derks JB, Rademaker CM, Bos AF, Van Den Berg P, Longini M, Buonocore G, Venegas M, Baquero H, Visser GH, Van Bel F 2009 Maternal allopurinol during fetal hypoxia lowers cord blood levels of the brain injury marker S-100B. Pediatrics 124: 350–357
Sauer H, Wartenberg M, Hescheler J 2001 Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem 11: 173–186
Busija DW, Leffler CW 1989 Dilator effects of amino acid neurotransmitters on piglet pial arterioles. Am J Physiol 257: H1200–H1203
Busija DW, Bari F, Domoki F, Louis T 2007 Mechanisms involved in the cerebrovascular dilator effects of N-methyl-d-aspartate in cerebral cortex. Brain Res Rev 56: 89–100
Meng W, Tobin JR, Busija DW 1995 Glutamate-induced cerebral vasodilation is mediated by nitric oxide through N-methyl-D-aspartate receptors. Stroke 26: 857–862, discussion 863
Busija DW, Meng W, Bari F, McGough PS, Errico RA, Tobin JR, Louis TM 1996 Effects of ischemia on cerebrovascular responses to N-methyl-D-aspartate in piglets. Am J Physiol 270: H1225–H1230
Busija DW, Wei M 1993 Altered cerebrovascular responsiveness to N-methyl-D-aspartate after asphyxia in piglets. Am J Physiol 265: H389–H394
Leffler CW, Mirro R, Shanklin DR, Armstead WM, Shibata M 1994 Light/dye microvascular injury selectively eliminates hypercapnia-induced pial arteriolar dilation in newborn pigs. Am J Physiol 266: H623–H630
Hobbs C, Thoresen M, Tucker A, Aquilina K, Chakkarapani E, Dingley J 2008 Xenon and hypothermia combine additively, offering long-term functional and histopathologic neuroprotection after neonatal hypoxia/ischemia. Stroke 39: 1307–1313
Thoresen M, Hobbs CE, Wood T, Chakkarapani E, Dingley J 2009 Cooling combined with immediate or delayed xenon inhalation provides equivalent long-term neuroprotection after neonatal hypoxia-ischemia. J Cereb Blood Flow Metab 29: 707–714
Pan Y, Zhang H, VanDeripe DR, Cruz-Flores S, Panneton WM 2007 Heliox and oxygen reduce infarct volume in a rat model of focal ischemia. Exp Neurol 205: 587–590
David HN, Haelewyn B, Chazalviel L, Lecocq M, Degoulet M, Risso JJ, Abraini JH 2009 Post-ischemic helium provides neuroprotection in rats subjected to middle cerebral artery occlusion-induced ischemia by producing hypothermia. J Cereb Blood Flow Metab 29: 1159–1165
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by grants from the National Scientific Research Fund of Hungary (OTKA, K68976, K63401). F.D. was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Rights and permissions
About this article
Cite this article
Domoki, F., Oláh, O., Zimmermann, A. et al. Hydrogen is Neuroprotective and Preserves Cerebrovascular Reactivity in Asphyxiated Newborn Pigs. Pediatr Res 68, 387–392 (2010). https://doi.org/10.1203/PDR.0b013e3181f2e81c
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181f2e81c
