
Abstract
Alterations in ATP metabolism have been proposed to be involved in the pathogenesis of cystinosis, the most common form of inherited Fanconi syndrome. A recent study showed normal activity of respiratory chain complexes I-IV with decreased ATP levels in cystinotic fibroblasts. Here, we show normal complex V expression and activity in mitochondria of cystinotic fibroblasts. This indicates that alterations in mitochondrial oxidative phosphorylation enzymes are not responsible for ATP decrease in cystinotic fibroblasts.
Similar content being viewed by others

Main
The autosomal recessive disorder cystinosis is caused by mutations in the CTNS gene (17p13.3), resulting in the defective lysosomal cystine transporter cystinosin (1). The consequential elevated cystine levels in the lysosomes lead to renal tubular dysfunction by mechanisms that are still elusive. Based on results of studies in cells loaded with cystine dimethyl ester, it was hypothesized that defects in mitochondrial ATP generating capacity are involved, resulting in decreased Na, K-ATPase activity (2,3). This ATP-driven proton pump generates a transmembrane Na+ gradient, required for tubular reabsorption of essential solutes such as amino acids, phosphate, and glucose in proximal tubules of the kidney. From this perspective, mitochondrial dysfunction is a possible cause of renal Fanconi syndrome in cystinosis (3).
Recently our group extensively studied ATP metabolism in cystinotic fibroblasts and demonstrated slightly decreased intracellular ATP levels, but an intact Na, K-ATPase activity, hormone stimulated mitochondrial ATP production and normal activity of respiratory chain complexes I-IV (4). The activity of mitochondrial enzyme complex V, the ATP synthase complex of the oxidative phosphorylation system, could not be measured at the time of these studies, because lack of sensitivity of the assay. At present, we complete this study in cystinotic fibroblasts by measuring complex V activity using an enzymatic assay. In addition, we examined the expression and assembly status of complex V by Western blot analysis in a blue native gel.
MATERIALS AND METHODS
Fibroblast culture.
Patient (n = 8) and control (n = 9) fibroblasts were obtained after informed consent and subsequently cultured as described previously (4). The study was approved by the Institutional Review Board of Radboud University Medical Centre. All patients were diagnosed with cystinosis by measuring elevated cystine levels in isolated polymorphonuclear cells (>0.5 nmol cystine/mg protein) by high performance liquid chromatography (HPLC) and confirmed by molecular analysis. Previous data showed elevated levels of cystine in cystinotic fibroblasts compared with controls (4.3 ± 1.1 versus 0.2 ± 0.1 nmol/mg protein) and decreased ATP levels in cystinotic fibroblasts (mean 35.9 (±16.7) versus 51.7 (±4.5) nmol/mg protein) (4). Approximately 10 × 106 cells were harvested using trypsin and divided in two samples for protein expression analysis and enzyme activity assay, respectively.
Expression of complex V.
A mitochondrial enriched fraction was prepared in phosphate-buffered saline (PBS) by incubating the cells for 10 min on ice in 2 mg/mL digitonin and subsequently centrifugation at 10000 × g for 10 min at 4°C (5). Supernatant was discarded and pellet was washed with PBS. The pellet was resuspended in aminocaproic acid (1.5 M) in Bis-Tris buffer (75 mM, pH 7.0) supplemented with 2% lauryl maltoside to dissolve the mitochondrial inner membranes, followed by a second centrifugation step at 10000 × g for 30 min at 4°C. An aliquot of the supernatant was used for protein determination using Protein Assay (Bio-Rad, Germany), followed by the addition of sample buffer containing Serva Blue G. Blue-native 5 to 15% gels were loaded with sample aliquots proportional to 20 μg protein and after polyacrylamide gel electrophoresis (PAGE) transferred onto a nitrocellulose membrane using standard methods. Complex V was detected using antibodies against the alpha subunit of ATPase (Invitrogen) and visualized using ECL Western Blotting substrate (Pierce, Thermo Scientific) after incubation with secondary goat-anti-mouse antibodies with HRP conjugate (Dako, Denmark). Finally, complex II was detected as loading control using antibodies against the complex II 70 kD subunit (SDHA; Invitrogen).
Complex V enzyme activity.
Mitochondrial fractions of control and cystinotic fibroblasts were prepared as described before (6). Complex V activity was determined in presence and absence of specific inhibitor oligomycin (8 mg/mL) in mitochondrial fractions incubated with ATP (5 mM) as substrate (7). Briefly, the ADP generated by complex V reacts with phosphoenolpyruvic acid (1 mM) to pyruvate in the presence of pyruvate kinase (1.5 U /mL). Pyruvate oxidizes with NADH (250 μM) to lactate and NAD+ in the presence of lactate dehydrogenase (2.5 U/mL). This reaction was monitored spectrophotometrically at 340 nm for 8 min and used to determine complex V activity. Complex V activities were normalized to complex IV enzyme activity, which was measured as described previously and expressed in mU/mU complex IV activity (8).
RESULTS
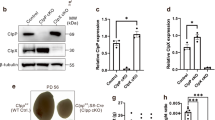
Blue native gel electrophoresis in combination with immuno blotting was used here to study the assembly and stability of the ATP generating complex V (5). Using native electrophoresis without reducing agents, complex V consisting of at least 16 subunits remains fully assembled. Western blot analysis of three control and four cystinotic mitochondrial enriched fractions showed similar complex V expression compared with complex II expression, indicating intact stability (Fig. 1A). The equal size of complex V bands and the absence of a band representing unassembled F1-ATPase, indicate complete assembly of complex V (7).

Complex V expression and activity in fibroblasts. (A) Expression of complex V and loading control complex II was determined in enriched mitochondrial fractions of control (n = 3) and cystinotic (n = 4) fibroblasts using blue native gel electrophoresis. (B) Complex V activity was measured in isolated mitochondria of control (n = 9) and cystinotic (n = 8) fibroblasts using an enzymatic assay and normalized to complex IV activity (B). Control and patient's samples loaded in figure A correspond to numbers indicated in figure B.
To study whether the activity of complex V in cystinotic fibroblast was intact, we performed an enzymatic analysis in isolated mitochondria and normalized the values using complex IV activity, which were earlier found to be normal in cystinotic fibroblasts (4). Enzymatic analysis of complex V was normal in all cystinotic fibroblast cell cultures (mean 0.82; SD 0.31 in control versus 0.65; SD 0.30 in cystinosis; p = 0.45, Fig. 1B). Additionally, we found no significant correlation between decreased ATP levels and complex V activity (data not shown).
DISCUSSION
The data presented here complete our previous study on ATP metabolism in cystinotic fibroblasts (4). In addition to the decreased ATP levels and normal Na, K-ATPase activity we found recently normal activity of complex I–IV in isolated mitochondria of cystinotic patients compared with healthy controls. In the present study, the ATP generating enzyme complex V of the oxidative phosphorylation system was investigated. We used an enzymatic method to measure hydrolase activity of complex V, which is used in diagnosing complex V deficient patients (7). Here, we found normal expression, intact assembly, and activity of complex V in cystinotic fibroblasts with elevated cystine levels compared with fibroblasts derived from healthy controls. Together with our previous data, the present study further indicates that mitochondrial ATP generating capacity in cystinotic fibroblasts is not altered. Consequentially, the decreased intracellular ATP levels in cystinotic fibroblasts are not caused by alterations in mitochondrial ATP production and require further explanation.
This raises the idea that ATP levels might be decreased by alterations in glycolysis, the main source for ATP production in cultured fibroblasts (9). An increased ATP consumption in cystinotic cells is an alternative explanation. However, the determination of ATP usage in intact cells is not feasible at the moment, as far as we know.
Alternative mechanisms involved in the pathogenesis of cystinosis can interfere with intracellular ATP levels. Cystinotic cells present alterations in glutathione metabolism (10–13), which can result in enhanced oxidative cell damage. Furthermore, increased cysteinylation of proapoptotic protein kinase Cδ, make cystinotic cells more sensitive for apoptotic stimuli (14). An increase in mitochondrial and cytosolic reactive oxygen species decreased intracellular ATP and increased sensitivity to apoptosis may all cooperate in the pathogenesis of cystinosis in a complex mechanism. Moreover, we still miss the link between cystine accumulation within lysosomes and down-stream pathogenetic events. Further investigations in human cystinotic proximal tubular cells (15,16) or in the ctns−/− mouse model (17) will generate new information in the next future.
It could be argued that measuring citric acid cycle intermediate lactate could be used to confirm mitochondrial alterations in patients with cystinosis. Although urinary excretion of lactate and hydroxybutirate is elevated in cystinotic patients, it remains questionable whether this can be attributed to mitochondrial dysfunction or reduced reabsorption due to altered proximal tubular function or due to both mechanisms (18). Thirumurugan et al. showed in their study increased lactate/creatinine ratio in several children with Fanconi syndrome, including cystinotic patients. Interestingly, a transplanted cystinotic patient had lactate/creatinine ratio comparable to nonFanconi renal controls. In patients with mitochondrial disorders urinary lactate was elevated in some, but not in all patients (19). This suggests that increased urinary lactate levels are not specific for identifying patients with mitochondrial disorders. Measuring ATP metabolism in patients with cystinosis by noninvasive magnetic resonance spectroscopy technique might answer the question whether ATP levels are decreased in vivo in cystinotic patients.
In conclusion, cystinotic fibroblasts with elevated cystine levels and decreased ATP levels have intact activity of all respiratory chain complexes (I-IV) and complex V. Whether decreased ATP levels are responsible for renal tubular dysfunction in cystinosis needs further investigation.
References
Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, Callen DF, Gribouval O, Broyer M, Bates GP, Van't Hoff W, Antignac C 1998 A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet 18: 319–324
Coor C, Salmon RF, Quigley R, Marver D, Baum M 1991 Role of Adenosine-Triphosphate (Atp) and Nak Atpase in the Inhibition of Proximal Tubule Transport with Intracellular Cystine Loading. J Clin Invest 87: 955–961
Baum M 1998 The Fanconi syndrome of cystinosis: insights into the pathophysiology. Pediatr Nephrol 12: 492–497
Levtchenko EN, Wilmer MJ, Janssen AJ, Koenderink JB, Visch HJ, Willems PH, Graaf-Hess A, Blom HJ, van den Heuvel LP, Monnens LA 2006 Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res 59: 287–292
Nijtmans LG, Henderson NS, Holt IJ 2002 Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods 26: 327–334
Bentlage HA, Wendel U, Schagger H, ter Laak HJ, Janssen AJ, Trijbels JM 1996 Lethal infantile mitochondrial disease with isolated complex I deficiency in fibroblasts but with combined complex I and IV deficiencies in muscle. Neurology 47: 243–248
Morava E, Rodenburg RJ, Hol F, De Vries M, Janssen A, van den Heuvel L, Nijtmans L, Smeitink J 2006 Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am J Med Genet A 140: 863–868
Cooperstein SJ, Lazarow A 1951 A microspectrophotometric method for the determination of cytochrome oxidase. J Biol Chem 189: 665–670
Robinson BH 1996 Use of fibroblast and lymphoblast cultures for detection of respiratory chain defects. Methods Enzymol 264: 454–464
Chol M, Nevo N, Cherqui S, Antignac C, Rustin P 2004 Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem Biophys Res Commun 324: 231–235
Levtchenko E, Graaf-Hess A, Wilmer M, van den Heuvel L, Monnens L, Blom H 2005 Altered status of glutathione and its metabolites in eystinotic cells. Nephrol Dial Transplant 20: 1828–1832
Laube GF, Shah V, Stewart VC, Hargreaves IP, Haq MR, Heales SJ, van't Hoff WG 2006 Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr Nephrol 21: 503–509
Mannucci L, Pastore A, Rizzo C, Piemonte F, Rizzoni G, Emma F 2006 Impaired activity of the gamma-glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatr Res 59: 332–335
Park MA, Pejovic V, Kerisit KG, Junius S, Thoene JG 2006 Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase Cdelta. J Am Soc Nephrol 17: 3167–3175
Laube GF, Haq MR, van't Hoff WG 2005 Exfoliated human proximal tubular cells: a model of cystinosis and Fanconi syndrome. Pediatr Nephrol 20: 136–140
Wilmer MJ, Graaf-Hess A, Blom HJ, Dijkman HB, Monnens LA, van den Heuvel LP, Levtchenko EN 2005 Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem Biophys Res Commun 337: 610–614
Cherqui S, Sevin C, Hamard G, Kalatzis V, Sich M, Pequignot MO, Gogat K, Abitbol M, Broyer M, Gubler MC, Antignac C 2002 Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol 22: 7622–7632
Thirumurugan A, Thewles A, Gilbert RD, Hulton SA, Milford DV, Lote CJ, Taylor CM 2004 Urinary l-lactate excretion is increased in renal Fanconi syndrome. Nephrol Dial Transplant 19: 1767–1773
Touati G, Rigal O, Lombes A, Frachon P, Giraud M, Ogier de Baulny H 1997 In vivo functional investigations of lactic acid in patients with respiratory chain disorders. Arch Dis Child 76: 16–21
Acknowledgements
We are grateful to the colleagues of the mitochondrial enzyme diagnostics of the Laboratory for Pediatrics and Neurology for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by a grant of the Cystinosis Research Network.
Rights and permissions
About this article
Cite this article
Wilmer, M., van den Heuvel, L., Rodenburg, R. et al. Mitochondrial Complex V Expression and Activity in Cystinotic Fibroblasts. Pediatr Res 64, 495–497 (2008). https://doi.org/10.1203/PDR.0b013e318183fd67
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e318183fd67
This article is cited by
-
Impact of atypical mitochondrial cyclic-AMP level in nephropathic cystinosis
Cellular and Molecular Life Sciences (2018)
-
Slow progression of renal failure in a child with infantile cystinosis
CEN Case Reports (2018)
-
The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives
Nature Reviews Nephrology (2017)
-
p62/SQSTM1 prominently accumulates in renal proximal tubules in nephropathic cystinosis
Pediatric Nephrology (2012)
-
A futile cycle, formed between two ATP-dependant γ-glutamyl cycle enzymes, γ-glutamyl cysteine synthetase and 5-oxoprolinase: the cause of cellular ATP depletion in nephrotic cystinosis?
Journal of Biosciences (2010)
