
Abstract
We present the clinical and laboratory features of a boy with a new syndrome of mitochondrial depletion syndrome and T cell immunodeficiency. The child suffered from severe recurrent infectious diseases, anemia, and thrombocytopenia. Clinically, he presented with severe psychomotor retardation, axial hypotonia, and a disturbed pain perception leading to debilitating biting of the thumb, lower lip, and tongue. Brain imaging showed hypoplasia of corpus callosum and an impaired myelinization of the temporo-occipital region with consecutive supratentorial hydrocephalus. Histologic examination of a skeletal muscle biopsy was normal. Biochemical investigation showed combined deficiency of respiratory chain complexes II+III and IV. MtDNA depletion was found by real-time PCR. No pathogenic mutations were identified in the TK2, SUCLA2, DGUOK, and ECGF1 genes. A heterozygous missense mutation was found in POLG1. The pathogenic relevance of this mutation is unclear. Interestingly, a lack of CD8+ T lymphocytes as well as NK cells was also observed. The percentage of CD45RO-expressing cells was decreased in activated CD8+ T lymphocytes. Activation of T lymphocytes via IL-2 was diminished. The occurrence of the immunologic deficiency in our patient with mtDNA depletion is a rare finding, implying that cells of the immune system might also be affected by mitochondrial disease.
Similar content being viewed by others


Main
Mitochondrial diseases are a complex family of disorders with clinical manifestations, which may be caused by mutations of nuclear DNA (nDNA) or mitochondrial DNA (mtDNA). Due to diverse genetic causes and occurrence in various developmental stages, tissues, or systems, there is a wide variability in clinical phenotype that spans all age groups (1). As many of the RC disorders involve brain and skeletal muscle, these disorders are also known as mitochondrial encephalomyopathies (2). Nevertheless, given the essential role of oxidative phosphorylation in tissue function, virtually all tissues and organs can be involved, accounting for a diverse array of multisystem disorders with a large variety of clinical symptoms in both childhood and adulthood.
MDS is defined as a reduction of mtDNA copy number compared with nDNA in different tissues, which leads to insufficient synthesis of RC complexes I, III, IV, and V (3). Autosomal recessive mutations of five nuclear genes have been identified in MDS patients with different clinical presentations (2,4–7). The onset of symptoms usually occurs during the first year of life and most patients die in early childhood. Thymidine phosphorylase (ECGF1) defects are associated with leukodystrophy and gastrointestinal disorders (8). Mutations in deoxyguanosine kinase (DGUOK) (9) and polymerase gamma (POLG1) (10,11) were described in the hepatocerebral form of MDS, whereas thymidine kinase 2 (TK2) mutations are associated with a mostly myopathic phenotype and CK elevation (12). Most recently, mutations in the succinyl-CoA synthase (SUCLA2) were identified in four infants of a single family reported with severe psychomotor retardation, epilepsy, hearing loss, Leigh-like lesions on MRI, and anemia (13). Mitochondrial depletion can affect single organs, typically skeletal muscle or liver, although in a later phase of the disease, multiple tissues are often involved (5–7).
Childhood myelodysplasia occasionally occurs as the initial symptom of a mitochondrial disease (14), and is usually associated with the Pearson's marrow-pancreas syndrome (15,16). This syndrome combines refractory anemia with ring sideroblasts and vacuolization of marrow precursors, and variable degrees of neutropenia and thrombocytopenia. We describe the clinical features of a child with a defect of the respiratory chain caused by mtDNA depletion with a unique T cell phenotype characterized by reduction in numbers of CD8+CD45RO+ T cells as well as of CD56+CD3– NK cells, and impaired T cell maturation and activation. To our knowledge, this is the first report of immunodeficiency of specific lymphocyte subsets associated with mitochondrial disease.
PATIENTS AND METHODS
Case Report.
The child was born at 39 wk of gestation to healthy, first-degree consanguineous parents. He had a healthy 1-y-old sister. At 16 wk of gestation, amniocentesis was performed following detection of hygroma colli on routine ultrasound examination. Levels of alpha-fetoprotein and acetylcholine esterase in amniotic fluid were normal, and chromosome analysis showed a normal male 46XY karyotype. The pregnancy was uneventful, and delivery was uncomplicated. At birth, he weighed 2610 g [<P10 (10th percentile)], was 49 cm long (P25), and had a head circumference of 35.5 cm (P50). Apgar scores were 8/10/10, and the umbilical arterial pH was 7.23. Postnatal tachycardia and dyspnea with need for oxygen administration lead to transfer to neonatal intensive care unit. Polycythemia, requiring hemodilution, as well as thrombocytopenia, requiring repeated transfusion of platelet concentrates, were noted immediately after birth. Clinically and biochemically, there were no signs of associated infection. A maternal TORCH [congenital infection with toxoplasmosis, other, rubella, cytomegalovirus (CMV), herpes simplex virus (HSV)] infection was excluded.
Neurologic examination revealed adynamia, hypotonia of the trunk, complete absence of deep tendon reflexes as well as primitive reflexes (Moro, palmar, and plantar grasp), and a disturbed pain perception with complete absence of pain expression. Cranial ultrasonography showed hydrocephalus internus. MRI showed hypoplasia of the corpus callosum, and an impaired myelinization of the temporo-occipital region with consecutive supratentorial hydrocephalus. Nerve conduction velocity demonstrated normal motor nerves, but impaired sensory conduction with reduced amplitude, consistent with combined axonal and demyelinating neuropathy. Repeated somatosensory-evoked potentials of the N. medianus demonstrated a disruption of ponto-cerebral conduction, and brainstem auditory-evoked potentials showed progressive slowing of conduction after entering the brainstem, indicative of a functional problem in central nervous myelination. EEG were normal. Facial features included a high forehead, short philtrum, low-set, dysplastic ears, and short neck, and partial syndactyly of toes II and III was present (Fig. 1).

Facial features and hepatosplenomegaly. The picture was taken at 5 mo of age.
At age 5 d, he developed septicemia with maculopapular exanthema, paronychia, and conjunctivitis. Staphylococcus aureus was cultured from a throat swab. Lesions resolved with standard antibiotic treatment. The boy was hospitalized until the age of 3 mo for recurrent episodes of bloody diarrhea, as well as septicemia and significant thrombocytopenia. Stool and blood cultures failed to detect any infectious agent. Isolated germs were Klebsiella pneumoniae from a skin and nose swab, and Pantoea agglomerans from the central venous catheter. The child was treated with several courses of standard intravenous antibiotics. At 3 mo of age, a first episode of significant bleeding of the tongue and ulcerations of the oral mucosa were noted. A blood count revealed anemia and thrombocytopenia requiring repeated red blood cell and platelet transfusions. The child was discharged from hospital at 3 mo of age with a prophylactic antibiotic and antimycotic medication. In addition, he was put on intravenous immunoglobulin (IVIG) substitution every 4 wk for treatment of hypogammaglobulinemia (Table 2).
Between the ages of 4 and 13 mo, the patient experienced six severe infectious episodes leading to hospitalization, three episodes of bloody gastroenteritis, three episodes of bronchopneumonia, and four episodes of septicemia, each with significant anemia and/or thrombocytopenia, requiring 12 erythrocyte and 6 platelet concentrate transfusions. Blood and stool cultures were repeatedly negative; the only germs isolated were S. aureus and Enterobacter cloacae from lesions of the thumb, Bacillus cereus and Pseudomonas aeruginosa from the eyes, E. cloacae from throat swabs, and S. epidermidis from an infected lymph node. The patient was frequently biting his thumb while sucking due to absent pain reaction, leading to progressive ulceration of the tongue and lip (Fig. 2). Severe statomotor retardation was noted at age 9 mo, with development corresponding to 2–3 mo of age. A muscle biopsy was performed at 11 mo of age.

Progressive ulceration of the tongue, first noted at age 3 mo. The pictures were taken at 5 mo of age.
At 13 mo, the boy was hospitalized for anemia and progressive deterioration of his clinical condition. His weight, height, and head circumference were below the 3rd percentiles for age (weight, 6.1 kg, i.e. 2.6 kg < P3; length, 69.2 cm, i.e. 4 cm < P3; head circumference, 43.5 cm, i.e. 1.5 cm < P3). Abdominal ultrasonography showed progressive homogenous hepatosplenomegaly. Several erythrocyte transfusions were given in the following months while the clinical condition of the boy was deteriorating further. Treatment with carnitine, nicotinamide, alpha-lipoic acid, vitamin C, CoQ (coenzyme Q), vitamin E, zinc, and vitamin D was started after the diagnosis of RC disease. Death occurred at 18 mo during an episode of septicemia. The child was admitted to hospital with Cheyne's stokes respiration, dilated fixed pupils, centralized circulation, increased muscle tone, hemorrhagic conjunctivitis, and bloody diarrhea. Resuscitation was attempted but without success.
Mild hyperlactatemia was noted on several occasions (range, 2.6–4.5 mM; norm < 2.5 mM). Extensive screening for metabolic disease was done, including pilocarpine iontophoresis, oral glucose tolerance test, acylcarnitine profile by tandem mass spectrometry, analysis of thyroid hormones, glucose-6-phosphate dehydrogenase, phenylalanine, galactose-1-phosphat-uridyltransferase, 17-OH progesterone, and biotinidase-activity, all of which were normal. A congenital disorder of glycosylation (CDG) was excluded by the presence of a normal isoelectric focusing of transferrins in serum. There was a normal pattern of amino acids in serum, and of organic acids as well as purine/pyrimidines in urine. There were no signs of cardiomyopathy, pancreatic dysfunction, or renal disease.
Written informed consent was obtained from the parents of the child.
Morphology and biochemistry of skeletal muscle.
Six-micrometer serial cross-sections of muscle biopsy specimens were obtained for histochemical stains according to standard procedures. A frozen part of the biopsy was used for biochemistry. The biopsy was kept at –80°C until analysis. RC complexes I–IV activities were determined in skeletal muscle as described (17).
DNA analysis.
DNA extraction from muscle, blood, and fibroblasts was done according to standard purification protocols (QIAGEN GmbH, Hilden, Germany). RFLP analysis of the frequent tRNA mutations and long-range PCR to detect mtDNA deletions were performed by standard methods. MtDNA and nDNA copy numbers and the mtDNA/nDNA ratio within tissues were determined by real-time PCR using a fluorescence temperature cycler (LightCycler, Roche Molecular Biochemicals, Mannheim, Germany). A LightCycler FastStart DNA Master SYBR Green I kit (Roche Molecular Biochemicals) was used with unlabeled primers as described (18). All samples were run in triplicate. Sequencing of the nuclear-encoded TK2 (12), SUCLA2 (13), POLG1 (19), DGUOK (9), and ECGF1(8) genes was performed as previously described. POLG1 cDNA primers can be provided on request. For RFLP analysis for c.3559C>T, the PCR product of exon 22 was digested with the restriction enzyme Age I.
Serological assays.
Specific antibodies against tetanus toxoid and Haemophilus influenzae were routinely measured by ELISA (Progen Biotechnik, Heidelberg, Germany). IgA, IgG, and IgM were assayed by nephelometry (Dade Behring Holding, Liederbach, Germany), and IgG-subclasses were analyzed by turbidimetry (The Binding Site, Birmingham, UK). Serum antibodies to pneumococcal conjugate vaccine (PCV)-serotypes 6B, 14, 19F, and 23F and nonPCV-serotype 5 were determined before and 7 and 28 d after the PPV (pneumococcal polysaccharide vaccine) booster vaccination. Serotype-specific IgG-antibody concentration was measured by a modified ELISA technique using Nunc CovaLink microtiter plates (Nunc GmbH. Wiesbaden, Germany) and serum lot 89-SF as reference (Food and Drug Administration, Bethesda, MD). Serum samples were preincubated with 10 μg/mL pneumococcal polysaccharide C (CPS, Statens Seruminstitut, Copenhagen, Denmark) for blocking of unspecific anti-CPS-antibodies. Reference serum 89-SF (kindly provided by Dr. C. Frash, Rockville, MD) was used for assay standardization. Sera with known high-antibody titers were used as reference and quality control sera (Endobulin HP, Immuno AG, Vienna, Austria) (20). Minimum antibody detection level was 0.1 μg/mL.
Immunophenotyping.
Disease-associated immunophenotyping was assessed by four-color flow cytometric determination of T cell subsets, natural killer (NK) cells, and B cells in peripheral blood (PB) samples on a Coulter Epics XL (Beckman Coulter GmbH, Krefeld, Germany). Briefly, 100 μL of PB were labeled for 20 min each with the 45/4/8/3 or 45/56/19/3 tetraChrome reagents (Coulter). In addition, naïve cells (CD45RA+CD4+, CD45RA+CD8+CD28+), memory cells (CD45RO+CD4+, CD45RO+CD8+), and activated cells (CD8+CD69+, CD8+HLA (human leucocyte antigen)-DR+, CD4+CD69+, CD4+HLA-DR+) were detected. All antibodies were obtained from Coulter Immunotech (Marseille, France), labeled with the corresponding four different antibodies conjugated with FITC, phycoerythrin (PE), phycoerythrin-Texas Red (ECD), and phycoerythrin-cyanine 5.1 (PC-5). The percentage of lymphocytes determined in these analyses was used to calculate the absolute lymphocyte counts in a dual-platform approach (21).
Cell stimulation and activation assay.
Peripheral blood mononuclear cells (PBMC) were separated from heparinized whole blood by density gradient centrifugation on Ficoll-Hypaque and washed three times with RPMI 1640 medium. Cells were cultured in 96-well flat-bottom plates in RPMI 1640 medium supplemented with 10% FCS, 1% penicillin/streptomycin, 1% HEPES, 2% glutamine, and 0.2% gentamicin at a concentration of 1 × 106 cells/mL. For induction of cell activation markers and proliferation, lymphocytes were incubated for 72 h in the presence of phytohemagglutinin (PHA) (1 mg/mL), IL-2 (100U/mL), and/or phorbol-12,13-dibutyrate (PBu2, 1 ng/mL) in conjunction with ionomycin (0.5 mg/mL). Before and after stimulation, immunophenotyping was used to determine the appropriate lymphocyte subset population as described above.
Cell proliferation.
The rates of DNA synthesis were assayed by measuring [3H]thymidine incorporation into PBMC. Cells were pulsed with [3H]thymidine (1.0 μCi/mL) over the last 18 h of the 72-h incubation period and the incorporated radioactivity was measured by scintillation counting.
RESULTS
Morphology and biochemistry of skeletal muscle.
Histologic investigation of skeletal muscle showed a normal muscle structure, no ragged red fibers, and normal succinate dehydrogenase (SDH) staining, but a generalized deficiency of the stain for cytochrome c oxidase (COX). Biochemical measurements of the respiratory chain enzymes showed combined deficiency of the complexes II+III and IV, related to both g NCP, and the mitochondrial marker enzyme citrate synthase (CS) (Table 1), whereas activity of complex I was normal. Complex II activity was not measured because of lack of muscle material. The nuclear encoded pyruvate dehydrogenase as well as citrate synthase had normal enzymatic activity. These findings are consistent with mtDNA depletion, and provide indirect evidence of normal SDH function. The activity of the complex IV in fibroblasts was normal.
DNA analysis.
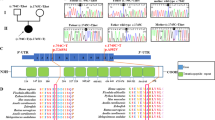
Screening of the muscle DNA with RFLP for the common tRNA mutations MELAS (mitochondrial myopathy, encephalopathy, lactacidosis, stroke) 3243, MERRF (myoclonic epilepsy and ragged red fiber disease) 8344, LEIGH/NARP (neuropathy, ataxia and retinitis pigmentosa) 8993G/C did not show any mutations. Long-range PCR excluded mtDNA deletions. Real-time PCR analysis of the patient's muscle DNA revealed a significant depletion of mtDNA. The mtDNA/nDNA ratio was 0.05 (normal range, 0.5–5.0), 10% of the lowest level found in control muscle DNA samples. Fibroblast DNA of the patient showed less severe, but also significant mtDNA depletion (mtDNA/nDNA ratio, 0.16; normal range, 0.5–2.5). Sequence analysis of the known mtDNA depletion related genes TK2, SUCLA2, DGUOK, and ECGF1 revealed no pathogenic mutations. A heterozygous base pair exchange, c.3559C>T was detected in POLG1, causing an exchange of a poorly conserved arginine to tryptophan at position 1187. This base pair exchange was not detected in 100 Turkish control alleles. RFLP analysis revealed that the father and both children (affected and unaffected) were heterozygous for c.3559C>T, while the mother was not carrying the mutation. POLG1 cDNA of the affected child was also heterozygous for c.3559C>T. No additional POLG1 mutation was detected in the affected child by complete sequence analysis of both DNA and cDNA.
Immunologic assessment.
Due to the increased number and severity of infectious episodes, immunodeficiency was suspected and the child's immune status was assessed repeatedly (Tables 2 and 3). The karyotype was 46XY with a normal chromosomal breakage rate. The thymus was of normal size. A bone marrow aspirate at the age of 3 wk showed a low megacaryocyte count with reduced but morphologically normal erythropoiesis. In particular, there were no sideroblasts or vacuolization of marrow precursors. Peripheral red blood cells were slightly hypochromic and microcytic, and reticulocyte counts were normal for age. Alloimmune thrombocytopenia and Wiskott-Aldrich syndrome were excluded. A search for parvovirus B19, CMV, HHV-6 in the bone marrow aspirate was negative. HIV1 and 2 infections were excluded. Septic granulomatosis was excluded by normal H2O2 production, leucocyte adhesion deficiency (LAD) type 1 was excluded by normal expression of CD11a-c and CD18, and ZAP (zeta associated protein)-70 cDNA sequence was normal. Hoyeraal-Hreidarsson syndrome was excluded by normal DKC1 (dyskeratosis congenital 1, dyskerin) gene. There were no clinical or biologic signs of allergy. There was no pattern of skewed X-inactivation of the HUMARA (X-linked human androgen receptor) locus in the patient's mother.
Humoral immunodeficiency.
Serum Ig levels were low for age despite regular IVIG substitution. IgG1 and IgG2 subclasses were also low for age; IgG3 and IgG4 were normal. Titers of antibodies against polysaccharide antigens, such as blood isohemagglutinin A (titers 1:2 at room temperature, 1:16 at indirect Coombs' test), and serum antibodies against S. pneumoniae were low, despite repeated vaccination. In contrast, titers of antibodies against protein antigens such as tetanus toxoid and H. influenzae were normal (Table 2).
Immuno phenotyping.
The total number and morphology of lymphocytes and polymorphonuclear cells were initially normal (Table 3). There was a progressive decline in absolute lymphocyte numbers, especially of CD8+CD3+ T cells and CD56+CD3– NK cells, which was first noted at age 19 d. After 1 mo, the number of B cells as well as CD3+CD4+ cells decreased. The number of CD8+CD3+ T cells and CD56+CD3– NK cells remained low. Expression of the memory marker CD45RO was low on CD8+ T cells, whereas it was normally expressed on CD4+ T cells.
T cell proliferation and activation.
Proliferative responses (Fig. 3) and cell activation (Fig. 4, A and B) to mitogens were normal in isolated PBMC. HLA-DR expression was slightly increased in PHA stimulated CD3+CD4+ cells (Fig. 4C), but normal in CD3+CD8+ cells (Fig. 4D). In contrast, activation via IL-2 was impaired in CD3+CD4+ cells (Fig. 4A) as well as in CD3+CD8+ cells (Fig. 4 B), as measured by expression of CD69. Additionally, after in vitro stimulation with phorbol ester, the percentage of CD45RO+CD8+ T lymphocytes was decreased (Fig. 5).

Proliferation. Isolated PBMC were incubated for 72 h in the presence of phytohemagglutinin (PHA, 1 mg/mL) or phorbol-12,13-dibutyrate (PBu2, 1 ng/mL) and ionomycin (0.5 mg/mL). Proliferation was assayed by measuring [3H]thymidine incorporation into the cells. Medium: □), PHA: ▪, PBu2/Ionomycin: □

Activation marker. Isolated PBMC were stimulated with PHA (5 μg/mL) or IL-2 (1000 IE/mL) for 96 h. Cell activation was measured by CD69 (A, B) or HLA-DR (C, D) expression on CD3+CD4+ and CD3+CD4+ cells using flow cytometry. Medium: □), PHA: ▪, JL-2: □

Memory marker. Activation of memory CD45RO+ cells was analyzed in CD3+CD4+ (□)) and CD3+CD8+ (▪) T cell subsets. PBMC were stimulated with PBu2 (1 ng/mL) and ionomycin (0.5 mg/mL) for 96 h. Expression of CD45RO was measured by flow cytometry.
DISCUSSION
We provide here a comprehensive clinical description of a child with an unknown syndrome of a mitochondrial disorder and T cell immunodeficiency. The patient presented with a unique severe phenotype, combining neurologic, hemato-immunologic, gastrointestinal, and skeletal muscle abnormalities. There were no signs of cardiac muscle abnormalities, endocrine, renal, or pancreatic involvement. The onset of the disease was very early, with no symptom-free period, suggesting antenatal expression of the disease. CNS involvement has been described in a few patients and includes corpus callosum agenesis or hypoplasia, porencephalic cysts, and enlarged cerebral ventricles (22), all of which were present in our patient. The craniofacial anomalies noted in the child have also been reported in other cases of RC deficiency (23). Malformation during embryogenesis might be either due to a lack of ATP or the alteration of an apoptotic event controlled by the mitochondrial machinery (24). Trunk hypotonia, hyporeflexia, peripheral neuropathy, statomotor retardation, and abnormal eye movements are common findings in other mitochondrial disorders (16). Gastrointestinal presentation of the disease was marked by severe chronic diarrhea with failure to thrive. In our patient, diarrhea was almost exclusively associated with septic episodes and accompanied by severe transfusion-dependent anemia.
In addition, the patient suffered from early severe and recurrent anemia and thrombocytopenia. Myelodysplasia has been described in patients with Pearson's marrow pancreas syndrome (15,25), which was ruled out in our patient by morphologically normal erythropoiesis, the absence of sideroblasts or vacuolization of marrow precursors, and by the absence of mtDNA deletion on long-range PCR. Other RC deficiencies associated with anemia are NDUFS1 (a nuclear-encoded Fe-S subunit of complex 1) (26), COXI (27), and COX10 (28), as well as SUCLA2 (13) mutations. However, none of these diseases are associated with immunodeficiency. Similar to the effect of the single deletion in blood cells in Pearson's marrow pancreas syndrome, mtDNA depletion might lead to anemia and thrombocytopenia. With real-time PCR, a significantly reduced mtDNA/nDNA ratio was detected in both muscle and fibroblast DNA of the patient, indicating the diagnosis of MDS. Involvement of immunologically active cells has so far not been described in association with mtDNA depletion. Unfortunately, the diagnosis of mtDNA depletion in the patient was post mortem. We could not perform mtDNA depletion studies in these tissues, however, we suggest that the mtDNA depletion in lymphocytes led to immunodeficiency in this case.
Pathogenic mutations in four of the five nuclear genes (TK2, SUCLA2, DGUOK, ECGF1) described in association with mtDNA depletion were excluded in our patient. A heterozygous base pair exchange, c.3559C>T was found in POLG1, causing an amino acid exchange on protein level. However, we believe that the heterozygous c.3559C>T mutation is not related to the disease phenotype for the following reasons: Arginine is poorly conserved at position 1187 in the C terminal part of the protein, within the polymerase domain, next to previously described pathogenic mutations (S1176L, D1184N), as well as polymorphisms (E1143G, R1146C, Q1236H). The severe early-onset phenotype and the depletion of mtDNA suggest an autosomal recessive disease and no second mutation was detected in both POLG1 DNA and cDNA. The consanguinity of the parents would also suggest an autosomal recessive disease but with a homoplasmic mutation. Although clinical symptoms of the patient might be partly compatible with POLG1 deficiency (muscular hypotonia, gastrointestinal symptoms, hepatopathy), the most important clinical hallmark of a POLG1-related mtDNA depletion syndrome, progressive epileptic activity, was excluded by normal EEG.
Immunodeficiency was first suspected on the basis of multiple clinical infectious diseases presented by the patient. He had numerous infections attributable to bacteria (Gram-negative bacilli, Gram-positive cocci) and fungi. No viral diseases were documented, suggesting that viral immunity was not significantly affected. The child had low serum Ig levels and did not produce antibodies specific for polysaccharide antigens. T and B cell responses to protein antigens and mitogens appeared normal except that serum Ig levels were low. The most striking immunologic phenotype was an early and persistent lack of CD8+ T cells and CD56+CD3– NK cells. The remaining CD8+ T cells had a unique phenotype with a severely impaired expression of the memory marker CD45RO after activation of cells. To our knowledge, there are no reports on immunodeficiency of specific lymphocyte subsets associated with mitochondrial diseases. Whether the immune defect is directly related to the mitochondrial depletion or whether this association is coincidental in our patient is not certain. It raises the question of whether involvement of the immune system in MDS needs to be considered. Future goals include understanding how the defective proteins perturb replication, why MDS affects only some tissues and spares others, and which other genes should be considered in patients with mtDNA depletion.
Abbreviations
- MDS:
-
mitochondrial DNA depletion syndrome
- POLG1:
-
polymerase gamma 1
- RC:
-
respiratory chain
- SUCLA2:
-
succinyl-CoA synthase 2
- TK2:
-
thymidine kinase 2
References
Thorburn DR 2004 Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis 27: 349–362
DiMauro S, Schon EA 2003 Mitochondrial respiratory-chain diseases. N Engl J Med 348: 2656–2668
Moraes CT, Shanske S, Tritschler HJ, Aprille JR, Andreetta F, Bonilla E, Schon EA, DiMauro S 1991 mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet 48: 492–501
Hirano M, Vu TH 2000 Defects of intergenomic communication: where do we stand?. Brain Pathol 10: 451–461
DiMauro S, Hirano M 2005 Mitochondrial encephalomyopathies: an update. Neuromuscul Disord 15: 276–286
Spinazzola A, Zeviani M 2005 Disorders of nuclear-mitochondrial intergenomic signaling. Gene 354: 162–168
Carrozzo R, Bornstein B, Lucioli S, Campos Y, de la Pena P, Petit N, Dionisi-Vici C, Vilarinho L, Rizza T, Bertini E, Garesse R, Santorelli FM, Arenas J 2003 Mutation analysis in 16 patients with mtDNA depletion. Hum Mutat 21: 453–454
Nishino I, Spinazzola A, Hirano M 1999 Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283: 689–692
Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, Eriksson S, Cohen N 2001 The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet 29: 337–341
Naviaux RK, Nguyen KV 2004 POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann Neurol 55: 706–712
Ferrari G, Lamantea E, Donati A, Filosto M, Briem E, Carrara F, Parini R, Simonati A, Santer R, Zeviani M 2005 Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gammaA. Brain 128: 723–731
Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O 2001 Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 29: 342–344
Elpeleg O, Miller C, Hershkovitz E, Bitner-Glindzicz M, Bondi-Rubinstein G, Rahman S, Pagnamenta A, Eshhar S, Saada A 2005 Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet 76: 1081–1086
Bader-Meunier B, Rotig A, Mielot F, Lavergne JM, Croisille L, Rustin P, Landrieu P, Dommergues JP, Munnich A, Tchernia G 1994 Refractory anaemia and mitochondrial cytopathy in childhood. Br J Haematol 87: 381–385
Rotig A, Cormier V, Blanche S, Bonnefont JP, Ledeist F, Romero N, Schmitz J, Rustin P, Fischer A, Saudubray JM 1990 Pearson's marrow-pancreas syndrome. A multisystem mitochondrial disorder in infancy. J Clin Invest 86: 1601–1608
Munnich A, Rustin P 2001 Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet 106: 4–17
Fischer JC, Ruitenbeek W, Gabreels FJ, Janssen AJ, Renier WO, Sengers RC, Stadhouders AM, ter Laak HJ, Trijbels JM, Veerkamp JH 1986 A mitochondrial encephalomyopathy: the first case with an established defect at the level of coenzyme Q. Eur J Pediatr 144: 441–444
Setzer B, Schlesier M, Walker UA 2005 Effects of didanosine-related depletion of mtDNA in human T lymphocytes. J Infect Dis 191: 848–855
Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, Papadimitriou A, Spelbrink H, Silvestri L, Casari G, Comi GP, Zeviani M 2002 Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol 52: 211–219
Zielen S, Broker M, Strnad N, Schwenen L, Schon P, Gottwald G, Hofmann D 1996 Simple determination of polysaccharide specific antibodies by means of chemically modified ELISA plates. J Immunol Methods 193: 1–7
Koehl U, Sorensen J, Esser R, Zimmermann S, Gruttner HP, Tonn T, Seidl C, Seifried E, Klingebiel T, Schwabe D 2004 IL-2 activated NK cell immunotherapy of three children after haploidentical stem cell transplantation. Blood Cells Mol Dis 33: 261–266
Samson JF, Barth PG, de Vries JI, Menko FH, Ruitenbeek W, van Oost BA, Jakobs C 1994 Familial mitochondrial encephalopathy with fetal ultrasonographic ventriculomegaly and intracerebral calcifications. Eur J Pediatr 153: 510–516
Cormier-Daire V, Rustin P, Rotig A, Chretien D, Le Merrer M, Belli D, Le Goff A, Hubert P, Ricour C, Munnich A 1996 Craniofacial anomalies and malformations in respiratory chain deficiency. Am J Med Genet 66: 457–463
von Kleist-Retzow JC, Cormier-Daire V, Viot G, Goldenberg A, Mardach B, Amiel J, Saada P, Dumez Y, Brunelle F, Saudubray JM, Chretien D, Rotig A, Rustin P, Munnich A, De Lonlay P 2003 Antenatal manifestations of mitochondrial respiratory chain deficiency. J Pediatr 143: 208–212
Greenberg PL, Young NS, Gattermann N 2002 Myelodysplastic syndromes. Hematology (Am Soc Hematol Educ Program) 136–161
Benit P, Chretien D, Kadhom N, de Lonlay-Debeney P, Cormier-Daire V, Cabral A, Peudenier S, Rustin P, Munnich A, Rotig A 2001 Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am J Hum Genet 68: 1344–1352
Gattermann N, Retzlaff S, Wang YL, Hofhaus G, Heinisch J, Aul C, Schneider W 1997 Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood 90: 4961–4972
Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, Shoubridge EA 2003 Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet 12: 2693–2702
de Vries E, de Bruin-Versteeg S, Comans-Bitter WM, de Groot R, Hop WC, Boerma GJ, Lotgering FK, van Dongen JJ 2000 Longitudinal survey of lymphocyte subpopulations in the first year of life. Pediatr Res 47( 4 Pt 1): 528–537
Acknowledgements
The authors thank the patient and his family and deeply respect how they coped with this fatal disease. We also thank Dr. D. Tews, Edinger-Institute Frankfurt, for histochemistry of the muscle, nerve, and skin biopsies. We thank Katrin Krug and Andrea Brinkmann for excellent technical support and Adrian Sewell for critical revision of the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reichenbach, J., Schubert, R., Horvàth, R. et al. Fatal Neonatal-Onset Mitochondrial Respiratory Chain Disease with T Cell Immunodeficiency. Pediatr Res 60, 321–326 (2006). https://doi.org/10.1203/01.pdr.0000233252.60457.cf
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000233252.60457.cf
This article is cited by
-
The immune system as a driver of mitochondrial disease pathogenesis: a review of evidence
Orphanet Journal of Rare Diseases (2022)
-
Contribution of nuclear and mitochondrial gene mutations in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome
Journal of Neurology (2021)
-
Clinical and Immunological Characterization of Combined Immunodeficiency Due to TFRC Mutation in Eight Patients
Journal of Clinical Immunology (2020)
-
Quantitative multiplex PCR of short fluorescent fragments for the detection of large intragenic POLG rearrangements in a large French cohort
European Journal of Human Genetics (2014)
-
Mitochondria as Oxidative Signaling Organelles in T-cell Activation: Physiological Role and Pathological Implications
Archivum Immunologiae et Therapiae Experimentalis (2013)
