
Abstract
Glutaryl-CoA dehydrogenase (GCDH) deficiency is a rare inborn disorder of l-lysine, l-hydroxylysine, and l-tryptophan metabolism complicated by striatal damage during acute encephalopathic crises. Three decades after its description, the natural history and how to treat this disorder are still incompletely understood. To study which variables influenced the outcome, we conducted an international cross-sectional study in 35 metabolic centers. Our main outcome measures were onset and neurologic sequelae of acute encephalopathic crises. A total of 279 patients (160 male, 119 female) were included who were diagnosed clinically after clinical presentation (n = 218) or presymptomatically by neonatal screening (n = 23), high-risk screening (n = 24), or macrocephaly (n = 14). Most symptomatic patients (n = 185) had encephalopathic crises, characteristically resulting in bilateral striatal damage and dystonia, secondary complications, and reduced life expectancy. First crises usually occurred during infancy (95% by age 2 y); the oldest age at which a repeat crisis was reported was 70 mo. In a few patients, neurologic disease developed without a reported crisis. Differences in the diagnostic criteria and therapeutic protocols for patients with GCDH deficiency resulted in a huge variability in the outcome worldwide. Recursive partitioning demonstrated that timely diagnosis in neurologically asymptomatic patients followed by treatment with l-carnitine and a lysine-restricted diet was the best predictor of good outcome, whereas treatment efficacy was low in patients diagnosed after the onset of neurologic disease. Notably, the biochemical phenotype did not predict the clinical phenotype. Our study proves GCDH deficiency to be a treatable disorder and a good candidate for neonatal screening.
Similar content being viewed by others


Main
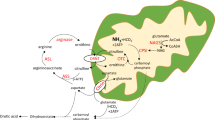
GCDH deficiency was first described in 1975 (1). The estimated prevalence is 1:100,000 newborns (2), but it is considerably higher in some genetic isolates (3,4). More than 150 disease-causing GCDH gene mutations have been identified (5–7). GCDH is a key enzyme in the degradative pathways of l-lysine, l-hydroxylysine, and l-tryptophan (8–10). The biochemical hallmark is an accumulation of GA and 3-OH-GA, in particular in the CNS (11–13). These pathologic dicarboxylic acids and their corresponding carnitine (glutaryl carnitine) and glycine ester (glutaryl glycine) can be detected by gas chromatography/mass spectrometry in body fluids (14), or MS/MS in dried blood spots (15,16). MS/MS-based determination of glutaryl carnitine has already been implemented into neonatal screening programmes in some countries (15–17).
Strikingly, acute encephalopathic crises, which are precipitated by intercurrent febrile illnesses and primarily affect the striatum (18–20), usually manifest during infancy (3,19,21). However, late-onset neurologic disease has recently been described (22,23). Before the onset of an encephalopathic crisis, the presentation of affected children is nonspecific, with macrocephaly being the most characteristic clinical feature (3,19,21). If diagnosed in a timely manner and treated with a special diet and l-carnitine supplementation as well as an intensified emergency therapy during intercurrent illness (24), the development of acute encephalopathic crises and striatal damage has been prevented in 65–95% of children (3,19,25). However, the neuroprotective effect of different protocols has not been studied systematically and the level of evidence in the majority of clinical studies has been low. As a result of variations in the natural history, and differences in the diagnostic criteria and therapeutic protocols, the outcome worldwide has been varied. This has hampered our understanding to such a degree that it is not certain whether the disorder is really treatable (4,6).
PATIENTS AND METHODS
Study population.
We studied 279 patients (119 female, 160 male) with GCDH deficiency diagnosed through 1) neonatal screening (n = 23), 2) screening of high-risk families (n = 18) or communities (n = 6), or 3) selective screening due to encephalopathic crises (n = 218) or macrocephaly (n = 14). Diagnosis was confirmed by mutation (5–7) and/or enzyme analyses (8,9).
Questionnaire-based survey.
Forty international metabolic centers were contacted, and 35 of them responded (response rate, 87.5%), identifying a total of 314 patients. Subsequently, from March 2003 to February 2004 a standardized questionnaire (available online at www.metabnet.de) was sent, asking questions on personal data, family history, socioeconomic status, pregnancy and delivery, diagnostic procedure, clinical presentation, neuroradiology, treatment protocol, biochemical monitoring, and outcome. Questionnaires were filled in by responsible physicians and were returned to the study office (University Children's Hospital, Heidelberg, Germany). Patients were made anonymous by initials, birth date, and gender, and double entries were excluded by these parameters. Data were only included if written informed consent was obtained from patients and/or parents. The study was approved by the institutional ethics committee of the University of Heidelberg (#314/2002). Patients with unconfirmed diagnosis of GCDH deficiency and with glutaric aciduria types II or III were excluded. We received 279 completed questionnaires (response rate, 88.9%); the remaining 35 patients were excluded because the information was incomplete or the diagnosis remained doubtful.
Definition of acute encephalopathic crisis, striatal lesion, and outcome scores.
Acute encephalopathic crisis was defined as acute onset of neurologic disease (e.g., dystonia) after an episode that was likely to precipitate a catabolic state (e.g., febrile illness) during infancy or childhood in the absence of any alternative cause (e.g., meningitis). Striatal lesions (e.g., atrophy or persistent abnormal signals of putamen and/or caudate) were determined by cranial magnetic resonance imaging or computer tomography.
For an evaluation of the neurologic outcome, we used a handicap score, referring to the overall estimation given by the caring physicians on the questionnaire (0 point = unaffected; 1 point = mild; 2 points = moderate; 3 points = severe; 4 points = very severe handicap) and a morbidity score summarizing four items (1 point = yes and 0 point = no for each item), i.e., loss of mobility, feeding problems, respiratory problems, and seizures, ranging from 0 points (asymptomatic) to 4 points (multiple morbidity). Both outcome variables were defined in advance of sending out the questionnaires, and each case was evaluated and scored in the study office using a defined scoring algorithm.
Statistical analysis.
Nonparametric tests (χ2 test, Mann-Whitney U test) were used to compare the outcome variables of patients with and without encephalopathic crises, clinically (classic versus insidious onset) and biochemically (high versus low excretors) defined subgroups, and deceased and surviving patients. We used Kaplan-Maier analysis to calculate the survival in symptomatic children up to age 25 y. Recursive partitioning as previously described by Breiman et al. (26) was used to determine predictors for the categorical outcome variables acute encephalopathic crisis, handicap score, and death. The analysis included independent variables, such as diagnostic procedure, dietary treatment, l-carnitine supplementation, riboflavin, gender, macrocephaly, perinatal complications, socioeconomic status of parents, biochemical phenotype, biochemical response to treatment, frequency of biochemical monitoring, and membership in parent groups. We chose this statistical procedure to identify relevant predictors that are useful to make clinical decisions in GCDH deficiency because recursive partitioning can handle numerical data that are highly skewed or multimodal, as well as categorical predictors with either ordinal or nonordinal structure.
RESULTS
Study population.
Overall, 279 patients from 37 nations on four continents [Europe (n = 184), America (n = 64), Asia (n = 26), and Africa (n = 5)] were identified. At time point of diagnosis, 61 of them were neurologically asymptomatic and 218 symptomatic. All symptomatic patients were identified by selective screening and received no specific therapy before diagnosis was made. The median age was 48 mo (range, 6–197 mo) in asymptomatic and 118 mo (range, 5–813 mo) in symptomatic patients. Most patients were born after the description of the two index cases in 1975 (Fig. 1A). Age at diagnosis differed markedly between asymptomatic (median, 0.5 mo; range, 0.25–87 mo) and symptomatic patients (median, 15 mo; range, 1–785 mo), and decreased sharply from the 1970s to the birth cohorts in the 2000s (Fig. 1B). The median follow-up after diagnosis was 75 mo (range, 2–331 mo).

Birth cohorts (A) and age at diagnosis (B) of included study patients. ○, single patient; ▪, mean age at diagnosis of five consecutive birth cohorts.
Genotype, biochemical and clinical phenotype.
Diagnosis was confirmed by mutation (n = 150) and/or enzyme analyses (n = 181), showing a sensitivity of 98.7% (mutation analysis) or 100% (enzyme analysis). Seventy-nine different mutations were identified, with R402W, the most frequent Caucasian mutation, accounting for 16% of mutant alleles. Despite the large number of GCDH gene mutations, homozygosity was found in 79 patients, reflecting a high frequency of known consanguinity (37%). Enzyme analysis revealed complete loss of GCDH activity (i.e., <1% residual activity) in the majority of patients (57.5%). Maximal GCDH residual activity of 30% was found in two symptomatic patients with compound heterozygosity for V400M and R227P.
Analysis of homozygous patients showed that the genotype consistently predicted the residual GCDH enzyme activity. A293T (median residual GCDH activity: 0% of control), A421V (0%), R88C (0%), R402W (0%), and E365 K (1%) accounted for a complete loss of GCDH activity, whereas P248L (2%), V148I (3%), IVS1 + 5g>t (4%), and V400M (10%) predicted minor to moderate residual activity. In analogy, GCDH gene mutations also predicted the amount of organic acids excreted in urine, in particular GA. In compound heterozygous patients, the biochemical phenotype was usually determined by the less severe mutation (data not shown). Thus, the genotype predicted the biochemical phenotype.
Next, we divided all patients into two biochemically defined subgroups called high excretors (≥100 mmol GA/mol creatinine; 67% of study patients) and low excretors (<100 mmol GA/mol creatinine; 33%) according to Baric et al. (14), and determined the clinical presentation and outcome in both subgroups. Statistical analysis (χ2 or Mann Whitney U test) revealed that the frequency of encephalopathic crises (high excretors: 73% of symptomatic patients; low excretors: 85%; χ2 (1) = 2.99; p = 0.084), the age at first crisis (mean age; high excretors: 11.2 mo; low excretors: 10.1 mo; W = 2232, p = 0.593), the handicap score (mean; high excretors: 2.3 pts; low excretors: 2.6 pts; W = 4211; p = 0.194), or the morbidity score (mean; high excretors: 1.7 pts; low excretors: 1.9 pts; W = 4071; p = 0.333) was not different in the biochemical subgroups. These results clearly showed that the biochemical phenotype did not predict the clinical phenotype.
Natural history and clinically defined subgroups. Macrocephaly was found in 74% of patients and was already present at birth in these patients, whereas no other characteristic signs or symptoms were evident before the manifestation of encephalopathic crises (Fig. 2A). Overall, 265 crises were reported in 185 patients. Most patients (70%) suffered only a single crisis, whereas repeat crises were less frequently (two crises: 22.5% of patients; three crises: 6.5%; four crises: 1%; five or more crises: none). The first encephalopathic crisis occurred at a median age of 9 mo (range, 1–37 mo; 95% of first crises by age 2 y). The oldest age at which a repeat crisis was found was 70 mo (Fig. 2B). A comparison of high and low excretors showed no difference in the age distribution of encephalopathic crises (Fig. 2C). Encephalopathic crises were frequently precipitated by nonspecified intercurrent febrile illness, gastrointestinal infections, and pneumonia (Fig. 2D). In most cases, the retrospective data analysis did not allow a precise calculation of the time period between the start of intercurrent febrile illness and the onset of an acute encephalopathic crisis, however, this time interval was usually suggested to be 24–72 h. In general, encephalopathic crises were accompanied by striatal lesions (Table 1) and a movement disorder (Fig. 2E), in particular dystonia (Fig. 2F). Only 6% of affected patients had no significant neurologic abnormalities after such crises.

Acute encephalopathic crises. (A) Neurologic presentation and psychomotor retardation before onset of encephalopathic crises; (B) age at onset of encephalopathic crises [□, all crises (n = 265); ○, 1st crisis (n = 185); ▵, 2nd crisis (n = 60); ▿, 3rd crisis (n = 17], ⋆, 4th crisis (n = 3)]; (C) age at first encephalopathic crisis in high (•) and low excretors (▵); (D) precipitation of encephalopathic crises; (E) frequency of movement disorders (MD) after 1st crisis (filled bars) or 2nd to 4th crises (open bars); and (F) types of movement disorders in patients with (filled bars) or without reported crises (open bars).
In 169 symptomatic patients, encephalopathic crisis was the predominant manifestation (78%, classic onset) and it was likely that, in an additional 5 patients, the onset was neonatal, which is not usually considered in GCDH deficiency. The five patients in our study presented with irritability and transient lactic acidosis during the first week of life. Alternative causes, such as asphyxia, sepsis, and respiratory distress, were excluded. All affected neonates recovered and did not manifest a movement disorder, however, they showed a delayed motor development. In the other symptomatic patients, no acute encephalopathic crises were reported. Thirty-four of them were classified as insidious onset (6,19) and five as late onset (22,23); five patients remained unclassified. Patients with late onset type were not treated specifically before diagnosis, and were diagnosed only after symptoms supervened. Patients with classic onset had striatal lesions more frequently and more severe motor handicap than patients with insidious-onset type (Table 2). By contrast, more than half the patients (n = 61) diagnosed before they had symptoms mostly remained well (n = 38), although some patients showed a transient neurologic symptomatology (n = 7) or had an encephalopathic crisis (n = 16).
Morbidity and mortality.
Forty-nine children died (median age, 79 mo; range, 5–490 mo), and Kaplan-Maier analysis of the survival rate in symptomatically diagnosed patients (n = 218) showed that an estimated 50% of cases died by age of 25 y (Fig. 3A). Although aspiration pneumonia was the most frequent cause of death, it remained unclear in the majority of patients (Fig. 3B). All deceased children had a history of encephalopathic crises, except for one who had a fulminant sepsis. Those who died had more severe striatal lesions (Table 3) and a more severe disability (Fig. 3C), and received dietary treatment and l-carnitine supplementation less frequently than surviving children (Table 3). Interestingly, the age at first crisis influenced the outcome (21). Regression analysis revealed that the severity of neurologic sequelae, which was evaluated by handicap score (estimate: −0.034 pts/mo; p < 0.01) and morbidity score (estimate: −0.035 pts/mo; p < 0.01), decreased with increasing age at first crisis. In addition, the age at first crisis was lower in deceased than in surviving children (Table 3).

Death and multi-morbidity. (A) Kaplan-Maier analysis of symptomatic patients (n = 218) with GCDH deficiency; (B) cause of death; and (C) degree of morbidity in deceased (filled bars, n = 49) and surviving patients (open bars, n = 230).
Age at diagnosis and mode of therapy predict the outcome.
Differences in the age at diagnosis and in treatment protocols among metabolic centers and countries have varied the outcome worldwide (Fig. 4). A comparison of patients from countries using protein (e.g., United Kingdom, Canada) or lysine (e.g., Germany, Spain) restriction (Fig. 4) showed that the frequency of encephalopathic crises and the degree of disability particularly varied in presymptomatically diagnosed patients (Fig. 4, A and C), whereas it was similar in symptomatic patients (Fig. 4, B and D). To unravel which single variables have a major influence on the outcome, we used recursive partitioning. With acute encephalopathic crisis (Fig. 5A), degree of disability (handicap score; Fig. 5B) or death (Fig. 5C) as dependent variables, this analysis showed that timely diagnosis in asymptomatic children, l-carnitine supplementation and lysine-restricted diet predicted a positive outcome. In contrast, the analysis showed no beneficial effect for protein-restricted diet and riboflavin although widely used. In symptomatic patients, l-carnitine supplementation was found to be beneficial (Fig. 5, B and C), whereas dietary treatment was less effective, if at all. Many other variables, including gender, macrocephaly, perinatal complications, socioeconomic status of parents, biochemical phenotype, biochemical response to treatment, frequency of monitoring, and membership in parent groups, did not relevantly influence the outcome. The benefit of emergency treatment could not be assessed because delay in starting the emergency treatment could not be determined precisely in the majority of patients, yet it appears to be crucial in determining the outcome.

Worldwide variability of neurologic outcome. Frequency of encephalopathic crises in presymptomatically diagnosed (A) or symptomatic patients (B). Degree of disability (handicap score) in presymptomatically diagnosed (C) and symptomatic patients (D) from different countries. Results from all study patients (asymptomatic/symptomatic: 61/218) and countries with the largest number of study patients, i.e., Germany (24/38), Spain (5/34), United Kingdom (6/33), and Canada (9/20), were demonstrated, and were compared with results from three recent studies (3,20,25). *Ireland (asymptomatic/symptomatic patients: 10/11); **the Amish Community (20/17); and ***Scandinavia (3/25). Note that treatment protocols differed between countries and metabolic centers: i) lysine-restricted diet plus l-carnitine supplementation was common in Ireland, Germany, and Spain); ii) protein-restricted diet plus l-carnitine and riboflavin were used in the Amish population, United Kingdom, and Canada); and iii) a combination of these protocols was used for Scandinavian patients.

Recursive partitioning. Recursive partitioning of the dependent variables “encephalopathic crisis” (A), “degree of disability” (as determined by the handicap score; B), and “death” (C) demonstrated that early diagnosis of neurologically asymptomatic infants (“asymptomatic”) and subsequent prevention of encephalopathic crises (“enceph. crisis”) by treatment with lysine-restricted diet (“lysine restriction”) and carnitine supplementation (“carnitine”) were the strongest predictors of a positive outcome in GCDH deficiency. The dark gray areas of each pie chart represent the percentage of patients in that subgroup who have suffered an encephalopathic crises (A) or deceased (C), or represent the degree of disability (B; white pie = no handicap [minimum]; dark gray pie = severe handicap [maximum]). The overall area of the pie chart indicates the size of each subgroup relative to the total population. To minimize the risk of overfitting information, the trees were cut after the second node.
DISCUSSION
In the present study, we described the variability of the natural history and outcome in GCDH deficiency. The key results of the present study were that 1) the outcome was dependent on the age at onset, and severity of acute encephalopathic crises during a vulnerable period of brain development; 2) the genotype predicted the biochemical phenotype but not the clinical phenotype; and 3) lysine restriction and l-carnitine supplementation in those diagnosed presymptomatically was neuroprotective.
A single crisis during infancy or childhood determines the outcome.
We confirmed previous studies demonstrating that the encephalopathic crises critically determine the extent of striatal damage and therefore the prognosis as this is responsible for the movement disorder, secondary complications, and reduced life expectancy. Encephalopathic crises occurred during a narrow time span, indicating an age-dependent vulnerability (21,27). Most patients had only a single crisis. Furthermore, the age at first crisis reciprocally influenced the morbidity and mortality of affected children confirming a previous study (21). We showed that simple functional items, such as mobility and feeding skills, are important prognostic factors. The value of these items giving an indication of the likely survival in children has previously been described for infantile cerebral palsy and will be helpful for counseling families (28). Although neurologic disease usually manifests acutely, we found evidence for chronic neurologic changes in two clinically defined subgroups, i.e., insidious- and late-onset type. In both subgroups, patients developed (progressive) neurologic disease in the absence of any reported acute crisis (6,19,23). Despite obvious clinical and neuroradiological differences, we cannot exclude the possibility that these subgroups represent no more than a variation of the natural disease course or the sequelae of a mild encephalopathic crisis. Furthermore, we found evidence for a neonatal onset of symptoms in a few patients but neurologic sequelae of these neonatal episodes were much milder than those observed following acute crises in infants or children.
Carnitine supplementation and lysine restriction are neuroprotective.
The organic acids that accumulate are potential neuronal and mitochondrial toxins (29,30). Post mortem studies (11–13) and one brain biopsy (23) demonstrated that GA and 3-OH-GA concentrations are 10- to 1000-fold higher in brain tissue than in plasma and are found in a similar concentration range in high and low excretors. These results suggest there is de novo synthesis of GA and 3-OH-GA in the brain and because transport of dicarboxylic acids across the blood brain barrier is strongly limited these acids are trapped. This notion is supported by the observation that lysine-restricted and tryptophan-reduced diet that reduces the flux of precursor amino acids to the brain resulted in near normalization of cerebral GA and 3-OH-GA concentrations in two patients (31,32). The implications of these studies are that 1) the cerebral concentrations of GA and 3-OH-GA are similar in high and low excretors and account for a similar risk for neurologic disease in both subgroups and that 2) treatment should be aimed at reduced production or increased removal of potentially toxic organic acids and CoA esters. We found that the frequency of encephalopathic crises was independent from the biochemical phenotype, confirming previous studies (6,8). In addition, the age at first crisis and severity of the clinical presentation was also not significantly influenced by the biochemical phenotype. In contrast to this, the outcome was influenced by the mode of treatment. We hypothesized that lysine intake and l-carnitine supplementation were the most relevant key variables, inasmuch as lysine is the quantitatively most important precursor amino acid for GA and 3-OH-GA, and l-carnitine supplementation increases the physiologic detoxification of acyl-CoA esters and prevents secondary l-carnitine depletion (19,33,34). In fact, we demonstrated that dietary treatment and l-carnitine supplementation were neuroprotective in presymptomatically diagnosed patients. In particular, it is the lysine-restricted diet using natural protein with a low lysine content and appropriate amino acid supplements (lysine-free, tryptophan-reduced) that has been shown to be effective. Although this effect might be best explained by effective restriction of cerebral lysine uptake, we cannot fully exclude a confounder bias, such as differences in the compliance of patients and parents, or in the personal experience of metabolic specialists. However, many other variables that might influence the outcome have been considered in the statistical analysis, but none of those had a significant influence on the outcome variables. This analysis also revealed no beneficial effect for riboflavin, although it is widely used. Of course, these results require confirmation from prospective studies.
In contrast to presymptomatically diagnosed patients, only l-carnitine supplementation was protective in symptomatic patients, whereas the efficacy of dietary treatment remained unclear. Furthermore, it remains to be elucidated whether treatment efficacy varies among specific subgroups of patients, such as insidious- or late-onset type. We could not estimate the neuroprotective value of emergency treatment, which is considered an important treatment strategy in GCDH deficiency (3,19,25) because the exact timing of the start of treatment could not be ascertained in the retrospective analysis of the episodes of acute illness.
Timely diagnosis is crucial for a good outcome.
Dietary treatment and l-carnitine supplementation prevented complications if started before encephalopathic crises occurred, whereas dietary treatment had only a limited effect in patients who already had symptoms. Therefore, early diagnosis is a conditio sine qua non to prevent neurologic disease in GCDH deficiency. Because the clinical presentation is not specific before the onset of encephalopathic crises, neonatal screening for GCDH deficiency is the only reliable method for the detection of presymptomatic patients. In this light, inclusion of GCDH deficiency into MS/MS-based neonatal screening programs is crucial to improve the outcome. Alternative strategies, such as DNA-based screening, should be considered for the screening for known low excretor cohorts with private mutations (4).
Management of patients with orphan diseases.
At present, the evidence base is usually low for the management of patients with orphan diseases, notably inherited metabolic diseases. Treatment is frequently based on experience with very small numbers of patients. This results in poor clinical understanding and highly variable experience in diagnosis, treatment, and counseling of rare and often very serious disorders. Our study has implications beyond one rare disease as it demonstrates the feasibility and power of a cross-sectional international study to delineate and answer fundamental questions in orphan diseases and, finally, to assist the development of guidelines. It is undisputed that randomized controlled studies would be preferable, however, in diseases like GCDH deficiency they would be extremely difficult, very time-consuming, and expensive because of the rarity of the condition and the large variation of the natural disease course. In conclusion, this study proved GCDH deficiency to be a treatable disorder and a good candidate for neonatal screening programs.
Abbreviations
- GA:
-
glutaric acid
- GCDH:
-
glutaryl-CoA dehydrogenase (EC 1.3.99.7)
- MS/MS:
-
tandem mass spectrometry
- 3-OH-GA:
-
3-hydroxyglutaric acid
References
Goodman SI, Markey SP, Moe PG, Miles BS, Teng CC 1975 Glutaric aciduria: a “new” disorder of amino acid metabolism. Biochem Med 12: 12–21
Lindner M, Kölker S, Schulze A, Christensen E, Greenberg CR, Hoffmann GF 2004 Neonatal screening for glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27: 851–859
Strauss KA, Puffenberger EG, Robinson DL, Morton DH 2003 Type I glutaric aciduria, part 1: natural history of 77 patients. Am J Med Gen Semin Med Genet 121: 38–52
Greenberg CR, Prasad AN, Dilling LA, Thompson JR, Haworth JC, Martin B, Wood-Steinman P, Seargeant LE, Seifert B, Booth FA, Prasad C 2002 Outcome of the first 3- years of a DNA-based neonatal screening program for glutaric acidemia type 1 in Manitoba and northwestern Ontario, Canada. Mol Gen Metab 75: 70–78
Goodman SI, Stein DE, Schlesinger S, Christensen E, Schwartz M, Greenberg CR, Elpeleg ON 1998 Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): review and report of thirty novel mutations. Hum Mutat 12: 141–144
Busquets C, Merinero B, Christensen E, Gelpi JL, Campistol J, Pineda M, Fernandez-Alvarez E, Prats JM, Sans A, Arteaga R, Marti M, Campos J, Martinez-Pardo M, Martinez-Bermejo A, Ruiz-Falco ML, Vaquerizo J, Orozco M, Ugarte M, Coll MJ, Ribes A 2000 Glutaryl-CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically, and biochemically distinct. Pediatr Res 48: 315–322
Zschocke J, Quak E, Guldberg P, Hoffmann GF 2000 Mutation analysis in glutaric aciduria type I. J Med Genet 37: 177–181
Christensen E, Ribes A, Merinero B, Zschocke J 2004 Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27: 861–868
Christensen E 1983 Improved assay of glutaryl-CoA dehydrogenase in cultured cells and liver: application to glutaric aciduria type I. Clin Chim Acta 129: 91–97
Fu Z, Wang M, Paschke R, Rao KS, Frerman FE, Kim JJ 2004 Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: structural bases of dehydrogenation and decarboxylation reactions. Biochemistry 43: 9674–9784
Goodman SI, Norenberg MD, Shikes RH, Breslich DJ, Moe PG 1977 Glutaric aciduria: biochemical and morphological considerations. J Pediatr 90: 746–750
Leibel RL, Shih VE, Goodman SI, Bauman ML, McCabe ER, Zwerdling RG, Bergman I, Costello C 1980 Glutaric acidemia: a metabolic disorder causing progressive choreoathethosis. Neurology 30: 1163–1168
Funk CB, Prasad AN, Frosk P, Sauer S, Kölker S, Greenberg CR, Del Bigio MR 2005 Neuropathological, biochemical and molecular findings in a glutaric acidemia type 1 cohort. Brain 128: 711–722
Baric I, Wagner L, Feyh P, Liesert M, Buckel W, Hoffmann GF 1999 Sensitivity and specificity of free and total glutaric and 3-hydroxyglutaric acid measurements by stable- isotope dilution assays for the diagnosis of glutaric aciduria type I. J Inherit Metab Dis 22: 867–881
Schulze A, Lindner M, Kohlmüller D, Olgemöller K, Mayatepek E, Hoffmann GF 2003 Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics 111: 1399–1406
Chace DH, Kalas TA, Naylor EW 2003 Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem 49: 1797–1817
Wilcken B, Wiley V, Hammond J, Carpenter K 2003 Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N Engl J Med 348: 2304–2312
Hoffmann GF, Trefz FK, Barth PG, Böhles HJ, Biggemann B, Bremer HJ, Christensen E, Frosch M, Hanefeld F, Hunneman DH, Jacobi H, Kurlemann G, Lawrenz-Wolf B, Rating D, Roe CR, Schutgens RB, Ullrich K, Weisser J, Wendel U, Lehnert W 1991 Glutaryl-coenzyme A dehydrogenase deficiency: a distinct encephalopathy. Pediatrics 88: 1194–1203
Hoffmann GF, Athanassopoulos S, Burlina AB, Duran M, de Klerk JB, Lehnert W, Leonard JV, Monavari AA, Müller E, Muntau AC, Naughten ER, Plecko-Starting B, Superti-Furga A, Zschocke J, Christensen E 1996 Clinical course, early diagnosis, treatment, and prevention of disease in glutaryl-CoA dehydrogenase deficiency. Neuropediatrics 27: 115–123
Kyllerman M, Skjeldal O, Christensen E, Hagberg G, Holme E, Lonnquist T, Skov L, Rotwelt T, von Dobeln U 2004 Long-term follow-up, neurological outcome and survival rate in 28 Nordic patients with glutaric aciduria type 1. Eur J Paediatr Neurol 8: 121–129
Bjugstad KB, Goodman SI, Freed CR 2000 Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type I. J Pediatr 137: 681–686
Bähr O, Mader I, Zschocke J, Dichgans J, Schulz JB 2002 Adult onset glutaric aciduria type I presenting with leukoencephalopathy. Neurology 59: 1802–1804
Külkens S, Harting I, Sauer S, Zschocke J, Hoffmann GF, Gruber S, Bodamer OA, Kölker S 2005 Late-onset neurologic disease in glutaryl-CoA dehydrogenase deficiency. Neurology 64: 2142–2144
Kölker S, Greenberg CR, Lindner M, Müller E, Naughten ER, Hoffmann GF 2004 Emergency treatment in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27: 893–902
Naughten ER, Mayne PD, Monavari AA, Goodman SI, Sulaiman G, Croke DT 2004 Glutaric aciduria type I, outcome in the Republic of Ireland. J Inherit Metab Dis 27: 917–920
Breiman L, Friedman JH, Olshen RA, Stone CJ 1984 Classification and Regression Trees. Chapman & Hall (Wadsworth, Inc.), New York,
Strauss KA, Morton DH 2003 Type 1 glutaric aciduria, part 2: a model of acute striatal necrosis. Am J Med Genet C Semin Med Genet 121: 53–70
Strauss DJ, Shavelle RM, Anderson TW 1998 Life expectancy of children with cerebral palsy. Pediatr Neurol 18: 143–149
Kölker S, Koeller DM, Okun JG, Hoffmann GF 2004 Pathomechanisms of neurodegeneration in glutaryl-CoA dehydrogenase deficiency. Ann Neurol 55: 7–12
Sauer SW, Okun JG, Schwab MA, Crnic LR, Hoffmann GF, Goodman SI, Koeller DM, Kölker S 2005 Bioenergetics in glutaryl-coenzyme A dehydrogenase deficiency: a role for glutaryl-coenzyme A. J Biol Chem 280: 21830–21836
Bennett MJ, Marlow N, Pollitt RJ, Wales JK 1986 Glutaric aciduria type 1: biochemical investigations and postmortem findings. Eur J Pediatr 145: 403–405
Kölker S, Hoffmann GF, Schor DS, Feyh P, Wagner L, Jeffrey I, Pourfarzam M, Okun JG, Zschocke J, Baric I, Bain MD, Jakobs C, Chalmers RA 2003 Glutaryl-CoA dehydrogenase deficiency: region-specific analysis of organic acids and acylcarnitines in post mortem brain predicts vulnerability of the putamen. Neuropediatrics 34: 253–260
Müller E, Kölker S 2004 Reduction of lysine intake while avoiding malnutrition—major goals and major problems in dietary treatment of glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27: 903–910
Mühlhausen C, Hoffmann GF, Strauss KA, Kölker S, Okun JG, Greenberg CR, Naughten ER, Ullrich K 2004 Maintenance treatment of glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27: 885–892
Acknowledgements
The authors thank the patients and their families for their participation and trust. We also thank B. Assmann, M.D. Bain, I. Baric, M.R. Baumgartner, G. Besley, O.A. Bodamer, F. Booth, R. Brackmann, R. Casey, A. Chan, M.L. Couce, A.P. Das, A.G.F. Davidson, A. Garcia-Cazorla, M. del Bigio, L. Dilling, U. John-Grafe, M. Leichsenring, Y.P. Lillquist, M. Martinez-Pardo, E. Mayatepek, A. Morris, C. Mühlhausen, H. Muscheid, A.N. Prasad, C. Prasad, M.L. Ruiz Falco, J. Schlage, L. Seargeant, D. Skladal, R. Surtees, M. Tomas, K. Ullrich, B. van Maldegem, and U. Wendel for reports on patients and fruitful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by a grant from the German Federal Ministry of Education and Sciences (BMBF # 01GM0305) and a grant from the “Kindness of Kids” Foundation, Munich, Germany (both to SK and GFH); by a grant from REDEMETH and INERGEN (Fondo de Investiagacion Sanitaria, Spain) to AR; and by financial support of travel expenses from Milupa Metabolics, Friedrichsdorf, Germany.
Rights and permissions
About this article
Cite this article
Kölker, S., Garbade, S., Greenberg, C. et al. Natural History, Outcome, and Treatment Efficacy in Children and Adults with Glutaryl-CoA Dehydrogenase Deficiency. Pediatr Res 59, 840–847 (2006). https://doi.org/10.1203/01.pdr.0000219387.79887.86
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.pdr.0000219387.79887.86
This article is cited by
-
Modeling Glutaric Aciduria Type I in human neuroblastoma cells recapitulates neuronal damage that can be rescued by gene replacement
Gene Therapy (2024)
-
Biochemical and molecular features of chinese patients with glutaric acidemia type 1 from Fujian Province, southeastern China
Orphanet Journal of Rare Diseases (2023)
-
Evaluierung und Optimierung des Neugeborenenscreenings mittels strukturierter Langzeitbeobachtung – am Beispiel der angeborenen Stoffwechselerkrankungen
Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz (2023)
-
Noninvasive fetal genotyping of single nucleotide variants and linkage analysis for prenatal diagnosis of monogenic disorders
Human Genomics (2022)
-
Biochemical and molecular features of Chinese patients with glutaric acidemia type 1 detected through newborn screening
Orphanet Journal of Rare Diseases (2021)
