
Abstract
Leukocytes are implicated in the pathogenesis of diarrhea-associated hemolytic uremic syndrome (D+ HUS). We hypothesized that increased circulating levels of granulocyte colony-stimulating factor (G-CSF), and the chemokines epithelial cell–derived neutrophil-activating protein-78 (ENA-78), growth related oncogen-α (GRO-α), macrophage inflammatory protein-1β (MIP-1β), and monocyte chemotactic protein-1 (MCP-1) are related to the severity of illness in Escherichia coli O157:H7 infections. We compared the circulating concentrations of these mediators in the course of E. coli O157:H7 enteritis, hemorrhagic colitis, and HUS. Our data show that, on admission, children with HUS presented 10-fold abnormally increased levels of G-CSF (p < 0.007), 3-fold increased MIP-1β concentrations (p < 0.001), and 2-fold lower values of ENA-78 (p < 0.0001). One week later, a further 4-fold decrease in ENA-78 concentration was noted (p < 0.0001) whereas MIP-1β levels returned to normal. HUS patients requiring peritoneal dialysis showed 6-fold increased G-CSF (p < 0.001) and 5-fold decreased ENA-78 (p < 0.001) levels. On admission, children with uncomplicated O157:H7 hemorrhagic colitis (HC) presented 3-fold abnormally increased concentrations of G-CSF (p < 0.001) and MIP-1β (p < 0.0001). Those with O157:H7 enteritis but no bloody stools showed higher rates of abnormal GRO-α, MIP-1β, and MCP-1 measurements than children with O157:H7 HC or HUS: GRO-α (50% enteritis, 36% HC, 17% HUS;p < 0.06), MIP-1β (40% enteritis, 22% HC, 11% HUS;p < 0.02), MCP-1 (77% enteritis, 20% HC, 18% HUS;p < 0.0001). The data indicates that GRO-α, MIP-1β, and MCP-1 are produced during E. coli O157:H7 enteritis, whether or not HC or HUS develops. Our data suggest that children with O157:H7 associated HUS may present abnormally increased circulating levels of G-CSF and decreased ENA-78 concentrations. The mechanisms responsible for leukocytes recruitment in O157:H7 infections are unclear and await further studies.
Similar content being viewed by others

Main
HUS is characterized by nonimmune hemolytic anemia, thrombocytopenia, and acute renal failure (1). D+ HUS may occur after a prodrome of enteritis due to Shiga toxin–producing Escherichia coli(2).
Several observations indicate that leukocytes are involved in the pathogenesis of D+ HUS, although their exact role remains unclear (3). Abnormally increased numbers of macrophages (4) and neutrophils (4, 5) have been found infiltrating the glomeruli of children with D+ HUS. Circulating leukocytosis is an independent risk marker for developing D+ HUS (6–8). Increased neutrophil (9, 10) and macrophage (10) counts are also associated with the severity of renal failure. HUS patients have activated circulating polymorphonuclear neutrophils (11–13), with increased circulating levels of elastases (14, 15) and alpha-1-antitrypsin (15). Enhanced leukocyte adhesion has also been demonstrated (16). Shiga toxin binds specifically to a distinct glycosphingolipid receptor on polymorphonuclear leukocytes (17). In the presence of human glomerular microvascular endothelial cells, Shiga toxin is transferred from neutrophils to renal endothelial cells, thereby suggesting that circulating neutrophils transport Shiga toxin from the gut to the kidney (17).
Chemokines are small (8–13 kD) cytokine-like molecules with proinflammatory activity, inducing unidirectional leukocyte migration toward a localized area of inflammation (18). Chemokines have one or two cysteine residues in their amino terminal domain. They are classified according to the presence or the absence of an amino acid between the first two cysteines: C-X-C or C-C (18). Specific neutrophil chemotactic activity is produced by C-X-C chemokines with ELR+. These include IL-8, ENA-78, and GRO-α. Chemokines from the C-C family, such as MCP-1 and MIP-1β, exert their activity on macrophages/monocytes, basophils, eosinophils, T cells, and natural killer cells, but are not chemotactic for polymorphonuclear neutrophils. Increased circulating levels of IL-8 have been reported in children with D+ HUS (4, 14, 19–21). Elevated MCP-1 (4) and G-CSF have also been observed (21, 22). Except for IL-8 and MCP-1, there is no other data on circulating chemokines in children with D+ HUS.
We hypothesized that increased circulating levels of G-CSF, ENA-78, GRO-α, MIP-1β, and MCP-1 are related to the severity of illness in E. coli O157:H7 infections. First, we analyzed variations over time in chemokine concentration among children with uncomplicated O157:H7 HC by serial blood samples after admission. Secondly, we determined whether abnormal chemokine levels were also present in children with uncomplicated O157:H7 enteritis who did not have bloody stools. We therefore compared the circulating chemokine levels among the following independent groups: 1) normal controls, 2) disease controls with chronic renal failure, 3) children with uncomplicated HC due to E. coli O157:H7, and 4) children with O157:H7-associated HUS. Finally, we determined whether the circulating levels are associated with the severity of HUS, as measured by the need for peritoneal dialysis.
METHODS
Clinical data.
Children aged less than 18 y old who presented with HUS at Sainte-Justine Hospital or at the Children's Hospital of Eastern Ontario, between April 1, 1996, and September 1, 2000, were eligible for the study. Enteritis was defined as the acute onset of diarrhea (>1 d) with or without abdominal pain. HC was defined as the acute onset of diarrhea with grossly bloody stools noted before medical consultation. HUS was defined (23) as a prodrome of enteritis or HC with the following ensuing criteria: a) thrombocytopenia (<150,000 U/L), b) microangiopathic hemolytic anemia (Hb below the third percentile for age and sex with schistocytes on blood smear) (24), and c) acute renal failure (serum creatinine >62 μM if aged <5 y old; >88 μM if between 6 and 12 years old; >88 μM and >102 μM for females and males aged >12 y old, respectively). HUS was partial if only one or two of the above criteria were met and not attributable only to dehydration on serial laboratory analysis. The normal control group included 15 patients matched for age and sex who had blood sample collection before undergoing an elective surgery for inguinal hernia or strabismus. The disease control group was composed of nine children on chronic ambulatory peritoneal dialysis with chronic renal failure due to reflux nephropathy (n = 4), renal dysplasia (n = 2), glomerulonephritis (n = 2), and nephrosis (n = 1). The median serum creatinine was 390 μmol/L (range 118–1000 μmol/L). They presented no signs of upper respiratory tract infections, and had normal temperature as well as negative urine and dialysate cultures before obtaining blood samples.
Age, sex, results of stool cultures, and the date of onset of enteritis and HUS were noted. In the latter group, the need for peritoneal dialysis was determined by the attending pediatric nephrologist, and all these children presented with oligoanuria (25). Among patients presenting with HUS, blood samples were obtained on admission, before starting peritoneal dialysis if needed. Moreover, we aimed to collect a second blood sample 1 wk later. Among children presenting with uncomplicated O157:H7 HC, serial blood samples were collected on admission, and then on d 1, 2, and 8 after enrollment. Serial samples were obtained in 46% (27 of 59) of patients in the O157:H7 HC group.
Laboratory data.
Sorbitol-negative colonies grown on MacConkey-sorbitol agar were subcultured onto blood agar and screened for serotype O157 by slide agglutination (Difco, Detroit, MI, U.S.A.). Colonies agglutinating with the antiserum were identified as E. coli by standard biochemical reactions. Identification of pathogen was available within 24–48 h of sample collection.
Blood samples were collected in an EDTA-containing tube and then centrifuged for 10 min at 4°C and 3000 ×g. Specimens were stored at −80°C until assayed. Measurements of G-CSF, ENA-78, GRO-α, MIP-1β, and MCP-1 were performed in duplicate on plasma by sandwich ELISA (R & D Systems, Minneapolis, MN, U.S.A.). Samples were diluted 1:2; G-CSF, 50 μL; ENA-78, 25 μL; GRO-α, 100 μL; MIP-1β, 75 μL; and MCP-1, 100 μL. Standard curves were established for each microtiter plate and measurements were then performed as previously described (19, 20, 26). Plates were developed and the OD of the reaction mix was quantified with an automated ELISA reader. We calculated the relative concentration of G-CSF and chemokines by comparing the mean OD obtained from duplicate wells to a semilog standard curve. The sensitivities were as follows: G-CSF >20 pg/mL, ENA-78 >15 pg/mL, GRO-α >10 pg/mL, MIP-1β >11 pg/mL, and MCP-1 >5 pg/mL.
Ethics.
Written informed consent was obtained from the parents of all children. The study was approved by the Ethics Committee of Sainte-Justine Hospital and of the Children's Hospital of Eastern Ontario.
Statistics.
Descriptive statistics are presented as mean ± SD for data with a normal distribution; median and range were used otherwise. The t test was used to compare continuous data with a normal distribution and the Mann-Whitney U test if the distribution was abnormal. An ANOVA for repeated measurements of mediator concentrations was performed in the subgroup of children with uncomplicated O157:H7 HC who had serial blood samples collected. The percentage of abnormal measurements were compared using a 2 × 3 contingency table in the following independent groups: children with O157:H7 enteritis without bloody stools, patients with O157:H7 HC, and those with HUS. The Kruskal-Wallis test was used to compare age and mediator concentrations between the following independent groups: 1) normal controls, 2) disease controls with chronic renal failure, 3) children with uncomplicated HC due to E. coli O157:H7, and 4) children with O157:H7-associated HUS. Orthogonal comparisons were then performed using the Dunn test. All statistical tests were two-tailed and p < 0.05 was considered significant; statistical significance for orthogonal comparisons was established at p < 0.0083. To control for time, children in groups 3 and 4 were also compared using the Mann-Whitney U test after matching for the time interval between the onset of enteric symptoms and the day of blood sample collection. The Kruskal-Wallis test was finally used to compare normal and disease control groups, and HUS patients stratified according to requirement for peritoneal dialysis. Linear regression analysis between mediator concentrations and serum creatinine, Hb level, platelet counts, and white blood cell, neutrophil, and monocyte counts were also done.
RESULTS
We performed 805 measurements among 109 children. Uncomplicated enteritis caused by E. coli O157:H7 occurred in 59 children, with 52 (88%) having bloody stools, whereas 7 (12%) had no blood in their stools. There were 26 patients with either full-blown (n = 23) or partial HUS (n = 3). Stool cultures showed evidence of E. coli O157:H7 in 77% (n = 20) of the patients with HUS. All patients who developed HUS had a diarrheal prodrome and 85% (22 of 26) had bloody stools.
Uncomplicated O157:H7 HC.
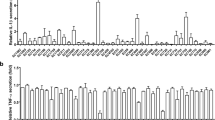
Figure 1 shows measurements of G-CSF, ENA-78, GRO-α, MIP-1β, and MCP-1 among the subgroup of 27 children with uncomplicated O157:H7 HC who had serial measurements. Levels of G-CSF (40 ± 31 pg/mL, p < 0.001) and MIP-1β (63 ± 29 pg/mL, p < 0.0001) were increased 3-fold on admission. We did not find any significant variations over time in mediator levels, although a trend toward a decreasing G-CSF concentrations was observed during the first week after admission (p < 0.1).

Circulating levels of G-CSF, ELR+ C-X-C chemokines (ENA-78 and GRO-α), and C-C chemokines (MIP-1β and MCP-1) in 27 children with uncomplicated HC due to E. coli O157:H7. This subgroup of patients had serial blood samples collection on admission and d 1, 2, and 8. Mean values are presented as (—). Compared with normal controls (NC), children with O157:H7 HC presented abnormally increased levels of G-CSF (*p < 0.001) and MIP-1β (†p < 0.0001). Variations over time in mediator levels were not significant, although a trend was noted for normalization of G-CSF concentrations on d 8 (p < 0.1).
Uncomplicated O157:H7 enteritis.
The mean circulating mediator levels were comparable among patients with uncomplicated O157:H7 enteritis whether or not they developed HC (data not shown). These subgroups had comparable delays between the onset of enteritis and blood sample collection (2.3 ± 2.0 versus 2.9 ± 2.0 d, respectively; NS). However, a trend for a lower leukocyte count on admission was noted in children with O157:H7 enteritis compared with those with HC (8.7 ± 2.7 versus 11.7 ± 4.3 × 109/L;p < 0.054). Patients with O157:H7 enteritis without bloody stools presented higher rates of abnormal measurements (i.e. values higher than the upper limit in normal controls) of GRO-α, MIP-1β, and MCP-1 than patients with O157:H7 HC or HUS: GRO-α (42% enteritis, 36% HC, 17% HUS;p < 0.05); MIP-1β (40% enteritis, 22% HC, 11% HUS;p < 0.02); MCP-1 (77% enteritis, 20% HC, 18% HUS;p < 0.0001). The highest percentages of abnormal G-CSF and ENA-78 measurements were found in patients with HUS: G-CSF (20% enteritis, 50% HC, 63% HUS;p < 0.009); ENA-78 (20% enteritis, 15% HC, 74% HUS;p < 0.0001).
O157:H7 HC and HUS.
The sex distribution was similar between: 1) normal controls, 2) disease controls, 3) children with O157:H7 HC, and 4) those with HUS. Disease controls with chronic renal failure (group 2) were older than those in any other groups (46 ± 12 mo, 153 ± 20 mo, 61 ± 45 mo, and 53 ± 8 mo, respectively;p < 0.0001). Age was comparable in patients with O157:H7 HC and those with HUS. The time intervals between the onset of enteritis and blood sample collection in children with O157:H7 HC and HUS were as follows: first sample 2.8 ± 1.9 versus 5.5 ± 3.1 d, p < 0.0001; second sample 10.9 ± 2.1 versus 11.8 ± 3.9 d, NS. The difference reflects the referral of HUS patients to a tertiary care center. The white blood cell counts on admission as well as on d 8 were lower in the HC than the HUS group: first sample 11.4 ± 4.1 versus 17.7 ± 7.2 × 109/L, p < 0.0001; and second sample 9.6 ± 3.8 versus 15.2 ± 7.1 × 109/L, p < 0.0002. Children with HUS showed higher neutrophil counts than those with O157:H7 HC on admission and on d 8: first sample 12.0 ± 7.2 versus 6.8 ± 3.6 × 109/L, p < 0.0003; and second sample 8.6 ± 6.1 versus 4.1 ± 2.6 × 109/L, p < 0.0002. They also presented increased monocyte counts: first sample 1.4 ± 0.7 versus 0.7 ± 0.4 × 109/L, p < 0.001; and second sample 1.0 ± 0.4 versus 0.5 ± 0.2 × 109/L, p < 0.0001. Leukocyte counts were all more severely abnormal on admission.
Figure 2 shows the circulating levels of G-CSF, ENA-78, GRO-α, MIP-1β, and MCP-1 among 1) normal controls, 2) disease controls with chronic renal failure, 3) children with uncomplicated O157:H7 HC, and 4) patients with HUS. Circulating levels in normal controls were as it follows: G-CSF, 14 ± 11 pg/mL; ENA-78, 1030 ± 711 pg/mL; GRO-α, 53 ± 19 pg/mL; MIP-1β 23 ± 25 pg/mL; MCP-1, 85 ± 34 pg/mL. On admission, patients with HUS had a 10-fold increase in levels of G-CSF (141 ± 201 pg/mL, p < 0.0003), a 3-fold increase of MIP-1β (60 ± 31 pg/mL, p < 0.001), and 2-fold lower levels of ENA-78 than normal controls (468 ± 662 pg/mL, p < 0.001). HUS patients had significantly higher concentrations of G-CSF (p < 0.007) and lower levels of ENA-78 (p < 0.0001) than any other group. MIP-1β levels were similarly increased 3-fold in O157:H7 HC (74 ± 35 pg/mL, p < 0.0001) and HUS (60 ± 31 pg/mL, p < 0.001), compared with normal controls. The mean concentrations of GRO-α were similar in all groups. MCP-1 levels were elevated (278 ± 103 pg/mL, p < 0.0002) only in the diseased control group with chronic renal failure. One week after admission, G-CSF levels remained significantly increased in HUS compared with children with uncomplicated O157:H7 HC (p < 0.02). A further 4-fold decrease in ENA-78 concentrations was noted among HUS subjects (284 ± 363 pg/mL, p < 0.0001). Moreover, higher G-CSF (52 ± 37 versus 39 ± 34 pg/mL;p < 0.02) and lower ENA-78 concentrations (658 ± 768 versus 1198 ± 699 pg/mL;p < 0.007) were also noted among 17 undialyzed patients with HUS than among 53 children with uncomplicated O157:H7 HC. Other mediator concentrations were comparable. Finally, to control for time, 20 patients with HUS and 20 children with uncomplicated O157:H7 were matched for the time interval between the onset of enteric symptoms and the day of blood sample collection (4.1 ± 5.5 d). Patients with HUS presented higher G-CSF levels than those with uncomplicated O157:H7 HC (159 ± 216 versus 35 ± 23 pg/mL, respectively;p < 0.005), as well as lower ENA-78 concentrations (618 ± 681 versus 1144 ± 640 pg/mL, respectively;p < 0.008). Other mediator concentrations were comparable between these two groups.

Circulating levels of G-CSF, ELR+ C-X-C chemokines (ENA-78 and GRO-α), and C-C chemokines (MIP-1β and MCP-1) in 15 normal controls (NC), 9 disease control patients on chronic ambulatory peritoneal dialysis with chronic renal failure (DC), 52 children with uncomplicated HC due to E. coli O157:H7 (HC), and 26 children with O157:H7 HUS. Mean values are presented as (—). The first samples obtained on admission are shown on the left and the last samples are presented on the right. Different concentrations were noted between groups on admission for each mediator, except for GRO-α. MCP-1 levels were only increased in the disease control group with chronic renal failure (***p < 0.0002). The concentrations of G-CSF on admission were increased 10-fold in HUS patients (*p < 0.007, compared with any other group) and remained higher on the last blood sample compared with children with O157:H7 HC (†p < 0.02). Two-fold decreased levels of ENA-78 were also noted among HUS patients (**p < 0.0001, compared with any other group), and a further 4-fold decrease was noted thereafter (††p < 0.0001). Three-fold increased levels of MIP-1β were found in children with O157:H7 HC and HUS compared with normal controls (***p < 0.0001 and p < 0.001, respectively), and these remained slightly increased in the last blood samples, whereas higher levels were observed in O157:H7 HC than in HUS (†††p < 0.004). The mean levels of GRO-α and MCP-1 in children with O157:H7 HC or HUS were comparable to those noted in normal controls (NS).
Peritoneal dialysis in HUS.
Thirty-five percent of children (9 of 26) with HUS required peritoneal dialysis for 20 ± 14 d. They were comparable for age and for the timing of blood sample collection to the undialyzed HUS patients. However, they showed higher white blood cell (20.7 ± 9.2 versus 14.9 ± 3.6 × 109/L;p < 0.04) and neutrophil counts (14.3 ± 10.4 versus 9.3 ± 3.4 × 109/L;p < 0.1). As shown in Figure 3, levels of G-CSF were increased 6-fold (220 ± 219 versus 37 ± 38 pg/mL;p < 0.001), whereas concentrations of ENA-78 (113 ± 141 versus 507 ± 638 pg/mL;p < 0.001) were decreased 5-fold in patients with HUS who required dialysis. Hb and platelet counts were unrelated to mediator levels. Serum creatinine was correlated with G-CSF (R2 = 0.2;p < 0.001) and ENA-78 (R2 = −0.15;p < 0.0001). G-CSF concentrations correlated with total white blood cell (R2 = 0.3;p < 0.0001), neutrophil (R2 = 0.35;p < 0.0001), and monocyte (R2 = 0.45;p < 0.0001) counts.

Circulating levels of G-CSF, ELR+ C-X-C chemokines (ENA-78 and GRO-α), and C-C chemokines (MIP-1β and MCP-1) in 15 normal controls (NC), 9 disease control patients on chronic ambulatory peritoneal dialysis with chronic renal failure (DC), 17 children with HUS who did not require peritoneal dialysis (Dial −), and 9 children with HUS who were dialyzed (Dial +). Mean values are presented as (—). Children with HUS who required peritoneal dialysis had 6-fold increased G-CSF levels (*p < 0.001; Dial+vs Dial− and Dial+vs controls, p < 0.001; Dial−vs controls, NS). They also presented 5-fold decreased levels of ENA-78 (†p < 0.001; ENA-78, Dial+vs Dial− and Dial+vs controls, p < 0.01; Dial−vs controls, NS). Abnormally increased concentrations of MCP-1 were noted in the disease control group with chronic renal failure (††p < 0.0001, compared with any other group).
DISCUSSION
C-X-C and C-C chemokines.
More than 40 chemokines have been identified in humans, and these may be produced by virtually all somatic cells (27). In children with D+ HUS, Van Setten et al.(4) reported an increased urinary excretion of IL-8 and MCP-1. There was no relationship between urinary and circulating chemokine concentrations, suggesting independent, local production within the kidney (4). Similar observations have been made for IL-6 (28). Van Setten et al.(4) found slightly increased circulating levels of MCP-1 in children with D+ HUS. However, this was noted in only two patients (4). We found normal mean MCP-1 concentrations in HUS with only four abnormal measurements. A limitation of our study may be that diseased controls for renal failure were significantly older than other patients. Moreover, we cannot exclude that peritoneal dialysis may have also modified circulating mediator levels measured on samples obtained 1 wk after HUS diagnosis. Nevertheless, we observed that children with O157:H7 HC or HUS had abnormally increased levels of MIP-1β on admission. These values remained high 1 wk later in the HC group only. Although the reason for this observation is unclear, there was no evidence that the time elapsed after the onset of symptoms was a confounding factor on mediator levels, inasmuch as HC and HUS patients were sampled 11 d after the onset of enteric symptoms. It may signify an appropriate recruitment of macrophages to eradicate infection. Unexpectedly, we found that children with uncomplicated O157:H7 enteritis without bloody stools had significantly higher rates of abnormal GRO-α, MIP-1β, and MCP-1 measurements than those with O157:H7 HC or HUS. This was noted in patients with enteritis who showed lower leukocyte counts than those with HC or HUS. Our data thus indicate that excessive synthesis of chemokines occurs during E. coli O157:H7 enteritis. It is unclear whether the circulating GRO-α, MIP-1β, and MCP-1 levels may reflect mucosal production within the gastrointestinal tract. This view is supported by recent data demonstrating basolateral secretion of C-X-C and C-C chemokines by human intestinal epithelial cells (29–31). Human monocytes stimulated with Shiga toxin also secrete cytokines such as IL-8, via a lipopolysaccharide-independent pathway (32), but there is no data on other chemokines. In the baboon model of HUS, Taylor et al.(33) found that an i.v. injection of Shiga toxin increased the urinary levels of tumor necrosis factor (TNF)-α and IL-6, as in the human disease (28). However, in contrast to children with D+ HUS (19–21, 28), circulating cytokines were undetectable (33). Normal circulating levels of TNF-α, IL-6, IL-8, IL-10, and G-CSF have been noted in asymptomatic carriers of E. coli O157:H7 (21). Available data thus indicate that an active enteritis, but not necessarely an HC or HUS, is associated with detectable cytokines and chemotactic cytokines in the circulation.
In this study, we observed decreased circulating levels of ENA-78 in patients with HUS and that lower values were noted in children who required dialysis. Compared with other chemokines, the pattern of ENA-78 secretion appeared as differentially regulated. This finding was unexpected inasmuch as chemokines are secreted for leukocyte recruitment to eradicate pathogens (27). ENA-78 was first isolated from type II pneumocyte-derived cells and was identified as an epithelial cell–derived neutrophil chemoattractant (34). The initial observation that alveolar type II epithelial cells (A549) constitutively release low amounts of ENA-78 has also been observed in human monocytes (35). Schnyder-Candrian et al.(35) have shown that cycloheximide, a protein synthesis inhibitor, may superinduce IL-8 mRNA in human monocytes stimulated with either lipopolysaccharides, TNF-α, or IL-1β. In contrast, ENA-78 mRNA synthesis was completely abrogated, indicating that IL-8 and ENA-78 exhibit different patterns of expression at the mRNA level in human monocytes (35). In human intestinal epithelial cells (HCT-8), Thorpe et al.(30) have shown that Shiga toxin 1 induces IL-8 and GRO-α protein synthesis. Moreover, they have shown that Shiga toxin 1 and TNF-α may superinduce GRO-α as well as ENA-78 mRNA synthesis (30). ENA-78 protein synthesis was not evaluated (30). Thus, decreased ENA-78 levels may arise either from differential transcription and/or translation.
An alternative explanation for decreased circulating ENA-78 may also be proposed. The Duffy antigen receptor for chemokines is a decoy receptor that is not coupled to trimeric G proteins, and ligand binding does not appear to evoke an intracellular signal. In humans, as in mice, it is mainly expressed on red blood cells, endothelial cells, and neurons (36). Data in transgenic mice overexpressing the Duffy antigen suggest it may have a role in the maintenance of homeostasis, perhaps by sequestering chemokines from circulation (36). Liu et al.(37) reported an up-regulation of mRNA and protein expression of the Duffy antigen receptor for chemokines in children with HIV-associated HUS as well as D+ HUS. The predominant localization of mRNA and protein was found in endothelial cells, although up-regulation in collecting duct epithelial cells and interstitial inflammatory cells was also noted (36). Whether this may account for lower circulating ENA-78 levels in HUS remains unclear, as it implies that the human Duffy antigen receptor may preferentially bind to ENA-78 compared other chemokines such as IL-8. Although IL-8 receptors A and B bind with different affinities to ENA-78, IL-8, GRO-α, and neutrophil activating polypeptide-2 (38), there is no data supporting that this is true of the Duffy antigen receptor. In the study of Chaudhuri et al.(39), GRO-α, IL-8, and neutrophil-activating polypeptide-2 bound to the human Duffy antigen receptor expressed on an erythroleukemia cell line with the same high affinity, but there was no binding for MIP-1α and MIP-β. Overall, the mechanisms responsible for decreased circulating ENA-78 levels in E. coli O157:H7-associated HUS are unclear and await further studies.
G-CSF.
We found that circulating G-CSF concentrations are correlated with the severity of illness during E. coli O157:H7 infections, as evaluated by the development of HUS and the requirement for peritoneal dialysis. Although patients with uncomplicated O157:H7 HC initially showed a 3-fold increase in G-CSF values, there was a trend toward normalization over the next week. Conversely, G-CSF seemed to remain increased in HUS.
Fibroblasts, lymphocytes, endothelial cells, and neutrophils can release substantial amounts of G-CSF (40). Monocytes and macrophages are nevertheless considered the main cellular sources upon activation with cytokines or lipopolysaccharide (40). G-CSF is a hematopoietic growth factor for neutrophils and monocytes (40). It is basically a proinflammatory mediator that primes neutrophils for chemotaxis and phagocytosis, thereby leading to the release of reactive oxygen species and lysosomal enzymes (40). Increased circulating levels of elastase (12, 14, 15) and alpha-1-antitrypsin (15) have been found in D+ HUS, as well as in HC (41). Vierzig et al.(22) showed that G-CSF serum concentrations strongly paralleled the immature to total neutrophil count in one patient. These data are consistent with the finding that G-CSF induces the release of immature neutrophils from the bone marrow into the circulation (42). This is also in agreement with previous data from our group showing that children with D+ HUS have abnormally decreased circulating levels of soluble l-selectin (19). The latter is down-regulated by G-CSF on CD34-positive hematopoietic progenitors (43).
In this study, we noted that G-CSF concentrations accounted for 45% of the variance in peripheral blood monocyte counts (R2 = 0.45), consistent with the finding that G-CSF indirectly stimulates the production of monocytes (44). Moreover, we observed that 35% of the variance in neutrophil counts was explained by differences in G-CSF levels. Vierzig et al.(22) reported that the peak in leukemoid reaction (50,000 cells/mm3) occurred while G-CSF concentrations had decreased below 50 pg/mL. Several mechanisms may account for these findings. In vitro, both immature and mature neutrophils may modulate exogenous G-CSF concentrations, through continuous receptor-mediated absorption and metabolism (45). Secondly, Liu et al.(46) also demonstrated that Shiga toxin delays neutrophil apoptosis. Moreover, children with D+ HUS have abnormally increased circulating levels of soluble Fas, an inhibitor of apoptosis, which may contribute to circulating leukocytosis (47). IL-8, GRO-α(48), and G-CSF (49) also inhibit neutrophil apoptosis. Leukocytosis is a key feature of D+ HUS and does not occur in atypical HUS, without a diarrheal prodrome. Walters et al.(9) previously suggested that leukocytosis reflects the severity of the precipitating enteric infection and the extent of tissue necrosis. Our data on circulating G-CSF levels are thus in agreement with this statement. Furthermore, it has been recently shown that G-CSF receptor is present on fetal enterocytes, indicating that it may also be important for intestinal growth and repair (50). In this regard, Hommes et al.(51) also showed that human recombinant G-CSF had a protective effect during experimental colitis in rabbits, with decreased production of proinflammatory eicosanoids and preserved synthesis of anti-inflammatory prostaglandin E2. The effect of recombinant G-CSF in models of O157:H7 infections remains to be evaluated.
In this study, we have shown that GRO-α, MIP-1β, and MCP-1 are produced at the early stage of E. coli O157:H7 enteritis, whether or not HC or HUS develop. Our data suggest that children with O157:H7-associated HUS may present abnormally increased circulating levels of G-CSF and decreased ENA-78 concentrations. The mechanisms responsible for leukocyte recruitment in O157:H7 infections are unclear and await further studies.
Abbreviations
- HUS:
-
hemolytic uremic syndrome
- D+ HUS:
-
diarrhea-associated hemolytic uremic syndrome
- HC:
-
hemorrhagic colitis
- G-CSF:
-
granulocyte colony-stimulating factor
- ELR+:
-
Glu-Leu-Arg motif
- ENA-78:
-
epithelial cell–derived neutrophil-activating protein-78
- GRO-α:
-
growth-related oncogen-α
- MCP-1:
-
monocyte chemotactic protein-1
- MIP-1β:
-
macrophage inflammatory protein-1β
References
Gasser C, Gauthier E, Steck A, Siebenmann RE, Oechslin R 1955 Hamolytisch-uramische syndrome: bilaterale nierenrindennekrosen bei akuten erworbenen hamolytischen anamien. Schweiz Med Wochenschr 85: 905–909
Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H 1985 The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis 151: 775–782
Proulx F, Seidman EG, Karpman D 2001 The pathogenesis of Shiga toxin associated hemolytic uremic syndrome. Pediatr Res 50: 1–9
Van Setten PA, Hinsbergh MV, Van Den Heuvel LPWJ, Preyers F, Dijkman HPBM, Assmann KJM, Ven Der Velden TJAM, Monnens LAH 1998 Monocyte chemoattractant protein-1 and interleukin-8 in urine and serum of patients with hemolytic uremic syndrome. Pediatr Res 43: 759–767
Inward CD, Howie AJ, Fitzpatrick MM, Rafaat F, Milford DV, Taylor CM 1997 Renal histopathology in fatal cases of diarrhea-associated haemolytic uraemic syndrome. Pediatr Nephrol 11: 556–559
Buteau C, Proulx F, Chaibou M, Raymond D, Clermont MJ, Mariscalco MM, Lebel MH, Seidman E 2000 Leukocytosis in children with Escherichia coli O157: H7 enteritis developing the hemolytic uremic syndrome. Pediatr Infect Dis J 19: 642–647
Bell P, Griffin PM, Lozano P, Christie DL, Kobayashi JM, Tarr PI 1997 Predictors of hemolytic uremic syndrome in children during a large outbreak of Escherichia coli O157: H7 infections. Pediatrics 100: e12
Wong CS, Jelacic S, Habeeb RL, Watkins S, Tarr PI 2000 The risk of the hemolytic uremic syndrome after antibiotic treatment of Escherichia coli O157: H7 infections. N Engl J Med 26: 1930–1936
Walters MD, Matthei IE, Kay R, Dillon MJ, Barratt TM 1989 The polymorphonuclear leukocyte count in childhood haemolytic uraemic syndrome. Pediatr Nephrol 3: 130–134
Coad NA, Marshall T, Rowe B, Taylor CM 1991 Changes in the postenteropathic form of the hemolytic uremic syndrome in children. Clin Nephrol 35: 10–16
Forsyth KD, Simpson AC, Fitzpatrick MM, Barratt TM, Levinsky RJ 1989 Neutophil-mediated endothelial injury in haemolytic uraemic syndrome. Lancet 2: 411–414
Hughes DA, Smith GC, Davidson JE, Murphy AV, Beattie TJ 1996 The neutrophil oxidative burst in diarrhea-associated haemolytic uraemic syndrome. Pediatr Nephrol 10: 445–447
King AJ, Sundaram S, Cendoroglo M, Acheson DW, Keusch GT 1999 Shiga toxin induces superoxide production in polymorphonuclear cells with subsequent impairment of phagocytosis and responsiveness to phorbol esters. J Infect Dis 179: 503–507
Fitzpatrick MM, Shah V, Trompeter RS, Dillon MJ, Barratt TM 1992 Interleukin-8 and polymorphoneutrophil leukocyte activation in hemolytic uremic syndrome of childhood. Kidney Int 42: 951–956
Kaplan BS, Mills M 1988 Elevated serum elastase and alpha-1-antitrypsin levels in hemolytic uremic syndrome. Clin Nephrol 30: 193–196
Morigi M, Micheletti G, Figliuzzi M, Imberti B, Karmali MA, Remuzzi A, Remuzzi G, Zoja C 1995 Verotoxin-1 promotes leukocyte adhesion to cultured endothelial cells under physiologic flow conditions. Blood 86: 4553–4558
Te Loo, Monnens LA, van Der Velden TJ, Vermeer MA, Preyers F, Demacker PN, van Den Heuvel LP, van Hinsbergh VW 2000 Binding and transfer of verocytotoxin by polymorphonuclear leukocytes in hemolytic uremic syndrome. Blood 95: 3396–3402
Rovin BH, Phan LT 1998 Chemotactic factors and renal inflammation. Am J Kidney Dis 31: 1065–1084
Proulx F, Litalien C, Turgeon JP, Mariscalco MM, Seidman E 1998 Inflammatory mediators in hemorrhagic colitis and hemolytic uremic syndrome. Pediatr Infect Dis J 17: 899–904
Litalien C, Proulx F, Mariscalco MM, Robitaille P, Turgeon JP, Orrbine E, Rowe PC, McLaine PN, Seidman E 2000 Circulating inflammatory cytokine levels predict severity of renal failure in hemolytic uremic syndrome. Pediatr Nephrol 35: 28–33
Murata A, Shimazu T, Yamamoto T, Taenaka N, Nagayama K, Honda T, Sugimoto H, Monden M, Matsura N, Okada S 1998 Profiles of circulating inflammatory and anti-inflammatory cytokines in patients with hemolytic uremic syndrome due to E. coli O157 infection. Cytokine 10: 544–548
Vierzig A, Roth B, Querfeld U, Michalk D 1998 A 12-year-old boy with fatal hemolytic uremic syndrome, excessive neutrophilia and elevated endogenous granulocyte-colony-stimulating-factor serum concentrations. Clin Nephrol 50: 56–59
Rowe PC, Milner R, Orrbine E, Klassen TP, Mackenzie AM, Wells GA 1997 A phase II randomized controlled trial of SYNSORB Pk for the prevention of hemolytic uremic syndrome in children with verotoxin producing E. coli gastroenteritis. Pediatr Res 41: 283A
VaughanIII VC, Litt IF 1992 Assessment of growth and development. In: Behrman RE (ed) Nelson Textbook of Pediatrics. WB Saunders, Philadelphia, pp 32–43
Brady HR, Brenner BM, Clarkson MR, Lieberthal W 2000 Acute renal failure. In: Brenner BM (ed) The Kidney. WB Saunders, Philadelphia, pp 1201–1262
Proulx F, Litalien C, Turgeon JP, Mariscalco MM, Seidman E 2000 Circulating levels of transforming growth factor beta 1 and lymphokines in children with hemolytic uremic syndrome. Am J Kidney Dis 35: 29–34
Rollins BJ 1997 Chemokines. Blood 90: 909–928
Karpman D, Andreasson A, Thysell H, Kaplan BS, Svanborg C 1995 Cytokines in childhood hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Pediatr Nephrol 9: 694–699
Kim JM, Oh YK, Oh HB, Cho YJ 2001 Polarized secretion of CXC chemokines by human intestinal epithelial cell in response to Bacteroides fragilis enterotoxin: NF-kappa B plays a major role in the regulation of IL-8 expression. Clin Exp Immunol 123: 421–427
Thorpe CM, Hurley BP, Lincicome LL, Jacewicz MS, Keusch GT, Acheson DW 1999 Shiga toxins stimulates secretion of interleukin-8 from intestinal epithelial cells. Infect Immun 67: 5985–5983
Yamasaki C, Natori Y, Zeng XT, Ohmura M, Yamasaki S, Takeda Y, Natori Y 1999 Induction of cytokines in a human colon epithelial cell line by Shiga toxin 1 and 2 but not by non-toxic mutant Shiga toxin 1 which lacks N-glycosidase activity. FEBS Lett 442: 231–234
Van Setten PA, Monnens LA, Verstraten RG, van den Heuvel LP, van Hinsbergh VW 1996 Effects of verocytotoxin-1 on nonadherent human monocytes: binding characteristics, protein synthesis and induction of cytokine release. Blood 88: 174–183
Taylor FB, Tesh VL, DeBault L, Li A, Chang ACK, Kosanke SD, Pysher TJ, Siegler RL 1999 Characterization of the baboon responses to Shiga-like toxin: descriptive study of a new primate model of toxic responses to Stx-1. Am J Pathol 154: 1285–1298
Walz A, Burgener R, Car B, Baggiolini M, Kundel SL, Strieter RM 1991 Structure and neutrophil-activating properties of a novel inflammatory peptide (ENA-78) with homology to interleukin 8. J Exp Med 174: 1355–1362
Schnyder-Candrian S, Walz A 1997 Neutrophil-activating protein ENA-78 and IL-8 exhibit different patterns of expression in lipopolysaccharide- and cytokine-stimulated human monocytes. J Immunol 158: 3888–3894
Du J, Luan J, Liu H, Daniel TO, Peiper S, Chen TS, Yu Y, Horton LW, Nanney LB, Strieter RM, Richmond A 2002 Potential role for Duffy antigen chemokine-binding protein in angiogenesis and maintenance of homeostasis in response to stress. J Leukoc Biol 71: 141–153
Liu XH, Hadley TJ, Xu L, Peiper SC, Ray PE 1999 Up-regulation of Duffy antigen receptor expression in children with renal disease. Kidney Int 55: 1491–1500
Ahuja SK, Murphy PM 1996 The CXC chemokines growth-regulated oncogene alpha, GRObeta, GROgamma, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J Biol Chem 271: 20545–20550
Chaudhuri A, Zbrzezna V, Polyakova J, Pogo AO, Hesselgesser J, Horuk R 1994 Expression of the Duffy antigen in K562 cells. J Biol Chem 269: 7835–7838
Hartung T, von Aulock S, Wendel A 1998 Role of granulocyte colony-stimulating factor in infection and inflammation. Med Microbiol Immunol 187: 61–69
Ishikawa N, Kamitsuji H, Nakayama A, Umeki Y 2000 Plasma levels of granulocyte elastase-alpha-1 proteinase inhibitor complex in children with hemolytic uremic syndrome caused by verotoxin-producing Escherichia coli. Pediatr Int 42: 637–641
Tamura M, Hattori K, Nomura H, Oheda M, Kubota N, Imazeki I, Ono M, Ueyama Y, Nagata S, Shirafuji N 1987 Induction of neutrophilic granulocytosis in mice by administration of purified human native granulocyte colony-stimulating factor. Biochem Biophys Res Commun 142: 454–460
Ohsaka A, Saionji K, Sato N, Mori T, Ishimoto K, Inamatsu T 1993 Granulocyte colony-stimulating factor down-regulates the surface expression of the human leukocyte adhesion molecule-1 on human neutrophils in vitro and in vivo. Br J Haematol 84: 574–580
Lord BI, Molineux G, Pojda Z, Souza LM, Mermod JJ, Dexter TM 1991 Myeloid cell kinetics in mice treated with recombinant interleukin-3, granulocyte colony-stimulating factor (CSF) or granulocyte-macrophage CSF in vivo. Blood 77: 2154–2159
Saito K, Nakamura Y, Waga K, Inoue F, Enokihara H, Nara N, Furusawa S 1998 Mature and immature myeloid cells decrease the granulocyte colony-stimulating factor level by absorption of granulocyte colony-stimulating factor. Int J Hematol 67: 145–151
Liu J, Akahoshi T, Sasahana T, Kitasato H, Namai R, Sasaki T, Inoue M, Kondo H 1999 Inhibtion of neutrophil apoptosis by verotoxin 2 derived from Escherichia coli O157:H7. Infect Immun 67: 6203–6206
Masri C, Proulx F, Toledano B, Clermont MJ, Liet JM, Mariscalco MM, Seidman E, Carcillo J 2000 Soluble Fas and soluble Fas-ligand in children with Escherichia coli O157: H7 associated hemolytic uremic syndrome. Am J Kidney Dis 36: 687–694
Dunican AL, Leuenroth SJ, Ayala A, Simms HH 2000 CXC chemokines suppression of polymorphonuclear leukocytes apoptosis and preservation of function is oxidative stress independent. Shock 13: 244–250
Matute-Bello G, Liles C, Radella F, Steinberg KP, Ruzinski JT, Hudson LD, Martin TR 2000 Modulation of neutrophil apoptosis by granulocyte colony-stimulating factor and granulocyte/macrophage colony-stimulating factor during the course of acute respiratory distress syndrome. Crit Care Med 28: 1–7
Calhoun DA, Lunoe M, Du Y, Christensen RD 2000 Granulocyte colony-stimulating factor is present in human milk and its receptor is present in human fetal intestine. Pediatrics 105: e7
Hommes DW, Meenan J, Dijkhuizen S, Ten Kate FJ, Tytgat GN, Van Deventer SJ 1996 Efficacy of recombinant granulocyte colony-stimulating factor (rhG-CSF) in experimental colitis. Clin Exp Immunol 106: 529–533
Acknowledgements
The authors thank Denise Levesque for technical assistance, Hughes Charron for patient recruitment at Sainte-Justine Hospital, and Lucie Hyde for data management at the Children's Hospital of Eastern Ontario.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Proulx, F., Toledano, B., Phan, V. et al. Circulating Granulocyte Colony-Stimulating Factor, C-X-C, and C-C Chemokines in Children with Escherichia Coli O157:H7 Associated Hemolytic Uremic Syndrome. Pediatr Res 52, 928–934 (2002). https://doi.org/10.1203/00006450-200212000-00019
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200212000-00019
This article is cited by
-
Species-specific enhancement of enterohemorrhagic E. coli pathogenesis mediated by microbiome metabolites
Microbiome (2019)
-
LPS-primed CD11b+ leukocytes serve as an effective carrier of Shiga toxin 2 to cause hemolytic uremic syndrome in mice
Scientific Reports (2018)
-
Pathogenic role of inflammatory response during Shiga toxin-associated hemolytic uremic syndrome (HUS)
Pediatric Nephrology (2018)
-
Age-Related Changes of Serum Soluble Interleukin 9 Receptor (sIL-9Rα) in Healthy Subjects
Indian Journal of Clinical Biochemistry (2012)
