
Abstract
A large number of molecular, cellular, and epidemiologic factors have been implicated in the regulation of bone development. A major unsolved problem is how to integrate these disparate findings into a concept that explains the development of bone as an organ. Often events on the organ level are simply presented as the cumulative effect of all factors that individually are known to influence bone development. In such a cumulative model it must be assumed that each bone cell carries the construction plan of the entire skeletal anatomy in its genes. This scenario is implausible, because it would require an astronomical amount of positional information. We therefore propose a functional model of bone development, which is based on Frost's mechanostat theory. In this model the genome only provides positional information for the basic outline of the skeleton as a cartilaginous template. Thereafter, bone cell action is coordinated by the mechanical requirements of the bone. When mechanical challenges exceed an acceptable level (the mechanostat set point), bone tissue is added at the location where it is mechanically necessary. The main mechanical challenges during growth result from increases in bone length and in muscle force. Hormones, nutrition, and environmental factors exert an effect on bone either directly by modifying the mechanostat system or indirectly by influencing longitudinal bone growth or muscle force. Predictions based on this model are in accordance with observations on prenatal, early postnatal, and pubertal bone development. We propose that future studies on bone development should address topics that can be derived from the mechanostat model.
Similar content being viewed by others

Main
Bone development is one of the key processes of intrauterine and postnatal growth. Indeed, major abnormalities in bone development are incompatible with survival. Elucidating the mechanisms of this process, therefore, is an important task in biology and medicine. Similar to other fields of biomedical investigation, current research in bone biology heavily relies on the reductionist approach, which excludes the physiologic context as far as possible and focuses on the role of individual factors (1). Methods based on this approach have led to spectacular new insights into the molecular and cellular events occurring during bone development. A rapidly increasing number of factors, commonly called determinants or regulators of bone development, have been implicated in this process. To the list of molecular and cellular factors must be added the many environmental and behavioral factors identified by epidemiology, such as nutritional aspects and physical activity.
Thus, the reductionist approach has been extremely useful in identifying individual parts of the developing bone's machinery. The problem is how to put the individual pieces back into place. This is an essential task when it comes to explaining bone development on the level that interests patients and physicians most—the organ level. Confronted with a maze of molecular and cellular pathways, one may easily reach the fatalistic conclusion that bone development is too complex to be understood. Alternatively, one may try to devise simplifying models of the relationship between the known organ-level features of bone development and the known molecular, cellular, and epidemiologic factors implicated in this process.
Possibly the simplest way to devise such a model is to present bone development as the cumulative effect of factors that individually are known to influence bone development. This could lead to a model similar to the one shown in Figure 1, which—with more or less variations in detail—appears to be widely used in the current literature. Organ-level bone development results from the osteotropic action of a variety of contributors. Although apparently straightforward, a weak point in this model is that it presents bone development as a process of blind steering. None of the proposed determinants of bone mass and architecture has any current information on how much bone has already accumulated and how much more is needed. This is similar to driving a car with eyes closed and ears plugged—hardly a good strategy for traveling safely. If such a model is adopted, it therefore must be argued that the genes know it all, i.e. the entire blueprint for constructing the skeleton is contained in the genes box of the model. Accordingly, bones must be assumed to self-assemble following an intrinsic genetic plan.

A cumulative model of bone development based on the combined action of factors that individually have been shown to influence bone development. In this model, none of the proposed regulators of bone receives any information on the outcome of the regulation. Thus, bone development would be the result of blind steering rather than regulation.
There is good evidence that the self-assembly hypothesis is correct as far as skeletal patterning during embryonic development is concerned (2). The shaping of bone templates during skeletal patterning occurs in a soft tissue. Thus, all participating cells can be coordinated through the diffusion of morphogens, which are molecules distributed in a gradient that alters the developmental fate of target cells in a concentration-dependent manner (2, 3). The resulting structures have a relatively simple geometry.
However, it is more difficult to explain skeletal development on the basis of a similar self-assembly process once mineralization has occurred. For example, cortical development is determined by changes on the periosteal and endocortical surfaces (4). The cells involved in this process cannot be coordinated by diffusible molecules, because they are separated by the mineralized bone cortex (4). Moreover, mineralized bone has an extremely complicated microarchitecture. The genome would have to harbor information on the final position of all structural elements (such as trabeculae and osteons) in the adult skeleton, and also on what each bone cell in the developing skeleton has to do to assemble these structures at the correct position. Models of mineralized bone development based on this self-assembly hypothesis therefore have to address the question of how the astronomical amount of positional information required for it could be stored in and released from the genome. A further problem is to explain how bone could adapt this genetically fixed plan to changing requirements, what these requirements are, and how they are detected.
A FUNCTIONAL MODEL OF BONE DEVELOPMENT: THE MECHANOSTAT
The cumulative concept outlined above inevitably presents the organ bone as the slave of its cells and molecules, inasmuch as it starts from the question “How do cells and molecules control or regulate bone development?” This is not the only possible way to start analyzing bone development. An alternative approach could be to ask the reverse question: “How does the organ bone control its cells and molecules to develop?” The following will illustrate this idea. Based on the reductionist approach, one might conclude that bone growth in width is controlled by the proliferation of periosteal osteoblast precursors. However, this is similar to saying that a car is moving because the wheels are controlled by the turning axes. Although both statements reasonably explain a phenomenon, they certainly catch just a small part of the truth. As far as the phenomenon of the moving car is concerned, asking the question “Who is the driver and where does he want to go?” could lead to complementary insights.
What then drives bone development and where is it headed for? To address the second part of the question first, a student of current bone literature might answer “The goal of bone development is to accumulate peak bone mass.” However, it is hard to see why a bone should be programmed to become as heavy as possible. Indeed, heavy bones may rather be a disadvantage for wild animals, because additional weight increases energy expenditure and decreases running speed (5). Maximizing the body's calcium stores by increasing skeletal weight probably does not carry a big functional advantage, as serum calcium is maintained stable even in severe osteopenia.
Obviously, the functionally most relevant property of a bone is not its weight, but its strength. Bone strength is critical for survival, because fracture of a major bone usually means death for an adult wild-living animal (5). As a consequence of this strong selection pressure, the evolutionary process should have led to mechanisms that maintain bone's mechanical integrity by whatever means are available. Thus, the aim of bone development should be to make bones as strong as necessary (5). In that perspective, increasing a bone's mass is not the aim of bone development, but rather one of the means to achieve this aim. Another means to increase bone stability is to adapt bone architecture, either on the macroscopic or on the microscopic level (6–8).
A viable model of functionally controlled bone development has to take into account a general principle of regulation, which applies to biology, engineering, and even to human organizations (9, 10) : Control requires not only the ability to act, but also needs information on the current state of affairs. Thus, a controlled system needs to have both effector and sensor mechanisms. The former performs an action, the latter generates feedback signals, which indicate whether the desired effect has been achieved or not. This automatically implies that there has to be information within the system about what the desired effect is.
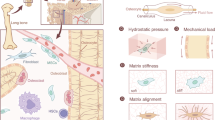
These requirements for a controlled system are met by Frost's mechanostat model (11) and related approaches (8, 10) (Fig. 2). It is proposed that the desired effect of bone homeostasis is to keep the mechanically induced deformation of bone (in biomechanical terminology called “strain”) close to a preset level, or set point. The deformation of a bone is a surrogate measure of its strength, because a strong bone will deform less than a weak bone when a mechanical challenge is applied. Bone deformation generates canalicular fluid flow (12), which could be monitored by osteocytes (13). When bone deformation exceeds a certain acceptable limit, osteocytes might sense this and send out signals, which could lead to adaptations in bone mass and architecture (14). These adaptations increase bone strength, and the mechanical strain returns to the acceptable level.

A functional model of bone development based on mechanostat theory. The central piece of bone regulation is the feedback loop between bone deformation (tissue strain) and bone strength. During growth this homeostatic system is continually forced to adapt to external challenges. Factors shown below modulate various aspects of the central regulatory system.
According to this model, changes in bone mass and architecture occur when bone stability is challenged and bone deformation exceeds an acceptable level. To put it differently, the required mechanical strength of a bone determines its mass and architecture, not vice versa. During growth, bone stability is continually threatened by two processes, the increase in bone length and the increase in muscle force. Longitudinal growth increases lever arms and bending moments and therefore leads to greater bone deformation (7, 15). Greater muscle force will also increase bone deformation during muscle contraction. Body weight alone puts relatively small loads on bones, but the effect of weight is amplified by muscle action (16, 17). These challenges create the need for adaptational changes in bone mass and architecture. This answers the first of the two questions asked at the start of this discussion—what drives bone development? In the mechanostat model, the drivers of bone development are two nonbone organs, growth plate and muscle.
In summary, the proposed model distinguishes two functionally different phases of bone development: The first is skeletal patterning, in which the basic shape of all bones is preformed according to a genetically determined plan. Spatial information at that stage is provided by morphogens and similar signaling molecules. The second phase starts when mineralization occurs in these templates and diffusing morphogens can no longer reach all cells involved in bone development. The spatial information about where mineralized bone needs to be added and where it should be removed is now provided by mechanical strain. This mechanical information is translated into biologic signals using the mechanostat mechanism. Bone mass and architecture are adapted to keep mechanical strain close to the set point. The mechanical stress required for this scenario comes from muscle contraction, which is present at the time when bone mineralization starts (18). Thus, in this model postembryonic bone development is controlled by the functional requirements of bone as an organ. The organ bone is not the slave, but the master of its cells and molecules, which it coordinates with the aim to maintain structural integrity.
Are mechanical factors more important for bone development than hormones and nutrition? The mechanostat model suggests that this question is similar to asking whether the steering wheel or the accelerator is the predominant regulator of car driving. As shown in Figure 2, mechanical and nonmechanical factors are not competitors. Nowhere in the model could one of these factors substitute for the other, because they have different roles. Hormones and nutrition influence the mechanical loads on growing bone by acting on longitudinal bone growth and muscle mass. Hormones and nutrition might also alter the mechanostat set point, or the width of the tolerance zone around the set point, and they could modify many aspects of osteoblast and osteoclast performance. However, hormones and nutrition cannot replace the guiding effect of mechanical strain on bone.
CLINICAL AND EXPERIMENTAL OBSERVATIONS RELATED TO THE MECHANOSTAT MODEL
A scientific model is useful, if it provides explanations for study results and helps to create new hypotheses. It is beyond the scope of this article to give a complete account of bone development with regard to mechanostat theory. Frost and others have discussed many aspects, such as the mechanism of endochondral ossification (19, 20), the influence of physical activity (16, 20), the set point defect in osteogenesis imperfecta (11, 21), and the possible race-related differences in the mechanostat set point (9, 22). In the following we briefly review some additional features of bone development in light of the mechanostat model.
Fetal Bone Development.
The hypothesis that fetal bone development should be driven by mechanical forces is certainly not widely accepted in the field of bone research. The fetus is usually thought to peacefully float in amniotic fluid and thus not to be exposed to mechanical forces. This view of the fetus is probably not shared by many women who have experienced pregnancy. In fact, regular fetal kicks against the uterine wall are a traditional clinical sign of fetal well-being (23), and the fetus is moving almost constantly even at early stages of development (18). The magnitude of the forces applied to the fetal skeleton during this intrauterine resistance training is not known. However, the forces are obviously sufficient to cause fractures when bone strength is decreased, such as in severe forms of osteogenesis imperfecta (24).
The hypothesis of mechanostat-controlled fetal bone development is in accordance with computer simulations of this process (8, 25) and is compatible with clinical and experimental observations. Neuromuscular disorders with intrauterine onset are associated with low bone mass at birth (26–28). Pharmacologic inhibition of muscle contraction during fetal development decreases periosteal expansion in rats (29).
Bone Development in Newborns and Premature Infants.
The proposed role of estrogen in the mechanostat system is to lower the set point on endosteal bone surfaces, thereby increasing the amount of endocortical bone (10, 30, 31). This could not only explain certain aspects of pubertal bone development in girls (see below), but is also of relevance for early postnatal events. During intrauterine life, the fetus is exposed to the high placental estrogen levels (32). Accordingly, endosteal surfaces should be very sensitive to mechanical strain, leading to small marrow cavities and high organ-level bone density. This is the case (33–35). Cutting off the placental estrogen supply at birth should increase the mechanostat set point on endosteal surfaces (31). Therefore, a substantial amount of bone next to marrow should now be interpreted as mechanically unnecessary by the mechanostat. This should lead to endocortical resorption and expansion of the marrow cavity, which in densitometric terms corresponds to a decrease in volumetric bone mineral density (Fig. 3). This mechanostat-based scenario is entirely consistent with observations in humans. There is indeed a postnatal increase in the size of the marrow cavity (35–37), which leads to a drop of approximately 30% in directly determined bone density within the first 6 mo of life (34, 35, 38).

Predicted pre- and postnatal time course of volumetric bone mineral density (BMD) in mature newborns and premature babies according to mechanostat theory.
At the same time, bones rapidly grow in length. From the perspective of mechanostat theory, the destabilizing effect of longitudinal growth should lead to the addition of bone tissue on periosteal surfaces, where the effect for stability is highest (7, 39). This is what happens (36, 40). Thus, the combined effect of decreased estrogen levels and increasing mechanical strain during the first postnatal months is a redistribution of bone tissue from the endocortical to the periosteal surface (35–37). This is a useful mechanism, because it optimizes the distribution of bone mass with regard to mechanical stability and decreases the amount of calcium that must be added from nutritional sources.
When birth occurs prematurely, the placental estrogen supply is cut off earlier. According to mechanostat theory, decreasing estrogen levels should lead to marrow cavity expansion and a decrease in organ-level bone density, similar to mature newborns (Fig. 3). Compared with mature newborns at birth, premature babies should have larger marrow cavities and lower bone density when they have reached expected term, because they have already started to adapt their bones to postnatal conditions (Fig. 3). However, the differences in bone density between term and preterm babies should be only transient, if the timing of birth did not affect the mechanostat set point.
These predictions about the effect of premature birth on the skeleton are in accordance with clinical findings. At expected term, premature babies indeed have a larger marrow cavity (41) and lower densitometric results than term newborns have at birth (42, 43). Although prematurity may have a deleterious effect on longitudinal bone growth (44–46), there is ample evidence from follow-up studies that the mass of the (shorter) bones is normal for size (44–50). This suggests that the mechanostat set point is not affected by premature birth. Thus, from the perspective of mechanostat theory, the so-called osteopenia of prematurity (not to be confused with rickets of prematurity) is the expression of physiologic postnatal adaptations.
Bone Development During Puberty in Girls.
When estrogen levels rise again in female puberty, endosteal bone surfaces are resensitized to mechanical strain, leading to endocortical apposition at many skeletal sites (51, 52). Adding bone on endocortical surfaces has a smaller effect on bone stability than adding the same amount of bone on the periosteal surface (7, 39). Consequently, postpubertal, premenopausal girls and women have more bone relative to their mechanical needs than males (53, 54). The purpose of this estrogen-dependent excess bone on endocortical surfaces could be to create a calcium reservoir, which can be tapped during pregnancy and lactation (55).
Growth in Length Precedes Increases in Bone Mass.
According to mechanostat theory, bone reacts to the challenges to its stability. As the response can only follow but not precede the challenge, bones are overloaded and thus fragile as long as growth continues (19, 56). The lag between growth in length and growth in strength should be exaggerated when longitudinal growth accelerates (19). In fact, the dissociation between bone's growth in length and in mass is a well-documented phenomenon during the pubertal growth spurt in both sexes (5, 52, 57–60), and could explain the increased fracture rate during that period in life (61–63). Because the timing of the maximal increase in bone length and muscle mass differs among musculoskeletal regions (64, 65), it is not unexpected that the increase in bone mass also follows a region-specific pattern during puberty (52).
STUDY TOPICS DERIVED FROM THE MECHANOSTAT MODEL
As discussed until now, the mechanostat model makes sense from a theoretical perspective and can explain many clinical and experimental observations. What questions should be addressed in further studies?
-
Studies analyzing bone development on the organ level: In osteopenic disorders, is low bone mass the result of decreased bone length, increased mechanostat set point, or decreased muscle force? What is the main target organ of each of the putative regulators of bone mass (estrogen, testosterone, GH, vitamin D, PTH, calcium intake, etc): muscle, growth plate, or bone? When there is a direct bone effect, does it alter the responsiveness to mechanical stimulation (and thus change the set point)? It is obvious that these questions can only be addressed in studies that are not limited to measuring bone density, but also take bone length and muscle force into account.
-
Studies analyzing bone development on the tissue, cell, and molecular levels: How is bone strain sensed? What are the constituents of the set point? By what mechanisms do hormones alter the set point? How is the set point affected in the various bone diseases? What are the effector signals? How do these signals recruit osteoclasts and osteoblasts for remodeling and modeling?
CONCLUSION
The mechanostat model is not just another item on the long list of regulators of bone development. Rather, it is a revolutionary concept, which puts the functional requirements of the organ at the center of bone development. It is revolutionary in the sense that it reverses the widely accepted order of things. Whereas prevailing models of bone development view the organ bone as the slave of its molecules and cells, the mechanostat model sees bone as their master. This model allows us to correctly predict many clinical and experimental observations. A whole new field of questions is waiting for answzers. It was not an exaggeration when Parfitt recently concluded that “unraveling the operation of the mechanostat…is probably the most important unsolved problem in skeletal biology”(22).
References
Bell JI 1999 Clinical research is dead; long live clinical research. Nat Med 5: 477–478
Erlebacher A, Filvaroff EH, Gitelman SE, Derynck R 1995 Toward a molecular understanding of skeletal development. Cell 80: 371–378
Hogan BL 1996 Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev 10: 1580–1594
Parfitt AM 1990 Bone-forming cells in clinical conditions. In: Hall BK (ed) The Osteoblast and Osteocyte, Vol 1. Telford Press, Caldwell, NJ, pp 351–429
Parfitt AM 1994 The two faces of growth: benefits and risks to bone integrity. Osteoporos Int 4: 382–398
Lanyon LE 1992 Control of bone architecture by functional load bearing. J Bone Miner Res 7( suppl 2): S369–375
Turner CH, Burr DB 1993 Basic biomechanical measurements of bone: a tutorial. Bone 14: 595–608
Carter DR, van der Meulen MCH, Beaupre GS 1996 Skeletal development: mechanical consequences of growth, aging and disease. In: Marcus R, Feldman D, Kelsey J (eds) Osteoporosis. Academic Press, San Diego, pp 333–350
Heaney RP 1995 Bone mass, the mechanostat, and ethnic differences. J Clin Endocrinol Metab 80: 2289–2290
Lanyon LE 1996 Using functional loading to influence bone mass and architecture: objectives, mechanisms, and relationship with estrogen of the mechanically adaptive process in bone. Bone 18: 37S–43S
Frost HM 1987 Bone “mass” and the “mechanostat”: a proposal. Anat Rec 219: 1–9
Qin L, Mak AT, Cheng CW, Hung LK, Chan KM 1999 Histomorphological study on pattern of fluid movement in cortical bone in goats. Anat Rec 255: 380–387
Aarden EM, Burger EH, Nijweide PJ 1994 Function of osteocytes in bone. J Cell Biochem 55: 287–299
Donahue HJ 2000 Gap junctions and biophysical regulation of bone cell differentiation. Bone 26: 417–422
van der Meulen MC, Ashford MW Jr, Kiratli BJ, Bachrach LK, Carter DR 1996 Determinants of femoral geometry and structure during adolescent growth. J Orthop Res 14: 22–29
Burr DB 1997 Muscle strength, bone mass, and age-related bone loss. J Bone Miner Res 12: 1547–1551
Martin RB, Burr DB, Sharkey NA 1998 Skeletal Tissue Mechanics. Springer Verlag, New York
de Vries JI, Visser GH, Prechtl HF 1985 The emergence of fetal behaviour: II. Quantitative aspects. Early Hum Dev 12: 99–120
Frost HM, Jee WS 1994 Perspectives: a vital biomechanical model of the endochondral ossification mechanism. Anat Rec 240: 435–446
Frost HM 1997 Why do marathon runners have less bone than weight lifters? A vital-biomechanical view and explanation. Bone 20: 183–189
Rauch F, Travers R, Parfitt AM, Glorieux FH 2000 Static and dynamic bone histomorphometry in children with osteogenesis imperfecta. Bone 26: 581–589
Parfitt AM 1997 Genetic effects on bone mass and turnover: relevance to black/white differences. J Am Coll Nutr 16: 325–333
Christensen FC, Rayburn WF 1999 Fetal movement counts. Obstet Gynecol Clin North Am 26: 607–621
Rowe DW, Shapiro JR 1998 Osteogenesis imperfecta. In: Avioli LV, Krane SM (eds) Metabolic Bone Disease and Clinically Related Disorders, 3rd Ed. Academic Press, San Diego, pp 651–695
Carter DR, Orr TE, Fyhrie DP, Schurman DJ 1987 Influences of mechanical stress on prenatal and postnatal skeletal development. Clin Orthop 219: 237–250
Burke SW, Jameson VP, Roberts JM, Johnston CED, Willis J 1986 Birth fractures in spinal muscular atrophy. J Pediatr Orthop 6: 34–36
Rodriguez JI, Palacios J, Garcia-Alix A, Pastor I, Paniagua R 1988 Effects of immobilization on fetal bone development: a morphometric study in newborns with congenital neuromuscular diseases with intrauterine onset. Calcif Tissue Int 43: 335–339
Rodriguez JI, Garcia-Alix A, Palacios J, Paniagua R 1988 Changes in the long bones due to fetal immobility caused by neuromuscular disease: a radiographic and histological study. J Bone Joint Surg Am 70: 1052–1060
Rodriguez JI, Palacios J, Ruiz A, Sanchez M, Alvarez I, Demiguel E 1992 Morphological changes in long bone development in fetal akinesia deformation sequence: an experimental study in curarized rat fetuses. Teratology 45: 213–221
Rodan GA 1991 Mechanical loading, estrogen deficiency, and the coupling of bone formation to bone resorption. J Bone Miner Res 6: 527–530
Frost HM 1999 On the estrogen-bone relationship and postmenopausal bone loss: a new model. J Bone Miner Res 14: 1473–1477
Forest MG, Ducharme JR 1993 Gonadotropic and gonadal hormones. In: Bertrand J, Rappaport R, Sizonenko PC (eds) Pediatric Endocrinology, 2nd Ed. Williams & Wilkins, Baltimore, pp 100–120
Rodriguez JI, Palacios J, Rodriguez S 1992 Transverse bone growth and cortical bone mass in the human prenatal period. Biol Neonate 62: 23–31
Trotter M, Peterson RR 1970 The density of bones in the fetal skeleton. Growth 34: 283–292
Trotter M, Hixon BB 1974 Sequential changes in weight, density, and percentage ash weight of human skeletons from an early fetal period through old age. Anat Rec 179: 1–18
Bernard J, Laval-Jeantet M 1962 Le rapport cortico-diaphysaire tibial pendant la croissance. Arch Franc Pediat 19: 805–817
Bonnard GD 1968 Cortical thickness and diaphysial diameter of the metacarpal bones from the age of three months to eleven years. Helv Paediatr Acta 23: 445–463
Trotter M 1971 The density of bones in the young skeleton. Growth 35: 221–231
Einhorn TA 1996 Biomechanics of bone. In: Bilezikian JP, Raisz LG, Rodan GA (eds) Principles of Bone Biology. Academic Press, San Diego, pp 25–37
Maresh MM 1961 Bone, muscle and fat measurements: longitudinal measurements of the bone, muscle and fat widths from roentgenograms of the extremities during the first six years of life. Pediatrics 28: 971–984
Beyers N, Alheit B, Taljaard JF, Hall JM, Hough SF 1994 High turnover osteopenia in preterm babies. Bone 15: 5–13
Greer FR, McCormick A 1986 Bone growth with low bone mineral content in very low birth weight premature infants. Pediatr Res 20: 925–928
James JR, Congdon PJ, Truscott J, Horsman A, Arthur R 1986 Osteopenia of prematurity. Arch Dis Child 61: 871–876
Helin I, Landin LA, Nilsson BE 1985 Bone mineral content in preterm infants at age 4 to 16. Acta Paediatr Scand 74: 264–267
Horsman A, Ryan SW, Congdon PJ, Truscott JG, Simpson M 1989 Bone mineral content and body size 65 to 100 weeks' postconception in preterm and full term infants. Arch Dis Child 64: 1579–1586
Fewtrell MS, Prentice A, Jones SC, Bishop NJ, Stirling D, Buffenstein R, Lunt M, Cole TJ, Lucas A 1999 Bone mineralization and turnover in preterm infants at 8–12 years of age: the effect of early diet. J Bone Miner Res 14: 810–820
Congdon PJ, Horsman A, Ryan SW, Truscott JG, Durward H 1990 Spontaneous resolution of bone mineral depletion in preterm infants. Arch Dis Child 65: 1038–1042
Schanler RJ, Burns PA, Abrams SA, Garza C 1992 Bone mineralization outcomes in human milk-fed preterm infants. Pediatr Res 31: 583–586
Lapillonne AA, Glorieux FH, Salle BL, Braillon PM, Chambon M, Rigo J, Putet G, Senterre J 1994 Mineral balance and whole body bone mineral content in very low-birth- weight infants. Acta Paediatr Suppl 405: 117–122
Hori C, Tsukahara H, Fujii Y, Kawamitsu T, Konishi Y, Yamamoto K, Ishii Y, Sudo M 1995 Bone mineral status in preterm-born children: assessment by dual-energy X-ray absorptiometry. Biol Neonate 68: 254–258
Garn SM 1972 The course of bone gain and the phases of bone loss. Orthop Clin North Am 3: 503–520
Bass S, Delmas PD, Pearce G, Hendrich E, Tabensky A, Seeman E 1999 The differing tempo of growth in bone size, mass, and density in girls is region-specific. J Clin Invest 104: 795–804
Schiessl H, Frost HM, Jee WS 1998 Estrogen and bone-muscle strength and mass relationships. Bone 22: 1–6
Schoenau E, Neu CM, Mokov E, Wassmer G, Manz F 2000 Influence of puberty on muscle area and cortical bone area of the forearm in boys and girls. J Clin Endocrinol Metab 85: 1095–1098
Kovacs CS, Kronenberg HM 1997 Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev 18: 832–872
Frost HM, Jee WS 1994 Perspectives: applications of a biomechanical model of the endochondral ossification mechanism. Anat Rec 240: 447–455
Krabbe S, Christiansen C, Rodbro P, Transbol I 1979 Effect of puberty on rates of bone growth and mineralisation: with observa2tions in male delayed puberty. Arch Dis Child 54: 950–953
Fournier PE, Rizzoli R, Slosman DO, Theintz G, Bonjour JP 1997 Asynchrony between the rates of standing height gain and bone mass accumulation during puberty. Osteoporos Int 7: 525–532
Blimkie CJ, Lefevre J, Beunen GP, Renson R, Dequeker J, Van Damme P 1993 Fractures, physical activity, and growth velocity in adolescent Belgian boys. Med Sci Sports Exerc 25: 801–808
Bailey DA, McKay HA, Mirwald RL, Crocker PR, Faulkner RA 1999 A six-year longitudinal study of the relationship of physical activity to bone mineral accrual in growing children: the university of Saskatchewan bone mineral accrual study. J Bone Miner Res 14: 1672–1679
Alffram PA, Bauer GC 1962 Epidemiology of fractures of the forearm: a biomechanical investigation of bone strength. J Bone Joint Surg Am 44-A: 105–114
Landin LA 1983 Fracture patterns in children: analysis of 8,682 fractures with special reference to incidence, etiology and secular changes in a Swedish urban population 1950–1979. Acta Orthop Scand Suppl 202: 1–109
Bailey DA, Wedge JH, McCulloch RG, Martin AD, Bernhardson SC 1989 Epidemiology of fractures of the distal end of the radius in children as associated with growth. J Bone Joint Surg Am 71: 1225–1231
Cameron N, Tanner JM, Whitehouse RH 1982 A longitudinal analysis of the growth of limb segments in adolescence. Ann Hum Biol 9: 211–220
Tanner JM, Hughes PC, Whitehouse RH 1981 Radiographically determined widths of bone muscle and fat in the upper arm and calf from age 3–18 years. Ann Hum Biol 8: 495–517
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rauch, F., Schoenau, E. The Developing Bone: Slave or Master of Its Cells and Molecules?. Pediatr Res 50, 309–314 (2001). https://doi.org/10.1203/00006450-200109000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200109000-00003
This article is cited by
-
Association between walking speed and calcaneus stiffness index in older adults
Journal of Bone and Mineral Metabolism (2023)
-
Einfluss von Pubertät und Hormonen auf die Knochenentwicklung
Gynäkologische Endokrinologie (2023)
-
Quantifying calcium changes in the fetal spine using quantitative susceptibility mapping as extracted from STAGE imaging
European Radiology (2022)
-
Characteristics of healthy German children and adolescents across tertiles of calcaneal stiffness index
Journal of Public Health (2022)
-
Comparison of DNA preservation between adult and non-adult ancient skeletons
International Journal of Legal Medicine (2022)
