
Abstract
In pathogenesis of celiac disease, the significance of prolamin peptide interactions with enterocytes is controversial. Changes in cellular metabolism induced by gliadin peptides, as well as uptake and presentation by enterocytes, are discussed. We analyzed peptide binding to enterocytic membranes as a potential key event. Binding capacities of brush border membranes isolated from small intestinal biopsies of untreated (n= 49) and treated celiac patients on a gluten-free diet (n= 30), as well as control subjects (n= 43), were measured with a dot blot chemiluminescence assay. Synthetic gliadin peptides comprising amino acid position 8–19 (G XIV) and 30–41 (G XI) of α-gliadins, a peptic-tryptic digest of gliadin (PT-GLI), and a synthetic zein peptide were used. Comparing treated celiac patients with controls, we observed significantly enhanced membrane-binding of PT-GLI [mean 122.4 densitometric units/μg (95% confidence interval 116.0–128.9) vs 108.9 (102.1–115.7)] and of zein peptide [50.2 (38.4–61.9) vs 28.8 (13.4–44.2)], but only slightly increased binding of the synthetic gliadin peptides G XIV [65.5 (60.6–70.5) vs 62.4 (56.3–68.5) and G XI [75.2 (69.8–80.6) vs 65.9 (55.2–76.5)]. Independent of patient group, membrane-binding capacities for celiac-active gliadin peptides exceeded those of the zein peptide. Thus, interaction of gliadin peptides with the apical enterocytic membrane was not found exclusively in celiac disease. Furthermore, increased binding capacities in treated celiac disease were not confined to celiac-active peptides. Quantitative differences in gliadin peptide binding as a primary characteristic in celiac disease might contribute to pathogenetic effects exerted on small intestinal epithelial cells.
Similar content being viewed by others


Main
The complex interplay of environmental and inherent factors in the pathogenesis of celiac disease is far from being understood. Celiac disease is induced by ingestion of prolamins and is characterized by small intestinal villous atrophy, crypt cell hyperplasia, and lamina propria infiltration by lymphocytes (1, 2). It has been proposed that the presentation of prolamin peptides by HLA class II molecules to T cells initiates immunopathogenesis (2, 3). There is increasing evidence that tissue transglutaminase plays a role in celiac disease. Enzymatic deamidation of gliadin peptides unmasks an epitope that binds to HLA-DQ2 and is recognized by gut-derived T cells (4). Furthermore, new antigenic complexes are created by cross-linking of gliadin with tissue transglutaminase (5).
Although celiac disease is viewed mainly as a T cell-mediated disorder, interactions between prolamin peptides and small intestinal epithelial cells might be involved in several ways (6). First, apical binding and uptake of prolamin fragments might show direct effects on enterocytes and lead to cell damage. HLA-DR overexpression (7), reduction of cell height (8), inhibition of biosynthesis of BBM hydrolases (9), and impaired cellular metabolism (10) have been demonstrated as effects of gliadin peptides on enterocytes, mediated in part by other cell types. Second, uptake, transcellular transport, and basolateral release of prolamin peptides by enterocytes might enable transglutaminase-mediated modification in the subepithelial region, followed by antigen presentation and T cell stimulation (4, 11). Third, presentation of modified peptides by enterocytes might cause T cell activation directly (12–14). In each case, initial binding to the apical BBM is supposed to be a prerequisite to the subsequent steps.
Conflicting results have been reported concerning gliadin peptide binding to enterocytes in celiac disease (15, 16). The aim of our study was therefore to clarify whether BBM-binding capacity for gliadin peptides is altered in celiac disease. Furthermore, we compared the BBM of patients with active disease with that of patients with treated disease to detect whether membrane-binding alterations in celiac disease are primary or secondary.
METHODS
Patients.
Small intestinal biopsies were obtained for routine diagnostic procedures from the following three patient groups:1) CON: 25 females and 18 males (0.4–14.3 y, median 3.1 y) with normal mucosa on a diet of gluten-containing food (diarrhea, failure to thrive);2) CD/A: 25 females and 24 males (0.3–18.3 y, median 4.1 y) with untreated active celiac disease, showing severe mucosal damage, according to Marsh (1), and 3) CD/R: 20 females and 10 males (2.8–69.2 y, median 46.4 y) with celiac disease in remission, showing intact intestinal mucosa on a gluten-free diet for at least 6 months.
The protocol of the study was approved by the local ethics committee.
BBM.
Small intestinal biopsies (wet weight 3–20 mg) were homogenized with a glass/Teflon homogenizer in 500 mM mannitol, 10 mM HEPES, pH 7.5. CaCl2 was added to reach a final concentration of 10 mM. After an incubation period of 15 min at 4°C, the homogenate was centrifuged (3,000 ×g, 15 min, 4°C) to separate aggregated membranes. After further centrifugation of the supernatant (30,000 ×g, 30 min, 4°C), the pellet of BBM vesicles was resuspended in 100 mM mannitol, 10 mM Tris, pH 7.4. For investigation of membrane morphology by electron microscopy, membrane vesicles were fixed with glutaraldehyde as previously described (17). The purity of BBM preparations was assessed by measuring the BBM enzymes sucrase (18) and alkaline phosphatase (test kit from Sigma Chemical Co., Deisenhofen, Germany). Enzyme units were defined as the amount of enzyme transforming 1 μmol of substrate/min under the experimental conditions. Specific enzyme activities were expressed as milliunits of enzyme/milligram of protein. Protein content was determined with the DC protein assay (Bio-Rad, Munich, Germany).
PT-GLI.
A crude mixture of gliadin peptides, free of contaminating proteases, was obtained by peptic-tryptic digestion of gliadin and was biotinylated with biotinamidocaproate-N-hydroxysulfosuccinimide ester (Sigma Chemical Co.), as described elsewhere (19).
Peptide synthesis.
Synthesis of gliadin peptides G XI and G XIV and zein peptide Z I was performed automatically by a solid phase peptide synthesizer (model 431 A, Applied Biosystems, Foster City, CA) using Fmoc (9-fluorenylmethyloxycarbonyl) chemistry on a small scale (1 mmol/Fmoc amino acid). Amino acid side chain protection during synthesis was as follow: Ser (t-butyl), Asn (trityl), and Gln (trityl).
Peptide G XI consisted of 12 amino acids at position 30–41 of α-gliadins (amino acid sequence: FPGQQQPFPPQQ) and peptide G XIV consisted of 12 amino acids at position 8–19 (LQPQNPSQQQPQ) (20). Zein peptide Z I (= control) was 14 amino acids long and covered the sequence of nontoxic zein SF4 at position 8–21 (sequence: LAPSAIIPQFLPPV).
Crude peptides were checked for amino acid sequences with a pulsed liquid sequencer (model 471 A; Applied Biosystems).
Biotinylation of synthetic peptides.
Synthesized crude peptides (10.0 mg) were dissolved in 2 mL of 0.1 M NaHCO3 buffer (pH 8.2). The solution of the biotinylation reagent was freshly prepared by dissolving biotinamidocaproic acid-3-sulfo-hydroxysuccinimide ester (Sigma Chemical Co., no. B 1022) in 400 μL DMSO; the amount of reagent was between 7.0 and 8.5 mg (molar ratio to peptides = 1:3). Both solutions were mixed and magnetically stirred for 4 h at room temperature (21).
Purification of biotinylated synthetic peptides.
The first step of purification was performed by RP-HPLC under the following conditions: reversed phase: Hypersil C18, 5 μm, 12 nm (Shandon, no. 30105); column: 4.6 × 240 mm; temperature: 50°C; injection: 240 μL of the biotinylated peptide solution; flow rate: 1.0 mL; detection: UV absorbance at 220 nm. The following elution system was used: (A) 0.01 M ammonium acetate (pH 7.0), (B) 0.01 M ammonium acetate (pH 7.0)/acetonitrile (60/40, vol/vol); gradient: linear, 0 min 35% B, 100 min, 60% B (peptide G XI) or 0 min 15% B, 30 min, 30% B, 50 min 42.5% B (peptide G XIV) or 0 min 70% B, 120 min 85% B (peptide Z I). The purified peptides were eluted between 55 and 72 min. The corresponding eluates were collected from 10 runs and freeze-dried.
In a second step, peptides obtained by RP-HPLC were purified by GP-HPLC under the following conditions: precolumn: Ultropac TSK × SWP (Pharmacia, Uppsala, Sweden); 7.5 × 75 mm; main column: Ultropak TSK × G 2000 SW (Pharmacia), 7.5 × 600 mm; room temperature; injection: 1 mg peptide dissolved in 500 μL 0.1 M acetic acid (G XIV and Z I) or 0.1 M acetic acid/ethanol (1:1, vol/vol) (G XI); flow rate: 0.5 mL/min (peptide G XIV) or 0.4 mL/min (other peptides); detection: UV absorbance at 220 nm. The following elution solvents were applied: 0.1 M acetic acid (G XIV) or 0.1 M acetic acid/ethanol (9:1, vol/vol) (Z I) or 0.1 M acetic acid/ethanol (4:1, vol/vol) (G XI). Peptides were eluted for 93–108 min, and corresponding eluates were collected from about 10 runs and were freeze-dried.
Mass spectrometry.
Ten micrograms of the purified peptides were dissolved in 1 mL 0.1% formic acid/methanol (1:1, vol/vol) and were analyzed by electrospray ionization mass spectrometry with an LC-Q spectrometer (Finnigan Mat, San José, CA).
Binding assay.
For measuring interactions of biotinylated peptides with brush border membranes, the dot blot chemiluminescence assay was performed essentially as described before (22), using 1 μg BBM protein/10 μL buffer for immobilization on nitrocellulose. Chemiluminescence signals of bound peptides produced by peroxidase-catalyzed oxidation of luminol were analyzed densitometrically. Binding intensities were calculated as densitometric units/μg BBM protein in relation to an internal standard.
Statistical methods.
BBM-binding capacities were described by means and 95% confidence intervals. Differences between control and celiac disease specimens were analyzed by nonparametric two-tailed Mann-Whitney test for unpaired groups. Values of enzyme activities and enrichment factors are given as mean ± SEM.
RESULTS
BBM characterization.
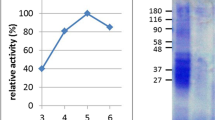
The apical membrane of enterocytes was successfully isolated independent of disease activity. Electron microscopy studies revealed that fractions of BBM formed closed right-side-out vesicles containing cytoskeleton elements inside. BBM vesiculation was not disturbed by changes in BBM composition in active celiac disease (Fig. 1). Enrichment factors for the BBM enzymes sucrase and alkaline phosphatase did not differ between patient groups (Table 1). Severe mucosal damage in untreated celiac disease was reflected by decreased specific enzyme activities, whereas reconstituted enzyme expression was demonstrated in specimens of patients in remission (Fig. 2). Membrane protein patterns obtained by PAGE did not differ between control subjects and treated patients (data not shown).

BBM were isolated as closed right-side-out vesicles independent of disease activity. (1) CON, (2) CD/A. For electron microscopy, membrane vesicles were fixed with glutaraldehyde. Bar = 60 nm.

Decreased specific activities of BBM enzymes sucrase and alkaline phosphatase in untreated celiac disease were reconstituted by gluten-free diet. Values are given as mean ± SEM.
Peptide characterization.
The amino acid sequences of the crude peptides synthesized with a solid-phase synthesizer were confirmed after cleavage from the resin by automatic Edman degradation. Peptides were biotinylated and purified 1) by RP-HPLC to remove, in particular, those by-products that still contained protecting groups and might falsify binding studies, and 2) by GP-HPLC to remove contaminants from RP-HPLC. Purified peptides were analyzed by mass spectrometry. The results were in agreement with calculated masses: G XI 1737.4 (calc. 1736.9), G XIV 1730.6 (1730.8), and Z I 1801.0 (1801.5).
Peptide binding to BBM.
The crude peptide mixture PT-GLI and the synthetic celiac-active gliadin peptides G XI and G XIV were used for measuring membrane-binding capacity. The synthetic nontoxic zein peptide Z I served as control.
BBM-binding capacity for PT-GLI was significantly higher in patients with celiac disease in remission than in control subjects (Fig. 3). Binding capacity was enhanced in active celiac disease, too, but did not reach significance over controls. Independent of disease activity, BBM-binding capacities for gliadin peptides of varying sizes (PT-GLI) exceeded that for the smaller synthetic ones (G XIV, G XI).

Peptide-binding capacities of BBM. Biotinylated synthetic peptides G XIV, G XI, and Z I were used in a concentration of 20 μg/mL; crude peptic-tryptic gliadin digest PT-GLI of 5 μg/mL. Binding intensities are expressed in densitometric units/μg BBM protein. Means and 95% confidence intervals are given. The p values for differences between CON and CD/A or CON and CD/R, respectively, were obtained by Mann-Whitney test for nonparametric data.
Differences in membrane-binding capacities for synthetic gliadin peptides between patient groups were smaller, compared with PT-GLI. Whereas binding capacities for G XIV did not differ significantly, BBM of patients with CD/R bound slightly more G XI, compared with CON (Fig. 3).
In addition, BBM of treated patients bound more G XI (mean: 75.2 densitometric units/μg BBM protein; 95% confidence interval: 69.8–80.6, n= 19) than G XIV (mean: 65.5 densitometric units/μg, 95% CI: 60.6–70.5, n= 20).
In contrast to gliadin peptides, membrane-binding capacity for the control zein peptide Z I was low. Interactions of Z I with BBM varied greatly and, in some cases, did not reach detectability (Fig. 3). In accordance with celiac-active peptides, however, binding of nontoxic Z I to BBM was enhanced in CD/A and CD/R.
Reliability of the binding assay was assessed by measuring lectin binding known to be altered in celiac disease. Because of cellular surface glycosylation disturbance, BBM binding of red kidney bean leukoagglutinin (PHA) specific for D-GalNAc/Gal was reduced in untreated celiac disease (mean: 204.8 densitometric units/μg, 95% CI: 146.6–262.9, n= 9) compared with controls (mean: 240.5 densitometric units/μg, 95% CI: 186.8–294.1, n= 11). In accordance with results of membrane protein pattern and enzyme activity analysis, membrane glycosylation in treated celiac disease as shown by PHA binding (mean: 264.7 densitometric units/μg, 95% CI: 127.5–401.9, n= 3) did not differ from that of controls.
DISCUSSION
Prolamins of the cereals wheat, rye, and barley are harmful for celiac disease patients. The putative celiac activity of wheat prolamins was ascribed to the tetrapeptide motifs PSQQ and QQQP, which are common for all toxic gliadin peptides studied (20). These common celiac-active amino acid sequences might take part in interactions of prolamin peptides with components of the apical membrane of small intestinal epithelial cells. In this respect, nontoxic prolamins not containing these sequences—for example, zein—might show different binding characteristics.
For the investigation of membrane-binding capacities in celiac disease, we chose toxic and nontoxic peptides. The synthetic gliadin peptides used in this study contained the motifs QQQP (G XI) and PSQQQP (G XIV). The peptic-tryptic digest of gliadin PT-GLI was an active crude peptide mixture comparable to Frazer's fraction III (23), but was devoid of contaminating trypsin (19). It was assumed that the synthetic zein peptide Z I did not to consist of celiac-active sequences.
We used an in vitro binding assay suitable for measuring specific ligand-membrane interactions (22). Because of limited amounts of BBM available from jejunal biopsies, variation of peptide concentrations or inhibition experiments were not possible. Specificity in terms of saturation and inhibition of gliadin peptide binding to membrane structures was previously shown for rat small intestinal BBM and apical membranes of the human epithelial cell lines Caco-2 and T84 (17, 24).
Our results suggest that although mucosal repair by gluten-free diet was achieved in the group of treated patients, celiac BBM were capable of binding more gliadin peptides than were the control membranes. This difference between CON and CD/R was significant for the peptide mixture PT-GLI. Earlier studies with peptic-tryptic gliadin digests have produced conflicting results. Colyer et al. (15) studied gliadin peptide binding to isolated enterocytes of treated celiac patients and healthy controls without detecting any difference. However, using formalin-fixed sections of small intestinal biopsies, Pittschieler et al. (16) demonstrated brush border binding of gliadin peptides exclusively in active celiac disease. Our findings of enhanced PT-GLI binding in active and treated celiac disease were supported by the data obtained with the synthetic gliadin peptide G XI. Although not reaching significance at the level of 5% error probability, there was a tendency for increased peptide binding in treated celiac disease, compared with controls.
In addition to enhanced gliadin peptide-binding capacity in celiac disease, two observations have been made. First, increased peptide-binding capacity in CD/R did not appear to be restricted to toxic gliadin peptides but was a general feature of the BBM, because it was also demonstrated for the nontoxic zein peptide Z I. Second, compared with binding intensities of the zein peptide, enhanced binding of synthetic gliadin peptides to the BBM of control patients indicated that differences in membrane binding of toxic and nontoxic peptides are not a celiac-specific phenomenon.
Because of identical labeling procedures and identical biotin:peptide ratios, binding intensities of the synthetic peptides were comparable to one another. However, the remarkable differences between PT-GLI and synthetic peptides with respect to their binding intensities should be discussed in a qualitative way, inasmuch as they might be due to differences in peptide size, extent of biotinylation, and interference of the label with binding properties, particularly of short peptides. Furthermore, deamidation of peptides could have occurred during peptic digestion. These questions should be addressed in further studies.
The differences in BBM-binding capacities observed for synthetic gliadin peptides and zein peptide are in agreement with in vitro toxicity studies. Whereas the celiac activity of peptide G XIV (position 8–19) was demonstrated by Cornell and Mothes (25), who used fetal chicken intestine and rat liver lysosomes as test systems, peptide 31–43 of A-gliadin, comparable to G XI 30–41, was found to be active in organ culture studies (26). No toxicity was observed with a peptic-tryptic digest of maize prolamin (26).
In contrast to previous studies (20), Shidrawi et al. (8) and Sturgess et al. (27) reported in vitro and in vivo toxicity only of the synthetic peptide A 31–49 and not of peptide C (3–21). It is tempting to speculate whether this difference of in vitro and in vivo toxicity tests is reflected to some extent by our membrane-binding assay, in that we observed stronger binding of G XI 30–41 to BBM of treated celiac disease patients, compared with G XIV 8–19.
In conclusion, we could show enhanced peptide-binding capacities of the small intestinal BBM in celiac disease. Because the increased binding capacities were not the result of mucosal damage and membrane alterations in untreated celiac disease, but were also observed in treated celiac disease, enhanced peptide binding could be termed a primary effect of celiac disease, in contrast to possible effects secondary to membrane alterations during the active disease process. Artifacts based on altered membrane composition or damages were excluded by analysis of membrane morphology, membrane protein pattern, glycosylation, and enzymatic activities. Age heterogeneity between our patient groups, especially CD/R compared with CON, might have contributed to the observed differences in peptide-membrane interactions. However, this age discrepancy is unavoidable as long as severe mucosal damage on gluten-containing food and intact intestinal mucosa on a gluten-free diet of at least 6-months' duration are inclusion criteria for CD/A and CD/R patients. Comparison of gliadin peptide-binding capacities of CD/R BBM of children and adults did not show an age-related difference. Besides, composition of the small intestinal brush border does not change after birth in humans, although complete functions of the mucosal barrier are not achieved before the age of 2 y (28).
Obviously, the underlying changes in membrane structure or ligand expression in celiac disease responsible for differential binding are too slight to be detectable with the methods used in this study to describe membrane reconstitution.
Clearly, BBM-binding characteristics demonstrated in vitro cannot be directly extrapolated to the pathophysiological processes occurring in the complex environment of the small intestine. However, results of altered peptide-binding capacities in celiac disease appear to be relevant if a threshold level of bound peptides is crucial for induction of consecutive cellular reactions. In the light of new findings concerning HLA-dependent activation of lamina propria lymphocytes by gliadin peptide presentation (4, 29, 30), the meaning of quantitative differences in membrane-binding capacities for intracellular antigen handling and nonprofessional antigen presentation by intestinal epithelial cells remains to be determined (6, 11, 12, 14).
Abbreviations
- BBM:
-
brush border membrane
- CD/A:
-
active celiac disease
- CD/R:
-
celiac disease in remission
- CON:
-
control group
- G XI:
-
synthetic gliadin peptide, position 30–41 of α-gliadins
- G XIV:
-
synthetic gliadin peptide, position 8–19 of α-gliadins
- PT-GLI:
-
peptic-tryptic gliadin digest
- RP-HPLC:
-
reversed-phase HPLC
- GP-HPLC:
-
gel permeation HPLC
- Z I:
-
synthetic zein peptide, position 8–21 of zein SF4
References
Marsh MN 1992 Gluten, major histocompatibility complex, and the small intestine. Gastroenterology 102: 330–354
Mäki M, Collin P 1997 Coeliac disease. Lancet 349: 1755–1759
Lundin KEA, Sollid LM, Anthonsen D, Norén O, Molberg O, Thorsby E, Sjöström H 1997 Heterogeneous reactivity patterns of HLA-DQ-restricted, small intestinal T-cell clones from patients with celiac disease. Gastroenterology 112: 752–759
Molberg O, McAdam SN, Körner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Noren O, Roepstorff P, Lundin KEA, Sjöström H, Sollid LM 1998 Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nature Med 4: 713–717
Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, Schuppan D 1997 Identification of tissue transglutaminase as the autoantigen of celiac disease. Nature Med 3: 797–801
Mayrhofer G 1995 Absorption and presentation of antigens by epithelial cells of the small intestine: hypotheses and predictions relating to the pathogenesis of coeliac disease. Immunol Cell Biol 73: 433–439
Maiuri L, Picarelli A, Boirivant M, Coletta S, Mazzilli MC, de Vincenzi M, Londei M, Auricchio S 1996 Definition of the initial immunologic modifications upon in vitro gliadin challenge in the small intestine of celiac patients. Gastroenterology 110: 1368–1378
Shidrawi RG, Day P, Przemioslo R, Ellis HJ, Nelufer JM, Ciclitira PJ 1995 In vitro toxicity of gluten peptides in coeliac disease assessed by organ culture. Scand J Gastroenterol 30: 758–763
Kreft D, Hauri HP, Belli DC, Bähler P, Naim HY, Keller KM, Lentze MJ 1997 The in vitro influence of gliadin peptides on the hydrolases of the duodenal brush border membrane in coeliac disease in remission. J Pediatr Gastroenterol Nutr 24: 452
Giovanni C, Mancini E, De Vincenzi M 1996 Inhibition of the cellular metabolism of Caco-2 cells by prolamin peptides from cereals toxic for coeliacs. Toxicol in Vitro 10: 533–538
Friis S, Dabelsteen E, Sjöström H, Norén O, Jarnum S 1992 Gliadin uptake in human enterocytes: differences between coeliac patients in remission and control individuals. Gut 33: 1487–1492
Zimmer KP, Poremba C, Weber P, Ciclitira PJ, Harms E 1995 Translocation of gliadin into HLA-DR antigen containing lysosomes in coeliac disease enterocytes. Gut 36: 703–709
Barbeau WE, Novascone MA, Elgert K 1997 Is celiac disease due to molecular mimicry between gliadin peptide-HLA class II molecule-T cell interactions and those of some unidentified superantigen?. Mol Immunol 34: 535–541
Van de Wal Y, Kooy YMC, van Veelen PA, Pena SA, Mearin LM, Molberg O, Lundin KEA, Sollid LM, Mutis T, Benckhuijsen WE, Drijfhout JW, Koning F 1998 Small intestinal T cells of celiac disease patients recognize a natural pepsin fragment of gliadin. Proc Natl Acad Sci USA 95: 10050–10054
Colyer J, Kumar PJ, Waldron NM, Clark ML, Farthing MJG 1987 Gliadin binding to rat and human enterocytes. Clin Sci 72: 593–598
Pittschieler K, Ladinser B, Petell JK 1994 Reactivity of gliadin and lectins with celiac intestinal mucosa. Pediatr Res 36: 635–641
Bolte G, Wolburg H, Beuermann K, Stocker S, Stern M 1998 Specific interaction of food proteins with apical membranes of the human intestinal cell lines Caco-2 and T84. Clin Chim Acta 270: 151–167
Dahlqvist A 1984 Assay of intestinal disaccharidases. Scand J Clin Lab Invest 44: 169–172
Bolte G, Osman A, Mothes T, Stern M 1996 Peptic-tryptic digests of gliadin: contaminating trypsin but not pepsin interferes with gastrointestinal protein binding characteristics. Clin Chim Acta 247: 59–70
Wieser H 1996 Relation between gliadin structure and coeliac toxicity. Acta Paediatr Suppl 412: 3–9
Bayer EA, Wilchek M 1990 Protein biotinylation. Methods Enzymol 184: 138–166
Bolte G, Knauss M, Metzdorf I, Stern M 1997 Dot blot chemiluminescence assay for studying food protein binding to small intestinal brush border membranes in vitro. J Biochem Biophys Methods 34: 189–203
Frazer AC, Fletcher RF, Ross CA, Shaw B, Sammons HG, Schneider R 1959 Gluten-induced enteropathy: the effect of partially digested gluten. Lancet 2: 252–255
Bolte G, Knauss M, Metzdorf I, Stern M 1998 Postnatal maturation of rat small intestinal brush border membranes correlates with increase in food protein binding capacity. Dig Dis Sci 43: 148–155
Cornell H, Mothes T 1995 Further studies of the in vitro activity of synthetic gliadin peptides in coeliac disease. Biochim Biophys Acta 1270: 168–172
Maiuri L, Troncone R, Mayer M, Coletta S, Picarelli A, De Vincenzi M, Pavone V, Auricchio S 1996 In vitro activities of A-gliadin-related synthetic peptides: damaging effect on the atrophic coeliac mucosa and activation of mucosal immune response in the treated coeliac mucosa. Scand J Gastroenterol 31: 247–253
Sturgess R, Day P, Ellis HJ, Lundin KEA, Gjertsen HA, Kontakou M, Ciclitira PJ 1994 Wheat peptide challenge in coeliac disease. Lancet 343: 758–761
Van Elburg RM, Uil JJ, De Monchy JGR, Heymans HSA 1992 Intestinal permeability in pediatric gastroenterology. Scand J Gastroenterol 27( Suppl): 19–24
Sjöström H, Lundin KEA, Molberg O, Körner R, McAdam SN, Anthonsen D, Quarsten H, Noren O, Roepstorff P, Thorsby E, Sollid LM 1998 Identification of a gliadin T-cell epitope in coeliac disease: general importance of gliadin deamidation for intestinal T-cell recognition. Scand J Immunol 48: 111–115
Molberg O, Kett K, Scott H, Thorsby E, Sollid LM, Lundin KEA 1997 Gliadin-specific, HLA DQ2-restricted T cells are commonly found in small intestinal biopsies from coeliac disease patients, but not from controls. Scand J Immunol 46: 103–109
Acknowledgements
We thank Dr. A. Osman and Dr. Th. Mothes, Institute of Clinical Chemistry and Pathologic Biochemistry, University of Leipzig, for peptic-tryptic gliadin digests, Dr. H. Wolburg, Institute of Pathology, University of Tuebingen, for electron microscopic studies, Dr. S. Noll, University Children's Hospital, Frankfurt, Dr. B. Moh, Olga-Hospital, Stuttgart, and Dr. D. Müller, University Children's Hospital, Leipzig, for supplying biopsy specimens, and Dr. P.-M. Weber, University Children's Hospital, Tuebingen, for preparing photographs.
Author information
Authors and Affiliations
Additional information
Supported by Deutsche Forschungsgemeinschaft (DFG Ste 305).
Rights and permissions
About this article
Cite this article
Bolte, G., Seilmeier, W., Wieser, H. et al. Enhanced Peptide-Binding Capacities of Small Intestinal Brush Border Membranes in Celiac Disease. Pediatr Res 46, 666 (1999). https://doi.org/10.1203/00006450-199912000-00010
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199912000-00010
