
Abstract
The type II alveolar epithelial cell synthesizes and secretes pulmonary surfactant. Terbutaline enhances phospholipid release from adult and fetal type II cells. Our hypothesis is that the actin network of microfilaments regulates the secretory activity of the type II cell. To examine the developmental regulation of the changes in actin subfractions associated with secretory activity, cultures of type II cells derived from adult and 19-d fetal rat lung were incubated with or without 10 μM terbutaline for 1, 30, and 60 min. Dose-response effects of terbutaline were examined in adult type II cells. Effects of phorbol ester were also examined. Globular (G-actin) and filamentous (F-actin) fractions were extracted from the cells and analyzed separately. Specified cellular equivalent volumes of each subfraction were analyzed by Western blotting, visualized by a color reaction, and quantified by densitometry. There was a decrease in the cytoskeletal F-actin pool along with an increase in the G-actin fraction within 1 min in adult type II cells exposed to terbutaline, indicating that depolymerization of F-actin occurs. Values returned to control levels by 60 min. In contrast, the decrease in F-actin, with a concomitant increase in G-actin, was maximal at 60 min in fetal cells exposed to terbutaline. There was a dose-dependent increase in actin depolymerization with maximal effects at 10 μM terbutaline. Phorbol ester also caused an increase in actin depolymerization. Depolymerization of the actin microfilament network may regulate transport and exocytosis of lamellar bodies in type II cells. We speculate that there is an early secretory mechanism that involves depolymerization of actin microfilaments and a late, actin-independent secretory mechanism present in adult type II cells. The timing of the response of the actin-dependent pathway is developmentally regulated. This may explain the developmental differences in the secretion of surfactant that we have previously shown.
Similar content being viewed by others


Main
The type II alveolar epithelial cell synthesizes and secretes pulmonary surfactant(1, 2). Terbutaline(3, 4) and a variety of other agents (such as purinergic receptor agonists, phorbol esters, cytochalasins, and leukotrienes) have been shown to be potent secretagogues of the surfactant phospholipids(5–10). The enhanced release of surfactant from the type II cell is associated with a substantial reduction in both the number and volume density of lamellar bodies per cell(11, 12). Morphologic evidence indicates that the secretion of lamellar bodies is accomplished by exocytosis. Freeze-etch replicas of the membranes of type II cell show depressions that may represent the sites of discharged lamellae(13). In addition, tonguelike folds are seen, which could be explained as the extensions of cytoplasm that surround the releasing lamellar bodies and that may flap over the exocytosis pit after discharge(13). We have recently shown that basal secretion of surfactant phospholipid increases with morphologic differentiation of the type II cell(4). Furthermore, we have also shown that secretion of phos-phatidylcholine by fetal cells is enhanced by terbutaline, and this process is developmentally regulated(14).
Actin exists in most cells in both a globular, or G form, as well as a filamentous, or F form. Studies in polymorphonuclear leukocytes have revealed that there are two distinct F-actin pools: F-actin which is insoluble in detergent and remains associated with the cytoskeletal framework (cytoskeletal F-actin), and a small pool of polymerized actin-containing aggregates and filaments that is soluble in detergent (noncytoskeletal F-actin)(15). The relative pool sizes of F- and G-actin as well as details of their interaction in type II cells are unknown. Shifts from one form to another may occur rapidly concomitant with changes in cellular function. Actin filaments appear to be involved in the secretion of surfactant. Cytochalasin D, which disrupts filamentous actin, has been shown to augment surfactant phospholipid release from type II cells by more than 3-fold(8). In fetal lung, Whitsett et al.(16) have shown that actin is phosphorylated by a cAMP-dependent protein kinase in vitro. Thus, the effects ofβ-adrenergic agonists on the actin cytoskeleton in type II cells may be mediated by cAMP.
Our hypothesis is that the actin network of microfilaments regulates secretion of surfactant phospholipid in type II cells, and this process is developmentally regulated. The purpose of this study was to investigate the effect of the secretagogue terbutaline on the distribution of actin subfractions (F- and G-actin) in type II cells derived from fetal and adult rat lung.
METHODS
Materials. Adult as well as timed pregnant Sprague-Dawley rats were obtained from Charles River Laboratories, Wilmington, MA. All culture media were purchased from Life Technologies, Inc., Grand Island, NY. All culture supplies and plastics were obtained from Costar, Cambridge, MA. Collagenase and trypsin were obtained from Worthington Biochemical Corp., Freehold, NJ. All other chemicals and reagents were purchased from Sigma Chemical Co., St. Louis, MO. The rat IgG was obtained from Calbiochem-Novabiochem Corp., La Jolla, CA.
Isolation of adult type II cells. The adult type II cells were obtained using the method of Dobbs et al.(17). Each rat was anesthetized using 1 mL of a pentobarbitone solution containing heparin [1.25 mL of pentobarbitone, 0.25 mL of heparin (1000 U/mL), 1 mL 0.9% saline]. After exposure of the contents of the thoracic cavity, the lungs were perfused twice via the right ventricle until the lungs were free of blood using solution II (NaCl 140 mM, KCl 5 mM, NaH2PO4·H2O 2.5 mM, HEPES 10 mM, CaCl2 2 mM, MgSO4·7H2O 1.3 mM; pH 7.4). Then, the lungs were lavaged extensively with solution I (NaCl 140 mM, KCl 5 mM, NaH2PO4·H2O 2.5 mM, HEPES 10 mM, glucose 6 mM, EGTA 0.2 mM; pH 7.4). The lungs were filled with 20 mL of solution II containing elastase (30 U/mL) and incubated in a water bath at 37°C for 5-10 min. Elastase instillation was repeated, and the tissue was incubated in this solution for an additional 20 min. Thereafter, the lungs were dissected from the tracheobronchial tree, minced in the presence of 4 mL of DNase (0.25 mg/mL) and 5 mL of fetal bovine serum, and filtered sequentially through 160- and 15-μm Nitex filters. The resulting cell suspension was centrifuged at 4°C at 180 × g for 10 min. The cell pellet was resuspended in Dulbecco's minimal essential medium, and the type II cells were purified by panning on IgG-coated bacteriologic dishes. The type II cells were plated onto 100-mm2 dishes, each containing approximately 13 × 106 cells and incubated in Dulbecco's minimal essential medium + 10% fetal bovine serum for 18-20 h at 37°C in 10% CO2/room air. Cultures contained 90-95% type II cells.
Isolation of fetal type II cells. The fetal type II cells were obtained by our previously published method(4). Briefly, the lungs of 19-d gestation fetal rats (term is 22 d) were removed, dissected free of connective tissue and nonparenchymal pulmonary tissue, and cultured as explants for 40-48 h in serum-free Waymouth's MB 752/1 medium with penicillin and streptomycin in humidified 95% O2/5% CO2 at 37°C. During this time, endothelial and blood cells do not survive, which is a crucial step in the enrichment of primary cultures of type II cells from fetal lung(4). The explant tissue was then harvested, and the cells dissociated using a solution of collagenase, trypsin, and DNase. The mixed cell suspension was subjected to three differential adhesions (75, 60, and 50 min) to remove contaminating fetal lung fibroblasts. The nonadherent suspension, containing an enriched population of type II cells, was collected and plated at 8-10 × 106 cells/dish in 2 mL of minimal essential medium containing penicillin (100 U/mL), kanamycin (100 μg/mL), and 2% fetal bovines serum. The cells were cultured for 20-22 h at 37°C in 5% CO2/room air. Cultures contained 90-95% type II cells of which >99% were viable as determined by exclusion of the vital dye, trypan blue.
Experimental protocol. After the overnight incubation, adult and fetal type II cells were exposed to 10 μM terbutaline for 1, 30, and 60 min. Controls were incubated in medium alone. We and others have previously shown that 10 μM terbutaline results in maximal stimulation of surfactant secretion in both adult and fetal type II cells, and this continues for at least for 2 h(3, 4, 14). To determine the dose-response relationship between terbutaline exposure and changes in actin subfractions, we incubated type II cells derived from adult rat lung in various concentrations of terbutaline for 1 min, based on the fact that maximal response in adult cells was seen at this time (see“Results”). To examine another secretagogue that acts through a different signal transduction pathway, we incubated type II cells in 10-7 M TPA for 1 min. Control dishes were incubated in medium alone.
Actin extraction. The actin cytoskeleton was extracted using a modification(18) of the method described by Watts and Howard(15). After exposure to terbutaline or medium(control), each plate of cells was washed twice with cold (4°C) PBS. The dishes were kept on ice. The cells were washed once with CSK solution (0.01 M PIPES, 0.3 M sucrose, 0.025 M NaCl, 1 mM EGTA, 5 mM MgCl2), followed by incubation of the cells with cold CSK + 0.2% Triton X-100. The CSK + Triton extract was saved as the Triton-soluble supernatant that contained G-actin and noncytoskeletal F-actin (a small pool of polymerized actin in aggregates and filaments that is sedimented by high speed centrifugation). The cytoskeletal framework containing F-actin remained attached to the dish.
Cytoskeletal F-actin isolation. After washing the plates once more with CSK, the Triton-insoluble cytoskeleton was scraped off the plates in 500 μL of 8 M urea and saved on ice. These samples were vortexed for 10-20 s and spun in a microcentrifuge for 5 min, and the supernatant was stored at-70°C. This contained the cytoskeletal F-actin fraction.
Noncytoskeletal F-actin isolation. The Triton-soluble extract was centrifuged at 108,000 × g for 50 min at 4°C (Beckman SW50.1 Ti). The supernatant was collected as the high speed supernatant (that contained G-actin) and processed as described below. The pellet, containing the noncytoskeletal F-actin fraction, was suspended in 50 μL of 8 M urea and stored at -70°C.
G-actin isolation. The high speed supernatant, containing G-actin, was precipitated overnight in 5 volumes of methanol at -70°C. It was then centrifuged at 10,000 × g for 1 h at 4°C. The precipitate, which contains the G-actin fraction, was suspended in 25 μL of 8 M urea and stored at -70°C.
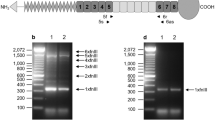
Measurement of actin subfractions. Cellular equivalent volumes of the subfractions were separated by SDS-PAGE(19) and analyzed by Western blotting using a monoclonal mouse anti-actin antibody. These amounts of the subfractions were not at saturating levels for Western analysis (see Fig. 1, lanes 2 through 4). Actin bands were visualized using 5-bromo-4-chloro-3-indolylphosphate p-toluidine salt and nitro blue tetrazolium chloride and then quantified using densitometry. The percentage of the total actin pool for each fraction was calculated by correcting the area of each peak obtained by the densitometer for the volume equivalent loaded on the gel for each actin subfraction.

Immunoblot characterization of anti-actin antibody. Ten micrograms of pure actin (lane 1), cytoskeletal F-actin isolated from 20× 106 type II cells (lanes 2-4), and whole type II cell homogenates (lane 5) were loaded onto SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were probed with anti-actin MAb and visualized as described in “Methods.” Lane 2, 1/64 of total cellular cytoskeletal F-actin content; lane 3, 1/32 of total cellular cytoskeletal F-actin content; lane 4, 1/16 of total cellular cytoskeletal F-actin content; lane 5, 1/32 of total cell homogenate.
Statistics. Data are expressed as percent of the total actin pool for each cell type under control or experimental conditions. Results reflect the mean values (± SEM) of three to four experiments. Percent change was calculated as treated minus control values; and negative values indicate a decrease in this fraction when exposed to terbutaline. Statistical analyses were done using the t test(20). Differences were considered significant when p < 0.05.
RESULTS
Specificity of the MAb to actin was confirmed because the antibody recognized actin purified from chicken gizzard (shown in lane 1,Fig. 1) and recognized total cellular actin in a type II cell homogenate without reacting to other proteins (visualized as a single band in lane 5, Fig. 1). Quantitative comparisons are possible because the assays were conducted using amounts of the cellular subfractions of actin that were not at saturating levels for the antigen-antibody system (various amounts of cytoskeletal F-actin shown in lanes 2-4 in Fig. 1). This was also true for noncytoskeletal F- and G-actin (data not shown).
The distributions of the actin subfractions were similar for the adult and fetal type II cells in the absence of terbutaline. The cytoskeletal F-actin fraction accounted for 86.1 ± 2.3%, noncytoskeletal F-actin comprised 5.5 ± 1.0%, and G-actin represented 8.4 ± 1.9% of the total cellular actin pool in the basal state (up to 60 min) in the adult type II cells. In the fetal type II cells the values were 87.7 ± 1.8%(cytoskeletal F-actin), 3.0 ± 0.9% (noncytoskeletal F-actin) and 9.7± 1.4% (G-actin), respectively.
There were no changes in the noncytoskeletal F-actin subfractions in both adult and fetal type II cells exposed to terbutaline. Significant changes in the cytoskeletal F- and G-actin subfractions were seen in both adult and fetal type II cells exposed to terbutaline, as depicted in Figures 2 and 3. In the adult type II cells (Fig. 2), there was no significant change in the distribution of the actin subfractions in the control group over time (1, 30, and 60 min). There was a significant reduction in the cytoskeletal F-actin fraction from 87 to 63% of total cellular actin (p < 0.02) with a concomitant increase in the G-actin fraction from 6.5 to 34% of total cellular actin (p < 0.003) after 1 min of exposure to terbutaline, as shown in Figure 2. The maximal response was seen at 1 min. A similar response was seen at 30 min, which was less in magnitude compared with that at 1 min, although still statistically significant (p < 0.02) compared with control values. At 60 min, the values were not different from the control group.

Alterations in the distribution of cytoskeletal F-actin and G-actin in adult type II cells exposed to 10 μM terbutaline for 1, 30, and 60 min. No significant changes were observed with noncytoskeletal F-actin(data not shown). Data (mean ± SEM, n = 3) are expressed as percentage of the total cellular actin pool. *p < 0.02vs control, **p < 0.003 vs control.#p < 0.03 vs 1 min, ##p < 0.02vs 1 min.

Alterations in the distribution of cytoskeletal F-actin and G-actin in fetal type II cells exposed to 10 μM terbutaline for 1, 30, and 60 min. No significant changes were observed with noncytoskeletal F-actin(data not shown). Data (mean ± SEM, n = 3-4) are expressed as percentage of the total cellular actin pool. *p < 0.0001vs control, **p < 0.04 vs control.#p < 0.009 vs 1 min, @p < 0.0001vs 1 min.
The response of the fetal cells to terbutaline is depicted in Figure 3. Once again, there was no significant change in the distribution of the actin subfractions over time in the control group. However, there was a biphasic response (seen at 1 and 60 min of exposure to terbutaline) in the changes of the actin subfractions. There was a significant(p < 0.0001) decrease in the cytoskeletal F-actin fraction from 88 to 77% of total cellular actin with a simultaneous increase in the G-actin fraction from 10 to 21% of total cellular actin at 1 min of exposure to terbutaline. After 30 min of exposure to terbutaline, the distribution of actin subfractions was similar in the control and terbutaline-exposed cells. After 60 min of exposure to terbutaline, there was a second significant(p < 0.04) decrease in the cytoskeletal F-actin subfraction from 87.5 to 63% of total cellular actin with a concomitant increase in the G-actin from 10 to 31% of total cellular actin (p < 0.04). Exposure of fetal cells to terbutaline resulted in maximal changes in G-actin and stable F-actin after 60 min.
Figure 4 shows that depolymerization of actin was dose-dependent. There was a 2.4-fold increase in G-actin in adult type II cells exposed to 0.01 μM terbutaline compared with controls. At 0.1 μM terbutaline, G-actin increased over 3-fold. The maximal response resulted in a 12-fold increase in G-actin at 10 μM terbutaline. There was a decrease in the response to 100 μM terbutaline, which was still 1.9-fold greater than control. This decrease in response to higher concentrations of terbutaline is also seen with secretion of phosphatidylcholine (see“Discussion”).

Concentration dependent increase in actin depolymerization in type II cells isolated from adult rat lung. Cells were exposed to various concentrations of terbutaline for 1 min and actin subfractions were analyzed. Data (means from two to three experiments) are expressed as treated/control ratio of G-actin pool.
We next asked whether the changes in actin subfractions in response to secretagogues would be observed using a secretagogue that acts through a different signal transduction pathway. We found that 10-7 M TPA caused an increase in G-actin in type II cells derived from adult rat lung after 1 min of exposure (115.3 ± 5.7% of control, n = 3, p= 0.05).
DISCUSSION
Studies in polymorphonuclear leukocytes have shown that there are two distinct F-actin pools: 1) a cytoskeletal-associated F-actin pool, which is Triton-insoluble, and 2) a noncytoskeletal F-actin pool, which is Triton-soluble(15). In polymorphonuclear leukocytes, the cytoskeletal F-actin pool accounts for 10-30% of total cellular actin, the noncytoskeletal F-actin pool accounts for 40-45% of the total cellular actin pool, and G-actin accounts for 30% of the total cellular actin(15). We have shown that cytoskeletal F-actin, noncytoskeletal F-actin, and G-actin subfractions of the total cellular actin pool are present in type II cells from adult and fetal rat lungs. We found that the cytoskeletal F-actin fraction accounted for 86.1 ± 2.3% of the total cellular actin pool, noncytoskeletal F-actin for 5.5 ± 1.0%, and G-actin 8.4 ± 1.9% in type II cells derived from adult rat lung after 20-24 h in culture. In the fetal type II cells the values were 87.7 ± 1.8% (cytoskeletal F-actin), 3.0 ± 0.9% (noncytoskeletal F-actin), and 9.7 ± 1.4% (G-actin), respectively. It is interesting to note that there is more of the cytoskeletal F-actin fraction in the type II cells compared with that in polymorphonuclear leukocytes. This could possibly reflect the different specialized functions with which the cells are involved, such as ameboid movement and phagocytosis (polymorphonuclear leukocytes) and secretion (type II cells).
After exposing cultures of type II cells derived from adult and fetal rat lung to terbutaline, we found an increase in the G-actin fraction concomitant with a decrease in the cytoskeletal F-actin fraction. This happens within 1 min of exposure to terbutaline. In the adult type II cells, the maximal effect was seen at 1 min of exposure to terbutaline. No major change was found in the noncytoskeletal F-actin fraction at any time examined. After 60 min, despite continued stimulation by terbutaline, the various actin pools show no significant difference compared with the controls. The fetal type II cells, on the other hand, behave in a different fashion. Significant changes are noted after exposure to terbutaline in the cytoskeletal F-actin (88% decreases to 77%) and G-actin fractions (10% increases to 21%) at 1 min. At 30 min no significant differences are noted in the actin fractions in the terbutaline-exposed versus the control fetal type II cells. However, the maximal effect was seen at 60 min of continuous stimulation by terbutaline. These changes in fetal cells after 60-min exposure to terbutaline are similar in magnitude to the changes in adult cells after 1-min exposure to terbutaline. Therefore, terbutaline causes depolymerization of cytoskeletal F-actin in type II cells, and these effects are developmentally regulated.
Tsilibary and Williams(21) demonstrated that actin filaments are found in intimate association with lamellar bodies, close to the cell surface of the type II cell. When stimulated with a β-adrenergic agonist (isoproterenol), more exocytic profiles were observed, the number of lamellar bodies were decreased, and the number of S1-labeled(subfragment 1 of myosin) actin filaments were markedly increased(21, 22). Pretreatment of the cells with cytochalasin D disrupts actin filaments and completely inhibits the isoproterenol-induced loss of intracellular lamellar bodies(22). This suggests that actin polymerization is associated with β-adrenergic stimulation and release of surfactant. On the contrary, we found that actin depolymerization is associated withβ-adrenergic stimulation. The difference in our results could be due to differences in methodology, such as in vivo versus in culture exposure to the β agonist or ultrastructural qualitative measurement of actin versus quantitative measurement of all cellular actin pools. It is possible that S1 labels both cytoskeletal F-actin and noncytoskeletal F-actin, the smaller of which is removed by our method.
Our results are consistent with those of Rice et al.(8) who found that cytochalasin D, which disrupts actin filaments, enhanced surfactant release from type II cells by more than 3-fold. In fetal lung, Whitsett et al.(16) have shown that actin is phosphorylated by a cAMP-dependent protein kinase in vitro. Thus, the effects of β-adrenergic agonists on the actin cytoskeleton in type II cells may be mediated by cAMP. Phosphorylation of actin inhibits polymerization of G-actin in vitro, supporting the hypothesis that actin phosphorylation may alter its function(23). Because actin depolymerization and polymerization are dynamic processes occurring simultaneously, factors that reduce polymerization will favor a net balance toward depolymerization. This also supports our finding that depolymerization occurs with β adrenergic stimulation.
β-Adrenergic agonists have been widely studied stimulants of surfactant secretion in type II cells derived from fetal and adult lung.β agonists enhance secretion of surfactant phospholipid in a time-dependent and dose-dependent manner(3, 4, 7). We have previously shown that the β agonist terbutaline stimulates secretion of surfactant phospholipid by fetal type II cells(4), and this process is developmentally regulated(14). In the current study we have shown that terbutaline stimulates actin depolymerization in adult and fetal type II cells. We also found a dose-dependent increase in actin depolymerization in type II cells in response to terbutaline, with maximal response at a concentration of 10 μM. This relationship exactly parallels the dose-dependent increase in phosphatidylcholine secretion in fetal(14) and adult type II cells(24). We(14) and others(24) have also found a decrease in phosphatidylcholine secretion by type II cells at 100 μM terbutaline.
It is possible that the changes in actin subfractions are not causally related to secretion of surfactant. We believe this to be unlikely, however, in view of the strong association between actin filaments and lamellar bodies(where surfactant is stored) in type II cells and the changes observed on stimulation with β-adrenergic agonists. The dose-dependent effects of terbutaline on both actin depolymerization and phosphatidylcholine secretion support our hypothesis that the actin network of microfilaments regulates secretion of surfactant phospholipid by type II cells. In view of the relationship observed between secretory activity and the microtubular-microfilamentous system in parotid as well as pancreatic islet cells(25–27), it is not unreasonable to suggest that the changes in actin subfractions that we found may be causally related to the secretion of surfactant from type II cells.
We found that TPA, which stimulates protein kinase C, also caused actin depolymerization. The small changes we observed are possibly due to the short exposure time which may not have allowed much entry of TPA into the cells to its site of action. Nevertheless, the stimulation of depolymerization by agents which activate protein kinase A (terbutaline) and protein kinase C(TPA) further supports our hypothesis that actin depolymerization may be the“final common pathway” for regulated phosphatidylcholine secretion by type II cells.
Insulin release in response to glucose concentration occurs as a biphasic phenomenon. This may be a result of the separation of insulin secretory granules within the cell into two pools(26). Malaisse et al.(27) have proposed that the “early component” corresponds to the mobilization of secretory granules already located in close vicinity to the microfilamentous cell web. The cell web is formed by a network of actin microfilaments, which may allow exocytosis of secretory granules when depolymerization occurs. They believe that the“late component” corresponds to the mobilization of secretory granules whose transport toward the cell membrane is under the control of the microtubular apparatus. It is likely, therefore, that the participation of the microtubular-microfilamentous system in insulin release indeed represents a physiologic basis for the functional segregation of secretory granules into two different pools(27).
In the adult type II cells, on stimulation with terbutaline, the maximal response is seen at 1 min. Because, as previously shown by us and others(3, 4), the secretion of surfactant continues for at least 2 h, it may be possible that at least two pools of surfactant exist: one released by an actin-dependent process (“early release” pool) and the other released by an actin-independent mechanism (“late release” pool). There is some pharmacologic evidence for two tissue pools of surfactant(28, 29). The first (what we refer to as the early release pool) is relatively small, turns over rapidly, and is under the control of the sympathetic nervous system(29). The second (the late release pool) is larger and is released in response to an increase in tidal volume(29). Power et al.(30) have isolated two subfractions of the lamellar bodies, each of which contains the full spectrum of surfactant phospholipids. There is evidence that the two fractions can be released differentially(31). It has been shown that, although salbutamol stimulates secretion of surfactant, the maximum amount released is smaller (what we refer to as the early release pool) and appears to emanate from a tissue pool that is more highly radiolabeled than that released by increasing tidal volume(28).
The response of the fetal cells to terbutaline is more complex. Changes in the actin subfractions are seen at both 1 and 60 min. The maximal effect is seen at 60 min. We have recently shown that secretion of surfactant phospholipid by fetal type II cells is developmentally regulated(4, 14). The later maximal effect of terbutaline on actin depolymerization in fetal cells might reflect the developmental immaturity of the actin-dependent mechanism of the release of surfactant.
We have found an association only between terbutaline-stimulation of type II cells and changes in actin. We have, as yet, not shown a cause-effect relationship between changes in actin subfractions and surfactant secretion. Although these experiments are snapshots in time, they allow us to theorize the possible role of actin in the release of the lamellar bodies by type II cells. It may be possible that the tonguelike folds of cytoplasm, seen at the time of release of the lamellar bodies during the process of exocytosis(13), may be changing shape with the depolymerization of cytoskeletal F-actin to G-actin allowing it to effect release of the lamellar bodies, similar to the release of insulin from the pancreatic β-cell(26). Recently, annexins have been shown to be involved in phosphatidylcholine secretion by type II cells(32). These proteins bind to cytoskeletal proteins as well as phospholipids. We speculate that depolymerization of F-actin may release the annexin-bound phospholipid, so that fusion of the lamellar body with the plasma membrane and exocytosis can occur.
In summary, we have demonstrated that actin subfractions (cytoskeletal F-actin, noncytoskeletal F-actin, and G-actin) exist in both fetal and adult type II cells. On exposure to a known secretagogue of surfactant, terbutaline, there is marked depolymerization of F-actin to G-actin. This process seems to be developmentally regulated.
Abbreviations
- HEPES:
-
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- PIPES:
-
piperazine-N,N′-bis(2-ethanesulfonic acid)
- TPA:
-
tetradecanoyl phorbol acetate
References
Kresch MJ, Gross I 1987 The biochemistry of fetal lung development. Clin Perinatol 14: 481–507
Rooney SA 1985 The surfactant system and lung phospholipid biochemistry. Am Rev Respir Dis 132: 439–460
Mettler NR, Gray ME, Schuffmann S, LeQuire VS 1981 β-Adrenergic induced synthesis and secretion of phosphatidylcholine by isolated pulmonary alveolar type II cells. Lab Invest 45: 575–586
Kresch MJ, Dynia DW, Gross I 1987 Culture of differentiated and un-differentiated type II cells from fetal rat lung. Biochim Biophys Acta 930: 19–32
Dobbs LG, Mason RJ 1978 Stimulation of secretion of disaturated phosphatidylcholine from isolated alveolar type II cells by 12-O-tetradecanoyl-13-phorbol acetate. Am Rev Respir Dis 118: 705–713
Gilfillan AM, Rooney SA 1987 Functional evidence for involvement for adenosine A2 receptor regulation of phosphatidylcholine secretion in cultured type II pneumocytes. J Pharmacol Exp Ther 241: 907–914
Griese M, Gobran LI, Rooney SA 1992 Ontogeny of surfactant secretion in type II pneumocytes from fetal, newborn, and adult rats. Am J Physiol 6: 262:L337–L343
Rice WR, Osterhoudt KC, Whitsett JA 1984 Effect of cytochalasins on surfactant release from alveolar type II cells. Biochim Biophys Acta 805: 12–18
Gilfillan AM, Rooney SA 1985 Arachidonic acid metabolites stimulate phosphati-dylcholine in primary cultures of type II pneumocytes. Biochim Biophys Acta 833: 336–341
Suwabe A, Mason RJ, Voelker DR 1991 Temporal segregation of surfactant secretion and lamellar body biogenesis in primary cultures of rat alveolar type II cells. Am J Respir Cell Mol Biol 5: 80–86
Olsen DB 1972 Neurohumeral-hormonal secretory stimulation of pulmonary surfactant in the rat. Physiologist 15: 230
Massaro D, Clerch L, Massaro G 1982 Surfactant secretion: evidence that cholinergic stimulation of secretion is indirect. Am J Physiol 243:C39–C45
Ryan US, Ryan JW, Smith DS 1975 Alveolar type II cells: studies on the mode of release of lamellar bodies. Tissue Cell 7: 587–599
Kresch MJ, Lima DM, Lu H 1996 Developmental regulation of phospholipid secretion by fetal type II pneumocytes. Biochim Biophys Acta 1299: 39–46
Watts RG, Howard TH 1992 Evidence for a gelsolin-rich, labile F-actin pool in human polymorphonuclear leukocytes. Cell Motil Cytoskeleton 21: 25–37
Whitsett JA, Hull W, Dion C, Lessard J 1985 cAMP dependent actin phosphorylation in developing rat lung and type II epithelial cells. Exp Lung Res 9: 191–209
Dobbs LG, Gonzalez R, Williams MC 1986 An improved method for isolating type II cells in high yield and purity. Am Rev Respir Dis 134: 141–145
Pachter JS 1992 Association of mRNA with the cytoskeletal framework: its role in the regulation of gene expression. Crit Rev Eukaryot Gene Expr 2: 1–18
Laemmli UK 1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685
Daniel WW 1974 Biostatistics: A Foundation for Analysis in the Health Sciences. Wiley & Sons, New York
Tsilibary EC, Williams MC 1983 Actin and secretion of surfactant. J Histochem Cytochem 31: 1298–1304
Tsilibary EC, Williams MC 1983 Actin in peripheral rat lung: S1 labeling and structural changes induced by cytochalasin. J Histochem Cytochem 31: 1289–1297
Grazi E, Magri E 1979 Phosphorylation of actin and removal of its inhibitory activity on pancreatic DNase I by liver plasma membranes. FEBS Lett 104: 284–286
Gilfillan AM, Lewis AJ, Rooney SA 1987 Effects of thiazinium chloride and other antihistamines on phosphatidylcholine secretion in rat type II pneumocyte cultures. Biochem Pharmacol 36: 277–281
Perrin D, Moller K, Hanke K, Soling H-D 1992 cAMP and Ca2+-mediated secretion in parotid acinar cells is associated with reversible changes in the organization of the cytoskeleton. J Cell Biol 116: 127–134
Grodsky G 1972 A threshold distribution hypothesis for packet storage of insulin and its mathematical modeling. J Clin Invest 51: 2047–2059
Malaisse WJ, Obberghen EV, Devis G, Somers G, Ravazzola M 1974 Dynamics of insulin release and microtubular-microfilamentous system. V. A model for the phasic release of insulin. Eur J Clin Invest 4: 313–318
Nicholas TE, Power JHT, Barr HA 1982 Surfactant homeostasis in the rat lung during swimming exercise. J Appl Physiol Respir Environ Exercise Physiol 53: 1521–1528
Barr HA, Nicholas TE, Power JHT 1988 Control of alveolar surfactant in rats at rest and during prolonged hyperpnoea: pharmacological evidence for two tissue pools of surfactant. Br J Pharmacol 93: 473–482
Power JHT, Barr HA, Nicholas TE 1988 Surfactant-associated 15- and 35-kDa proteins are concentrated in different organelles in rat lung tissue. Exp Lung Res 13: 209–224
Nicholas TE, Power JHT, Barr HA 1987 Differential release of two surfactant-containing fractions in rat lung. Fed Proc 46: 813
Liu L, Wang M, Fisher AB, Zimmerman UP 1996 Involvement of annexin II in exocytosis of lamellar bodies from alveolar epithelial type II cells. Am J Physiol 270:L668–L676
Author information
Authors and Affiliations
Additional information
Supported by grants from the American Lung Association (M.J.K.) and the American Heart Association (M.J.K.).
Rights and permissions
About this article
Cite this article
Bhandari, V., Lu, H., Pachter, J. et al. Actin Depolymerization Is Developmentally Regulated in Rat Type II Cells Exposed to Terbutaline. Pediatr Res 41, 166–171 (1997). https://doi.org/10.1203/00006450-199702000-00002
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199702000-00002
