
Abstract
Hyperprostaglandin E syndrome (HPS), the prenatal variant of Bartter's syndrome, is characterized by a marked and selective stimulation of prostaglandin E (PGE2) synthesis. In the study group HPS patients showed increased urinary levels of PGE2, an index of renal, and of 11α-hydroxy-9,15-dioxo-2,3,4,5,20-pentanor-19-carboxyprostanoic acid(PGE-M), an index of systemic PGE2 synthesis of 470% and of 570%, respectively. In addition, plasma concentration of PGE-M was also elevated 6.3-fold when compared with a control group. The urinary levels of other prostanoids were unaltered. During indomethacin treatment in both groups prostanoid excretion rates were suppressed to similar levels. To investigate the origin of stimulated prostanoid biosynthesis in HPS patients CD14+ monocytes were isolated from plasma samples, and the prostanoid synthesis was analyzed. The pattern and amounts of metabolites synthesized from endogenous arachidonic acid pools did not vary significantly between monocytes of the HPS and the control group. Thromboxane A2 (TXA2) was formed as the major prostanoid product. Using PGH2 as an exogenous substrate, again no difference in PGE2 biosynthesis was observed, indicating no difference in PGE-synthetic activity between both groups. Additionally, mRNA expression analysis of CD14+ monocytes via RT-PCR delineated the constitutive expression of cyclooxygenase-1, cyclooxygenase-2, and thromboxane synthase mRNA in cells from HPS patients and controls without statistical differences between these two groups. In conclusion, our data show that monocytes are not the source for the increased PGE2 biosynthesis in children with HPS, and a genetic defect in PGE synthesis can be excluded as the primary event in the pathogenesis in HPS.
Similar content being viewed by others

Main
The HPS or antenatal variant of Bartter's syndrome is known as an inherited, autosomal recessive disorder with renal as well as systemic clinical symptoms(1). The renal aspect is characterized by iso- or hyposthenuric polyuria, leading to polyhydramnios and premature birth, salt wasting associated with hypokalemic metabolic alkalosis, hypercalciuria with subsequent nephrocalcinosis, and hyperprostaglandinuria(2–6). Systemic manifestations related to this disease are recurrent diarrhea and vomiting, episodes of fever and convulsions, progressive osteopenia, normal blood pressure despite hyperreninismus, and a failure to thrive. The excessive formation of PGE2 is indicated as an important pathologic role in HPS, as the long-term administration of indomethacin, an inhibitor of PG formation, significantly improves HPS symptoms and is associated with normal growth(2, 7, 8).
The intracellular target of nonsteroidal antiinflammatory drugs is cyclooxygenase, a rate-limiting enzyme in the arachidonic acid cascade, forming the endoperoxide PGH2, which is then metabolized into PGs and TX by specifically associated enzymes. Recently it has been recognized that two isoforms of this enzyme exist: a constitutively expressed cyclooxygenase-1 and an inducible isoform, cyclooxygenase-2 [for review, see Otto and Smith(9)]. Whereas the enzymes synthesizing PGD2, PGF2α, TXB2, and PGI2 have also been identified biochemically and genetically, the nature of the PGE2-forming protein remains elusive.
The elevation in PGE2 formation and the associated renal and far more the extrarenal clinical symptoms point toward the cyclooxygenase or the PGE synthase as a possible genetic defect in this hereditary disease. Further, the therapeutical benefit of cyclooxygenase inhibition implies that in HPS patients an alteration in the biosynthetic capacity may be responsible for the excessive PGE2 formation. Several biochemical changes may explain this observation: 1) an up-regulation of cyclooxygenase enzymes increasing substrate availability for the metabolism toward PGE2,2) an increase in the PGE2-synthetic activity, 3) an altered prostanoid profile favoring the formation of PGE2 in specific cells, or 4) a continuous stimulation of the PGE2-forming system in specific cells.
To investigate these possibilities we analyzed the biosynthesis of prostanoids and the expression of responsible enzymes in blood monocytes derived from HPS patients in comparison with control subjects. We decided to study monocytes for two reasons: first, monocytes are spread throughout the body, including gastrointestinal tract, brain, and bone, and therefore could function as a cell population representing so-called index cells for the total organism, and second, these cells are easily accessible from blood by the effective method of magnetic activated-cell sorting.
METHODS
Subjects. Nine children [age, 8 mo to 14 y (median, 4 y); weight, 8.6-59 kg (median, 15.7 kg); size, 74-157 cm (median, 103 cm); 4 female and 5 male patients] with clinical and biochemical determinants of HPS were enrolled, according to the following criteria: premature birth associated with polyhydrammios, hypokaliemia (<3.3 mmol/L), fractional excretion rate of K+ > 15%, hyposthenuria (<350 mosm/kg), hypercalciuria(>0.15 mmol/kg/d), normomagnesemia (0.7-1.1 mmol/L), increase in renin(100% above upper limit), and increase in urinary PGE2 (100% above upper limit). Six of these patients [age, 3-14 y (median, 4 y); weight, 15.6-59 kg (median, 16 kg); size, 101-157 cm (median, 113 cm); 4 female and 2 male patients] were enrolled in the study omitting indomethacin. As controls we did not include healthy age-matched children for ethical reasons. Instead six healthy female volunteers [age, 16-36 y (median, 22 y); weight, 45-63 kg(median, 54 kg); size, 164-171 cm (median, 166 cm)] served as controls. The investigations with children and healthy adult human donors were performed according to the principles of the revised Declaration of Helsinki. Approval of the local Ethics Committee was obtained, and each volunteer or the parents gave written informed consent before the study was started.
Study design. HPS patients were on long-term indomethacin treatment with two single doses/d (3 mg/kg of body weight). They achieved a median plasma level of 1088 ng/mL 4 h after indomethacin application (>500 ng/mL is regarded as the effective level)(10). Four hours after the last application, a sample of 4 mL of EDTA-anticoagulated blood was taken by peripheral venous puncture and further processed for monocyte isolation as described subsequently. At 72 h after the last dose of indomethacin, another 4-mL blood sample was taken and processed under the same conditions. Controls were kept for 2 d on indomethacin treatment (3 mg/kg of body weight), and the time of blood taking was similar to that of HPS patients. For determination of urinary prostanoids, during the study urine samples were collected every 6 h and processed for prostanoid determination by the mean of GC-MS/MS. Data were presented as prostanoid excretion rates per 24 h.
Isolation of blood monocytes. A total volume of 4 mL of EDTA-anticoagulated blood was taken for preparation of blood monocytes. The plasma was separated from the cellular components by centrifugation. The pellet was adjusted and resuspended to a total volume of 6 mL with PBS/EDTA(pH 7.3) and layered over 3 mL of Ficoll-Hypaque 1.077 (Biochrom, Berlin, Germany). After centrifugation the resulting interphase with the mononuclear cell fraction was collected and diluted with PBS/EDTA. The mononuclear cell fraction was washed twice and afterward resuspended in a final volume of 50μL of PBS/EDTA. Anti-CD14 antibodies, 30 μL, coupled onto microbeads(Miltenyi Biotech, Bergisch Gladbach, Germany) were added, and cells were incubated for 30 min at 8 °C. After dilution with PBS/EDTA/BSA (pH 7.3) the cell suspension was placed on a preequilibrated separation column for depletion of the CD14-bearing monocytes. CD14+ monocytes were expulsed from the column apart from the magnet with a rubber stamp. The use of only single density gradient centrifugation is not capable of producing a comparable specific cellular enrichment (data not shown). Furthermore, the monocyte isolation procedure by conventional cellular adherence on tissue culture flasks takes several hours and in turn increases the risk of the artificial induction of cyclooxygenase-2 transcription and protein translation. The viability determined by trypan blue exclusion was >95%, and cells were directly subjected to the stimulation assay. Morphologic fluorescence-activated cell sorter analyses of monocytes after magnetic-activated cell sorting separation revealed a cell purity of>98%.
Stimulation assay of blood monocytes. To obtain prostanoid formation of endogenous arachidonic acid pools 3 × 105 CD14+ monocytes were resuspended in PBS/Ca2+ buffer (pH 7.3) and were stimulated with 2 μM A23187 or vehicle (DMSO) at 37 °C for 10 min in the presence of 5 mM CaCl2. The incubation was terminated by centrifugation of the cell suspension at 4 °C. The cell-free supernatants were stored at -80 °C until prostanoid measurement by GC-MS/MS. PGE2-synthetic activity was measured by the exogenous addition of PGH2 (Cayman Chemicals, Gif, France) as described recently(11).
Prostanoid measurement. The stable hydrolysis products of prostacyclin and TX, 6-keto-PGF1α, and TXB2, as well as the PGs PGE2, PGD2, PGF2α, and PGE-M were determined in the cell-free supernatants and in urine and plasma samples by GC-MS/MS as described by Schweer et al.(12).
Indomethacin determination. The plasma levels of indomethacin were determined by HPLC as described in Seyberth et al.(13).
RNA preparation and RT-PCR. Total RNA was isolated from CD14+ monocytes using the guanidinium thiocyanate method with acidic phenol(14). Reverse transcription and PCR were performed as described recently(15, 16). Briefly, 1 μg of total RNA was used for target-specific reverse transcription with Superscript RT (GIBCO BRL, Eggenstein, Germany) and the primers PCOX1R1 (5′-AGAGGGGCTCTGTGGATGGTCG-3′) and PCOX2R1(5′-ACAGTTCAGTCGAACGTTC-3′). PCR amplification was performed using cDNA with the primer PCOX1F1 (5′-CAGCCCTTCAATGAGTACCG-3′) and PCOX1R2 (5′-AGGCCCATCATCCTGACTGGCAT-3′) for COX-1 and PCOX2F1(5′-AATGAGTACCGCAAACGCTT-3′) and PCOX2R2(5′-TAATCATCTAGTCCGGAGCGG-3′) for COX-2. The reactions were cycled in a profile of 30 s at 94 °C, 30 s at 56 °C, and 30 s at 72 °C after a 5-min denaturing step at 95 °C.
In an identical way the primer PTXSHR1(5′-GCGGGATACGATCTTGATATAGACACC-3′) for TX synthase and PBAR1(5′-CTAGAAGCATTTGCGGTGGAC-3′) for β-actin were used to reverse transcribe the RNA. PCR amplification was performed for TX synthase with PTXSHR2 (5′-GGA TTCTAGCTGCAGCGGTAC-3′) and PTXSHF1(5′-AGATTCACACGGGAGGCAGCT-3′), for β-actin with PBAR1 and PBAF1 (5′-CATCACCATTGGCAATGAGCG-3′).
Amplification products were analyzed by 1.5% agarose gel electrophoresis and ethidium bromide staining. The identity of the fragments was evaluated by their molecular mass and dideoxy sequencing. Furthermore, samples were assayed at various dilutions to ensure proportionality in the yield of PCR products. Luminescence ratios of the PCR products for cyclooxygenase-1, cyclooxygenase-2, and TX synthase relative to the β-actin were evaluated as quotient numbers.
Statistics. Analysis of variance with repeated measurements were performed to test the main effects (prostanoid production and inhibition by indomethacin) using the appropriate between- and within-subject error terms. Results are expressed as single point demonstration and median values. Statistical differences were obtained by Wilcoxon signed-rank test for related samples. p values <0.05 were considered significnat.
RESULTS
Prostanoid measurement in urine and plasma from HPS patients versus control. Because one of the main biochemical characteristics in HPS is an elevation of PGE2, we analyzed the urinary concentrations of PGE2 and urinary as well as plasma levels of its catabolic metabolite PGE-M during indomethacin treatment and after its withdrawal.
The results of the urinary excretion of PGE2 and PGE-M, the predominant metabolites of the cyclooxygenase biosynthesis in HPS patients and controls, are given in Figure 1. Withdrawal of indomethacin in HPS patients for 3 d caused a significant and selective elevation of PGE-M from 378 to 1454 ng/h/1.73 m2 body surface(p < 0.05) and of PGE2 from 5 to 70 ng/h/1.73 m2 body surface (p < 0.05) (Fig. 1A). Simultaneously, a decrease of indomethacin in plasma from 1661 ng/mL (median) to undetectable levels after 72 h of withdrawal was observed (data not shown). Prostanoids different from PGE-M and PGE2 like TXB2 and its metabolites 2,3-dinor-TXB2 and 11-dehydro-TXB2, PGF2α, and 6-keto-PGF1α showed only marginal differences with or without indomethacin in the urine of HPS patients (data not shown). Also in the control group indomethacin caused a significant decrease in the urinary excretion of PGE2 from 20 to 6 ng/h/1.73 m2 (p < 0.05) and of PGE-M from 253 to 214 ng/h/1.73 m2 (p < 0.05) (Fig. 1B).

Prostanoid biosynthesis in urine from HPS-patients vs controls. Changes in urinary excretion of PGE2 and PGE-M in the presence and 72 h after withdrawal of oral indomethacin in HPS children(A) (n = 6) and control subjects (B)(n = 6). Urine was collected for 24 h. Data are given as single case values, and the median is indicated by the filled circle.
To further demonstrate the in vivo production of PGE2 we performed GC-MS/MS analysis of plasma samples from controls and HPS patients. PGE-M served as an indicator for systemic PGE2 formation, because direct measurement of PGE2 is difficult to perform in plasma owing to unspecific elevation of prostanoids that results from local tissue trauma during blood sampling(17). As shown in Table 1 PGE-M of HPS patients and controls were significantly suppressed by indomethacin to similar levels. In the absence of indomethacin, PGE-M in plasma samples of HPS patients was 9-fold increased compared with a 1.5-fold increase in the control group.
In vitro stimulation of CD14+monocytes and associated prostanoid biosynthesis. Monocytes represent a major source for PG formation in blood. Platelets are known to synthesize mainly TXA2 after stimulation, and other blood cells, such as lymphocytes and granulocytes, are devoid of cyclooxygenase enzymes and therefore unable to synthesize prostanoids.
To analyze prostanoid synthesis we used blood monocytes from HPS patients and control subjects. Owing to the limited availability of blood samples from very young children, a specific and efficient approach to isolate monocytes was necessary. For this purpose we performed cell isolation with an anti-CD14 affinity column (magnetic-activated cell sorting) to gain a highly pure fraction of blood monocytes. The CD14 antigen, which represents the LPS receptor, is expressed on the cell surface of blood monocytes.
In the following, monocytes were stimulated with 2 μM Ca2+ ionophor A23187 to analyze the endogenous prostanoid biosynthesis (Fig. 2). The major metabolite of stimulated cells of controls was TXB2 (median, 167.1 pg/105 cells), whereas PGE2 (median, 10.5 pg/105 cells) and PGD2 (median, 14 pg/105 cells) were synthesized in much lower concentrations (Fig. 2B). The formation of PGF2α and 6-keto-PGF1α in monocytes was negligible. Monocytes prepared from volunteers treated with oral indomethacin for 2 d (2 mg/kg of body weight) demonstrated a complete suppression of eicosanoid production in vitro (>99% for all metabolites measured).

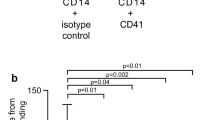
Prostanoid biosynthesis in CD14+ monocytes from HPS patients vs controls. CD14+ monocytes were isolated from peripheral blood by the method of magnetic-activated cell sorting. CD14+ monocytes(3 × 105) were stimulated in PBS/5 mM CaCl2 with 2 μM A23187 for 10 min at 37 °C. Stimulation assays were terminated with centrifugation at 4 °C. Resulting cell-free supernatants were prepared for prostanoid measurements with GC-MS/MS. Data are given as median values, indicated by the filled circle, and single case determinations. A, HPS (n = 6); B, control (n = 6).
Monocytes purified from HPS patients under indomethacin treatment showed only marginal levels of PGE2, PGD2, and TXB2 formation after ionophore stimulation. At 72 h after the last application of indomethacin, the in vivo indomethacin concentration had decreased to undetectable levels, and the urinary as well as the systemic PGE-M concentration reached untreated levels as shown above (see Fig. 1A and Table 1). Monocytes prepared under this condition produced TXB2 as a major product (median, 205.5 pg/105 cells) compared with PGE2 (median, 16.2 pg/105 cells) and PGD2 (median, 18.4 pg/105 cells) (Fig. 2A). A relative distribution of all five metabolites measured could be referred to as follows: TXB2 >> PGE2 = PGD2 > PGF2α > 6-keto-PGF1α. There was no statistically significant difference between values for prostanoid metabolites from stimulated CD14+ monocytes of HPS patients versus controls. Moreover, there was no difference in the sum of all prostanoids produced by monocytes between the HPS patients and control subjects with or without indomethacin treatment and also the amounts of prostanoids in the flow-through fraction containing CD14- cells, including lymphocytes and platelets, were not significantly different between the two groups (data not shown).
To exclude that the cyclooxygenase activity was rate-limiting and thereby restricting the formation of PGE2, we analyzed the PGE2-synthetic activity in monocytes by the addition of the substrate PGH2. The exogenous addition of PGH2 circumvents the cyclooxygenase step. However, no difference was observed in such stimulated monocytes of HPS patients compared with controls (Table 2).
Reverse transcriptase-PCR analysis of cyclooxygenase-1, cyclooxygenase-2, and TX synthase mRNA from CD14+monocytes. To exclude differences in expression that might be important for stimulatory conditions not mimicked by A23187 mRNA expression of the enzymes involved was studied. For this purpose RT-PCR analysis using target-specific reverse-transcribed mRNA purified from CD14+ monocytes of HPS patients and controls was performed to compare cyclooxygenase-1, cyclooxygenase-2, and TX synthase expression (Fig. 3A). The amplification of the specific fragments was normalized on the expression of β-actin, used as an internal standard. The data obtained give evidence that cyclooxygenase-1 and TX synthase mRNA were constitutively expressed at similar expression rates in monocytes from HPS and control volunteers (Fig. 3B). Interestingly, the transcription of the cyclooxygenase-2 in CD14+ monocytes from HPS and control persons was also detectable at a lower level in both study groups, but with no significant difference. With the administration of indomethacin, expression of cyclooxygenase-1, cyclooxygenase-2, and TX synthase mRNA in CD14+ monocytes from both study groups was not changed (data not shown).

RT-PCR of cyclooxygenase-1, cyclooxygenase-2, and TX synthase in CD14+ monocytes from HPS vs control subjects. CD14+ monocytes were extracted for total RNA, and 1 μg was subjected to reverse transcription (RT) with specific primer sets for cyclooxygenase-1, cyclooxygenase-2, and TX synthase together with β-actin as internal control. A, a representative presentation of the PCR fragments from one patient and one control person (COX, cyclooxygenase;TXS, TX synthase); B, graphic presentation of the amplification products normalized on β-actin (n = 6 for both groups).
DISCUSSION
Most clinical features observed in patients suffering from HPS are thought to be a result of the biologic activity of renal and/or extrarenal PGE2. Inhibition of tubular reabsorption of salt and free water, prevention of hypertension despite high plasma renin activity, mediation of fever, secretory diarrhea, vomiting, osteolysis, and hypercalciuria may be consequences of an excessive PGE2 formation(6, 18). This is supported by the effective therapy with indomethacin, an inhibitor of the prostanoid-synthesizing pathway. The targets of this drug are the enzymes cyclooxygenase-1 and -2; the former is suggested to be constitutively expressed, whereas the latter can be induced by different stimuli such as growth factors, oncogenes, or cytokines and is referred to as an inflammatory response gene(9). However, different recent studies point also toward a basal expression of this enzyme in specific cell types, thereby indicating an engagement of cyclooxygenase-2 in functional tasks(19, 20). A possible hypothesis to explain the altered prostanoid profile in HPS patients was therefore the assumption of an alteration in the biosynthesis system,e.g. an increase in the PGE2-synthesizing ability.
Both PGE2 and PGE-M are the main prostanoids in HPS. In contrast to this observation, isolated monocytes synthesized mainly TXA2 and, more important, no difference in the product pattern was observed between HPS patients and control subjects. A similar observation was made on the genetic level with a comparable expression of the relevant mRNA species for cyclooxygenase and TX synthase. Noteworthy, the application of indomethacin was without any effect on mRNA expression.
Although the precise nature of PGE2 synthase is not clarified and therefore is beyond our investigation on the genetic level, we assume that also at this step no change has to be expected, concluding from the stimulatory experiments with PGH2. This led us to exclude that urinary excretion of PGE2 and PGE-M after withdrawal of indomethacin treatment in children with HPS is the result of a genetic alteration in the expression of prostanoid enzymes. Therefore we speculate that continuous stimulation of the PGE2-synthetic system in specific cell types of organs, including in kidney, must be responsible for the high systemic and renal PGE2 production. The nature of this stimulus, e.g. cytokines, other arachidonic acid metabolites, or defective proteins, remains to be clarified. In line with this assumption is our observation of increased levels of secretory phospholipase A2 and cytokines in plasma of HPS patients (our unpublished observation).
Very recently, mutations in the NKCC2 gene(21), encoding the renal bumetanide-sensitive Na/K/2Cl cotransporter, and in the ROMK gene(22), encoding the inwardly rectifying K+ channel of the thick ascending limb of Henle's loop, were shown to cause HPS in some, but not all, patients. Therefore, regardless of the precise mechanism, a possible explanation for the PGE2 formation in patients with HPS may be an isolated renal transport defect on the luminal epithelial surface of the thick ascending limb of Henle, somehow resulting in stimulation of cyclooxygenase-1 in tubular epithelial cells. Cyclooxygenase-1 is known to be expressed mainly in the cellular structure of the kidney(23), and PGE2 synthesized via this enzyme contributes to the regulation of salt and water reabsorption [for an overview, see Breyer and Badr(24)]. In this context it is worth mentioning that high PGE2 concentrations, which may be reached in local renal structures, can act positively on the expression of cyclooxygenase-2, as recently shown for mesangial cells(25). Therefore, continuous de novo synthesis of cyclooxygenase enzyme could explain the incessant ability to form PGE2, despite the fact that cyclooxygenase is a suicide enzyme(26), being self-inactivated during catalysis. Moreover, electrolyte channels and cotransporters are also distributed in organs other than the kidney, and this might thereby explain the extrarenal consequences in HPS.
In summary, from our data a genetic disorder in enzymes of the arachidonic acid metabolism seems to be an unlikely explanation for the high synthesis and turnover of PGE2 in HPS. At the moment we do not know which mechanism(s) connect(s) the stimulation of prostanoid biosynthesis to the mutational dysfunction of a channel or cotransporter protein defect in the thick ascending loop of Henle. Further studies are necessary to gain more insight into the functional relationship between the different pathophysiologic events.
Abbreviations
- CD:
-
cluster of differentiation
- GC-MS/MS:
-
gas chromatography-tandem mass spectrometry
- HPS:
-
hyperprostaglandin E syndrome
- PG:
-
prostaglandin
- PGE-M:
-
11α-hydroxy-9,15-dioxo-2,3,4,5,20-pentanor-19-carboxyprostanoic acid
- TX:
-
thromboxane
References
Seyberth HW, Königer SJ, Rascher W, Kühl PG, Schweer H 1987 Role of prostaglandins in hyperprostaglandine E syndrome and in selected renal tubular disorders. Pediatr Nephrol 1: 491–497.
Seidel C, Reinalter S, Seyberth HW, Schärer K 1995 Pre-pubertal growth in the hyperprostaglandin E syndrome. Pediatr Nephrol 9: 723–728.
Fanconi A, Schachenmann G, Nüssli R, Prader A 1971 Chronic hypokalaemia with growth retardation, normotensive hyperrenin-hyperaldosteronism (“Bartter's syndrome”), and hypercalciuria. Helv Paediatr Acta 2: 144–163.
McCredie DA, Rotenberg E, Williams AL 1974 Hypercalciuria in potassium-loosing nephropathy: a variant of Bartter's syndrome. Aust Paediatr J 10: 186–295.
Proesmans W, Devlieger H, Van Assche A, Eggermont E, Vandenberghe K, Lemmens F, Sieprath P, Lijnen P 1985 Bartter syndrome in two siblings-antenatal and neonatal observations. Int J Pediatr Nephrol 6: 63–70.
Seyberth HW, Rascher W, Schweer H, Kühl PG, Mehls O, Schärer K 1985 Congenital hypokalemia with hypercalciuria in preterm infants: a hyperprostaglandinuric tubular syndrome different from Bartter syndrome. J Pediatr 107: 694–701.
Dillon M, Shah V, Mitchell MD 1979 Bartter's syndrome: 10 cases in childhood. Q J Med 191: 429–446.
Proesmans W, Massa G, Vanderschueren-Lodeweyckx M 1988 Growth from birth to adulthood in a patient with the neonatal from of Bartter syndrome. Pediatr Nephrol 2: 205–209.
Otto JC, Smith WL 1995 Prostaglandin endoperoxide synthases-1 and -2. J Lipid Mediat Cell Signal 12: 139–156.
Rane A, Oelz O, Frölich JC, Seyberth HW, Sweetman BJ, Watson JT, Wilkinsom GR, Oates JA 1978 Relationship between plasma concentration by indomethacin and its effect on prostaglandin synthesis and platelet aggregation in man. Clin Pharmacol Ther 23: 658–668.
Nüsing R, Ullrich V 1992 Regulation of cyclooxygenase and thromboxane synthase in human monocytes. Eur J Biochem 206: 131–136.
Schweer H, Watzer B, Seyberth HW 1994 Determination of seven prostanoids in 1 mL of urine by gas chromatography-negative ion chemical ionization triple stage quadrupole mass spectrometry. J Chromatogr 652: 221–227.
Seyberth HW, Knapp G, Wolf D, Ulmer HE 1983 Introduction of plasma indomethacin level monitoring and evaluation of an effective threshold level in very low birth weight infants with symptomatic patent ductus arteriosus. Eur J Pediatr 141: 71–79.
Chomczynski P, Sacchi N 1987 Single-step method of RNA isolation by acid guanidine thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159.
Klein T, Nüsing RM, Pfeilschifter J, Ullrich V 1994 Selective inhibition of cyclooxygenase 2. Biochem Pharmacol 48: 1605–1610.
Nüsing RM, Mohr S, Ullrich V 1995 Activin A and retinoic acid synergize in cyclooxygenase-1 and thromboxane synthase induction during differentiation of J774.1 macrophages. Eur J Biochem 227: 130–136.
Schweer H, Kammer J, Kühl PG, Seyberth HW 1986 Determination of peripheral plasma prostanoid concentration: an unreliable index of “in vivo” prostanoid activity. Eur J Clin Pharmacol 31: 303–305.
Seyberth HW, Soergel M, Köckerling A 1997 Hypokalaemic tubular disorders: the hyperprostaglandin E syndrome and the Gitelman-Bartter syndrome. In: Davison AM, Cameron JS, Grünfeld J-P, Kerr DNS, Ritz E (eds) Oxford Textbook of Clinical Nephrology. Oxford University Press, Oxford, pp 1085–1094.
O'Neill GP, Ford-Hutchinson AW 1993 Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett 330: 156–160.
Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD 1994 Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest 94: 2504–2510.
Simon DB, Karet FE, Hamdan JM, Dipietro A, Sanjad SA, Lifton RP 1996 Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet 13: 183–188.
Simon DB, Karet FE, Rodriguezsoriano J, Hamdan JH, Dipietro A, Trachtman H, Sanjad SA, Lifton RP 1996 Genetic heterogeneity of Bartter's syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 14: 152–156.
Smith WL, Bell TG 1978 Immunohistochemical localization of the prostaglandinforming cyclooxygenase in renal cortex. Am J Physiol 235:F451–F457.
Breyer MD, Badr KF 1996 Arachidonic acid metabolites and the kidney. In: Brenner MB (ed) The Kidney. WB Saunders, Philadelphia, pp 754–788.
Nüsing RM, Klein T, Pfeilschifter J, Ullrich V 1996 Effect of cyclic AMP and prostaglandin E(2) on the induction of nitric oxide- and prostanoid-forming pathways in cultured rat mesangial cells. Biochem J 313: 617–623.
Markey CM, Alward A, Weller PE, Marnett LJ 1987 Quantitative studies of hydroperoxide reduction by prostaglandin H synthase. J Biol Chem 262: 6266–6279.
Acknowledgements
The authors thank S. Mahr and B. Watzer for technical assistance in the GC-MS/MS analysis and S. Noll for organization of the control group.
Author information
Authors and Affiliations
Additional information
Supported by grants of the Deutsche Forschungsgemeinschaft (Se 263/11-1, Nu 73/2-1) and by the Stiftung P.E. Kempkes.
Rights and permissions
About this article
Cite this article
Nüsing, R., Schaub, T., Klein, T. et al. Prostanoid Biosynthesis by Blood Monocytes of Children with Hyperprostaglandin E Syndrome. Pediatr Res 42, 241–246 (1997). https://doi.org/10.1203/00006450-199708000-00019
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199708000-00019
This article is cited by
-
Pathophysiology and clinical presentations of salt-losing tubulopathies
Pediatric Nephrology (2016)
