
Abstract
In a genetic mouse model of human phenylketonuria we have examined the offspring of hyperphenylalaninemic mothers for the presence of cardiovascular defects, an important feature of the pathology of the human maternal phenylketonuria syndrome. Beginning at 14.5 d after conception (75% through gestation), a variety of cardiovascular defects became apparent among the progeny of the hyperphenylalaninemic females. These defects ranged from mild to serious and correlated with the maternal but not the fetal Pah genotype. Nearly all of the defects were vascular, however, whereas the most reported in humans so far have been cardiac. The predisposing biochemical condition in this mouse disease model seems to be the same as in the human disease; elevated maternal blood phenylalanine levels concentrated across the placental barrier to produce a teratogenic developmental environment. This model for congenital cardiovascular defects should enhance two related areas of research. 1) It should allow a more thorough investigation of the relationship between maternal diet and maternal phenylketonuria birth defects, and 2) it should provide an experimental tool to gain insight into the normal process of cardiovascular development.
Similar content being viewed by others

Main
The inborn error of metabolism PKU (McKusick no. 261600) was among the very first demonstrated to have a heritable component(1, 2). The investigation and characterization of this disease in the intervening years has been punctuated by discoveries of related syndromes [for a recent review, see Scriver et al.(3)]. Among these is a syndrome called maternal PKU in which a pregnant PKU woman on unrestricted dietary PHE intake is at very high risk of having a child with severe birth defects(4–6). Among these are mental retardation, microcephaly, intrauterine growth retardation, congenital heart defects, and dysmorphic facies(4, 7, 8). Although this phenomenon is not yet well characterized, it is suggested that PHE and/or its metabolites function as direct-acting teratogens(9–14).
This maternal syndrome of PKU is a growing problem because the effective treatment of PKU patients through early limitation of dietary PHE has led to a sizable population of PKU women, many of whom are no longer in strict compliance with dietary PHE limitation(15, 16). Strikingly, the fetus at risk is typically heterozygous for the PKU mutation, a genotype that, by itself, is not associated with a disease phenotype.
Efforts to produce animal model systems with which to study human maternal PKU have yielded some insights into the disease process(9, 17). However, the causative factors of these model systems are sufficiently different from the human disease syndrome that questions remain regarding the accuracy of the observed effects compared with human maternal PKU.
With high efficiency chemical mutagenesis, six different mutant mouse lines have been produced that share a common feature: a heritable inability to metabolize PHE efficiently(18–20). Among these are two lines, Pahenu2 and Pahenu3, that are very similar to human classical PKU in both initial genetic cause and ultimate phenotypic effects. These lines exhibit a deficiency in the same enzyme that is lacking in human PKU, they show equivalent levels of hyperphenylalaninemia, and they are mutated at the same genetic locus. More importantly, this deficiency confers hypopigmentation and neurologic phenotypes that appear similar to human PKU(20). In addition to the similarities already reported, we now report the incidence of birth defects among offspring to hyperphenylalaninemic Pahenu2 mutant females, which suggests that this mutant line will provide a helpful model for the human maternal PKU syndrome.
METHODS
Production of fetuses for examination. All animal manipulations have been approved by an Institutional Review Board, and all animals were obtained from a colony maintained by one of the authors (J.D.M.).
Homozygous mutant females were maintained on laboratory chows containing typical amounts of protein [Richmond Standard 5001 and 5015 (PMI Feeds, St. Louis, MO) and Prolab 2000 (Agway, Ithaca, NY) laboratory mouse diets have 23, 17, and 18% crude protein, respectively]. All of these laboratory chows produce blood PHE levels similar to those reported previously for the Pahenu2 line(20) (data not shown). While being maintained on these diets, these females were caged with males of varying genotype. In most cases, females were checked daily for the presence of a copulatory plug. Noon on the day of the plug was designated 0.5 DPC. In some cases, the plug was not detected, but fetuses were collected shortly before birth and their gestational stage was estimated by appearance.
Pregnant females were killed, and the fetuses were excised from the uterus and separated from the placenta and embryonic membranes. In some cases, Pelikan India ink was injected into the heart using a pulled capillary tube to provide additional contrast with surrounding tissues. The pipette was inserted into the umbilical vein or directly into the left ventricle, and the ink was ejected by gentle positive pressure until it filled all of the visible vasculature. Embryos were immersion-fixed in neutral buffered formalin overnight, dehydrated in 95% and 100% ethanol baths, and cleared in methyl salicylate:benzyl benzoate (50:50). Some dissection was done after clearing to observe the great arteries and their branches.
Determination of maternal and fetal blood PHE. Mutant females were maintained on the Richland Standard 5001 diet from before conception and were killed when their litters were near term. A blood sample was obtained from the mother immediately, then the individual fetuses were dissected from the uterine lining and exsanguinated to recover a fetal blood sample. Blood samples were centrifuged to yield plasma, and PHE levels were determined using the spectrofluorometric method of McCaman and Robins(21).
RESULTS
A total of 15 fetuses at 19.5 DPC from wild type mothers were analyzed as controls for the background incidence of heart defects. No abnormalities were noted.
Two litters of 11 fetuses each from mutant females were collected shortly after midgestation. The first was collected at 11.5 DPC and the second at 12.5 DPC. None of these showed abnormalities in patterning of the great arteries(Table 1).
By 14.5 to 15.5 DPC, some of the fetuses showed variation in the patterning of the great arteries. In one litter with 12 fetuses collected from a mutant female at 14.5 DPC, 5 of 12 had notable cardiovascular patterning defects. In one, the left carotid artery was displaced leftward, whereas in another the left carotid branch arose from the brachiocephalic artery rather than independently from the aorta. One had a simple hypoplastic aorta, whereas a second with hypoplastic aorta had an enlarged pulmonary trunk and ductus, and enlarged vertebral and left subclavian arteries at their origins from the aorta. The variations ranged from minor to severe, hence several showed these minor discrepancies, whereas one fetus in the litter showed a double outlet right ventricle with normal patterning of the great arteries. In a second such litter of 11 fetuses, only three were abnormal. Two were discolored, indicating suboptimal cardiovascular function but no overt abnormalities in cardiovascular patterning, whereas the third had a small pulmonary trunk.
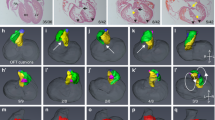
At later stages of development, similar variations were found. At 17.5 DPC, 19 fetuses were examined from two litters: seven showed detectable cardiovascular abnormalities. At 18.5 DPC, 29 fetuses were examined, and cardiovascular alterations were detected in nine. Finally, at 19.5 DPC, among 24 fetuses, cardiovascular abnormalities were detected in 10. Again, great variability was observed, and each pattern was unique in that individual. Some malformations were minor, and a few displayed major abnormalities. A single fetus had interrupted aorta, and a littermate showed transposition of the great arteries. Another fetus lacked a left lung and the heart was displaced to the left but showed no other cardiovascular defects. Photomicrographs displaying several representative cardiovascular defects from this study are shown in Figure 1.

Panels a, b, and c show the great arteries derived for aortic arches 3 (brachiocephalic, B; and carotid, LC, RC) and 4 (arch of the aorta, Ao), and the 7th intersegmental (subclavian, (*) asterisk). In panel a the normal pattern can be seen. Note the bifurcation of the brachiocephalic arch in to the right carotid and right subclavian. This branch point was frequently misplaced as can be seen in panel b. Panel c shows the ultimate displacement as the right subclavian does not branch from the brachiocephalic, but is retroesophageal and can be seen appearing from behind the esophagus. Panels d and e illustrate a normal outflow tract in comparison with pulmonary stenosis, respectively. The pulmonary trunks and ductus arteriosis are indicated by the arrow.
Two other features of mouse maternal PKU pathology emerge from this data set. Although examination of test fetuses at the earliest gestational time points revealed no defects, at later points the incidence of cardiovascular malformations ranged from 27 to 46% (Table 2). For all time points, this is remarkably greater than the 12-15% incidence of congenital heart defects reported for human maternal PKU offspring(4).
As in human maternal PKU, neither the paternal nor (by extension) the fetal genotype seems to be important in determining the incidence of cardiovascular malformations. Regardless of the paternal genotype, the incidence of cardiovascular defects among fetuses of at least 14.5 DPC stays relatively constant (Table 3).
Many of the defects noted (i.e. aberrations in patterning of the great arteries) were of a type that would not be apparent before 12.5 DPC, because the system is still remodeling to its final phenotype. Primary heart defects were not common but were observed occasionally. However, cardiovascular function was obviously compromised in a number of fetuses because of their poor color. The nature of this dysfunction was not immediately apparent but may be due to a primary myocardial defect.
Mutant mouse mothers concentrate PHE across the placental barrier about 2-fold (Table 4).
DISCUSSION
These early findings strongly suggest that, as in human PKU, there is an important maternal component to mouse PKU. However, the spectrum of heart defects seen in these fetal mice appears somewhat distinct from those seen in human maternal PKU offspring. Although these mouse fetuses are examined in such a way that both cardiac and vascular lesions would likely have been detected, most of the fetal mouse heart defects were vascular, whereas most human maternal PKU heart defects so `far reported are cardiac. It is difficult at present to say whether this observed difference is apparent or real, because human infants and our mouse fetuses were subjected to such different means of cardiovascular assessment. For this study, the cardiovascular system of the mouse fetuses was injected with ink followed by histologic clearing of all soft tissues to reveal the entire cardiovascular system in whole mount. Such analysis can reveal even very subtle constrictions, dilations, and defects in patterning. In contrast, human infants are subjected to much less invasive assessment techniques. Further analysis of both the human and mouse syndromes will be needed to determine the basis of these observed differences.
Like human PKU mothers, mutant mouse mothers concentrate their already greatly elevated levels of PHE across the placental barrier. The concentration gradients seen in this mutant lie in the upper reaches of the range of values that have been reported for humans [reviewed in Hanley et al.(6)]. Because the mother's blood PHE levels also exceed the 1200 μM level accepted as an indicator of classical human PKU, it seems likely that the defects arising in this disease model share the same etiology as human maternal PKU.
The need for animal models of human maternal PKU has led to a considerable body of previous research aimed at mimicking this disease syndrome. Such models have been produced in two general ways: 1) producing maternal hyperphenylalaninemia by administering PHE with inhibitors of PHE catabolism(22, 23) and 2) culturing mouse or rat fetuses in the presence of PHE or its metabolites(13, 24). This research has led to some important insights into the pathology associated with maternal PKU. However, there are notable shortcomings inherent in these methods that have hampered efforts to draw clear conclusions from studies involving such models. In the case of offspring born to females treated with PHE catabolism inhibitors, there are elevated levels of tyrosine subsequently induced that are dissimilar from that in the PKU disease syndrome. Further uncertainties arise regarding the contribution of unrelated side effects induced by the inhibitors. In the case of cultured fetuses, uncertainties arise due to the artificial nature of the culture conditions into which the fetus has been placed. It is well known that the placental barrier plays an important role in determining the composition of the milieu in which gestation occurs. By removing this barrier and exposing the fetuses directly to the culture medium, they may be exposed to atypical metabolites or atypical concentrations of metabolites. By reproducing more closely the actual human maternal PKU condition, we have excluded many of the uncertainties of former techniques.
The production of genetic mouse models of PKU should permit rigorous experimentation by providing the following experimental advantages. 1) Test and control animals that are genetically identical in every respect except the PKU mutation can be carried through each experiment and compared to reveal the effects of the mutation. 2) The environmental conditions, in particular the diet, can be completely controlled and easily manipulated to evaluate their contribution to the incidence of birth defects. 3) Large numbers of mice can easily be produced to provide statistically strong evidence for the relationship between maternal diet and fetal birth defects.
Owing to the comprehensive characterization of mouse embryonic and fetal development, this disease model should also provide a valuable experimental tool for the examination of basic features of cardiovascular development. Because of a long history of the study of mouse embryonic and fetal development, mouse cardiovascular development has been examined descriptively to very high resolution(25). Further, it has been demonstrated in the chick embryo that important anatomical features of the developing cardiovascular system are dependent on the influx and normal function of a special neural crest cell population(26–29). This PKU mouse has cardiovascular abnormalities that resemble those in the chick produced by ablation of the premigratory neural crest. Thus the model may be useful for defining the connection between cardiovascular anomalies and the correct migration and/or function of neural crest cells.
Abbreviations
- PKU:
-
phenylketonuria
- PHE:
-
phenylalanine
- DPC:
-
days postcoitum
References
Folling A 1934 Uber Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbingdung mit Imbezillitat. Hoppe-Seylers Z Physiol Chem 227: 169–176.
Penrose LS 1935 Inheritance of phenylpyruvic amentia(phenylketonuria). Lancet 2: 192–194.
Scriver CR, Eisensmith RC, Woo SLC, Kaufman S 1994 The hyperphenylalaninemias of man and mouse. Annu Rev Genet 28: 141–165.
Lenke RR, Levy HL 1980 Maternal phenylketonuria and hyperphenylalaninemia. N Engl J Med 303: 1202–1208.
Levy H, Waisbren S 1983 Effects of untreated maternal phenylketonuria and hyperphenylalaninemia on the fetus. N Engl J Med 309: 1269–1274.
Hanley WB, Clarke JTR, Schoonheyt W 1987 Maternal phenylketonuria (PKU)-a review. Clin Biochem 20: 149–156.
Lipson A, Beuhler B, Bartely J, Walsh D, Yu J, O'Halloran M, Webster W 1984 Maternal hyperphenylalaninemia fetal effects. J Pediatr 104: 216–220.
Rouse B, Lockhart L, Matalon R, Azen C, Koch R, Hanley W, Levy H, Dela Cruz F, Friedman E 1990 Maternal phenylketonuria pregnancy outcome: a preliminary report of facial dysmorphology and major malformations. J Inherit Metab Dis 13: 289–291.
Loo YH, Rabe A, Potempska A, Wang P, Fersko R, Wisniewski HM 1984 Experimental maternal phenylketonuria: an examination of two animal models. Dev Neurosci 6: 227–234.
Tuchman M, Fisch RO, Ramnaraine ML, Krivit W 1984 Acidic metabolites of phenylalanine in plasma of phenylketonurics. Biochem Med 34: 203–206.
Michals K, Lopus M, Gashkoff P, Matalon R 1986 Phenylalanine metabolites in treated phenylketonuria children. J Inherit Metab Dis 9: 212–214.
Kaufman S 1989 An evaluation of the possible neurotoxicity of metabolites of phenylalanine. J Pediatr 114: 895–900.
Denno KM, Sadler TW 1990 Phenylalanine and its metabolites induce embryopathies in mouse embryos in culture. Teratology 42: 565–570.
Dorland L, Poll-The BT, Duran M, Smeitink JAM, Berger R 1993 Phenylpyruvate, fetal damage, and maternal phenylketonuria. Lancet 341: 1351–1352.
Schuett VE, Gurda RF, Brown ES 1980 Diet discontinuation policies and practices of PKU clinics in the United States. Am J Public Health 70: 498–503.
Waisbren SE, Hamilton BD, St. James PJ, Shiloh S, Levy HL 1995 Psychosocial factors in maternal phenylketonuria: women's adherence to medical recommendations. Am J Public Health 85: 1636–1644.
Roux C, Rey F, Lyonnet S, Nizard S, Mulliez N, Munnich A 1991 An animal model for maternal phenylketonuria. J Med Genet 28: 718–719.
Bode V, McDonald JD, Guenet JL, Simon D . 1988 hph-1: a mouse mutant with hereditary hyperphenylalaninemia induced by ethylnitrosourea mutagenesis. Genetics 118: 299–305.
McDonald JD, Bode VC, Dove WF, Shedlovsky A 1990 Pahhph-5: a mouse mutant deficient in phenylalanine hydroxylase. Proc. Natl Acad Sci USA 87: 1965–1967.
Shedlovsky A, McDonald JD, Symula D, Dove WF 1993 Mouse models for human PKU. Genetics 134: 1205–1210.
McCaman MW, Robins E 1962 Fluorometric method for the determination of phenylalanine in serum. J Lab Clin Med 59: 885–889.
Lipton MA, Gordon R, Guroff G, Udenfriend S 1967 p- Chlorophenylalanine-induced chemical manifestations of phenylketonuria in rats. Science 156: 248–250.
Delvalle JA, Dienel G, Greengard O 1978 Comparison ofα-methylphenylalanine as inducers of chronic hyperphenylalaninemia in developing rats. Biochem J 170: 449–459.
Roux C, Rey F, Lyonnet S, Nizard S, Mulliez N, Munnich A 1991 An animal model for maternal phenylketonuria. J Med Genet 28: 718–719.
Kaufman MH 1992 The Atlas of Mouse Development. Academic Press, London
Kirby ML, Gale TF, Stewart DE 1983 Neural crest cells contribute to aorticopulmonary septation. Science 220: 1059–1061.
Nishibatake M, Kirby ML, Van Mierop LH 1987 Pathogenesis of persistent truncus arteriosis and dextroposed aorta in the chick embryo after neural crest ablation. Circulation 75: 255–264.
Bockman DE, Redmond ME, Kirby ML 1989 Alteration of early vascular development after ablation of cranial neural crest. Anat Rec 225: 209–217.
Kirby ML, Waldo KL 1995 Neural crest and cardiovascular patterning. Circ Res 77: 211–215.
Acknowledgements
The authors thank Kerry Bulla for expert technical assistance.
Author information
Authors and Affiliations
Additional information
Supported in part by the National Science Foundation Grant EPS-9550487. Additional support came from the Wesley Medical Research Foundation, Wichita, KS.
Rights and permissions
About this article
Cite this article
McDonald, J., Dyer, C., Gailis, L. et al. Cardiovascular Defects among the Progeny of Mouse Phenylketonuria Females. Pediatr Res 42, 103–107 (1997). https://doi.org/10.1203/00006450-199707000-00016
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199707000-00016
